
The Development of Plant Genome Sequencing Technology and Its Conservation and Application in Endangered Gymnosperms
Abstract
:1. Introduction
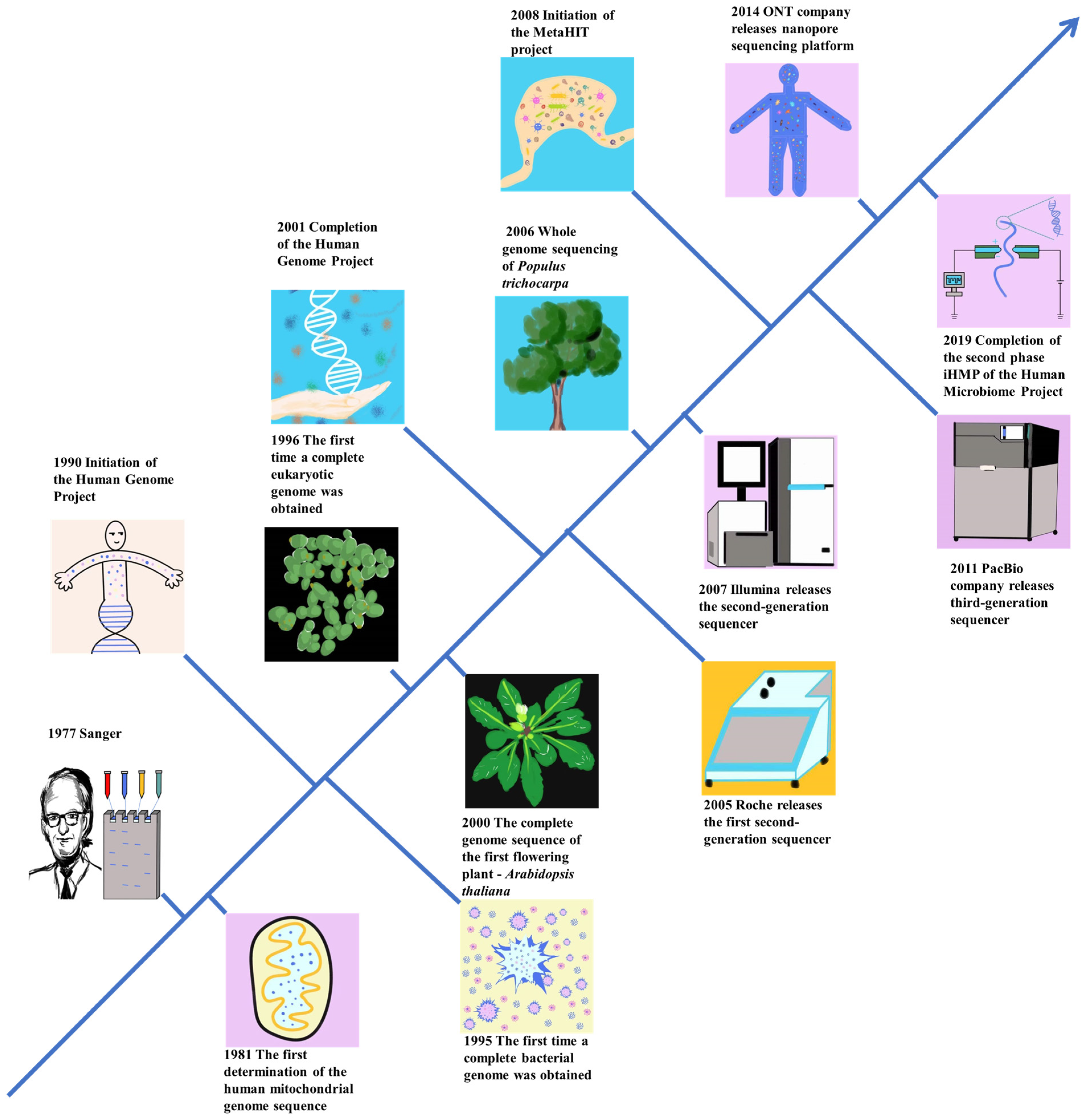
2. Advancements in Genome Sequencing Technology and Its Evolution
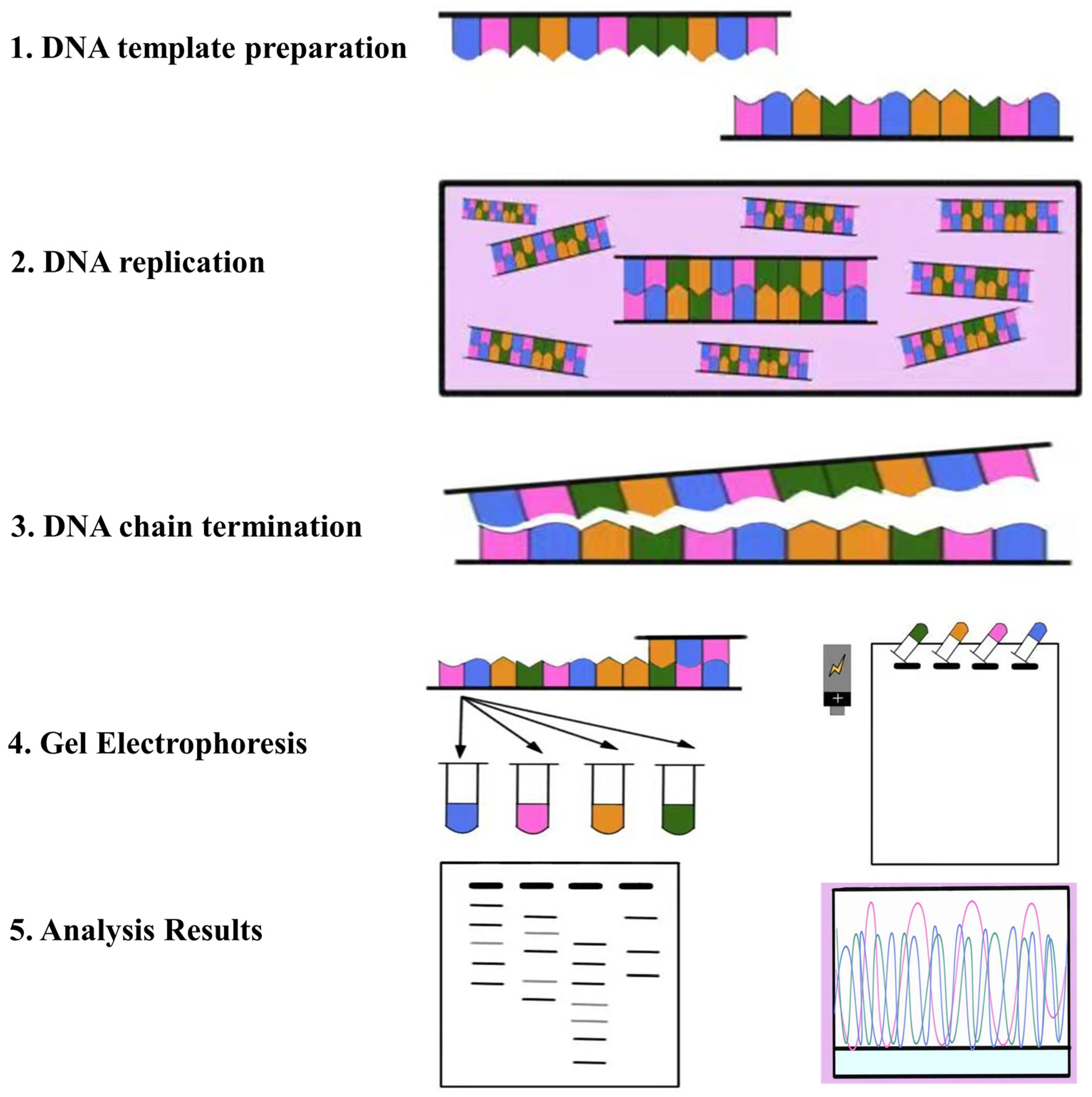
2.1. Pioneering Generation Genome Sequencing Technology
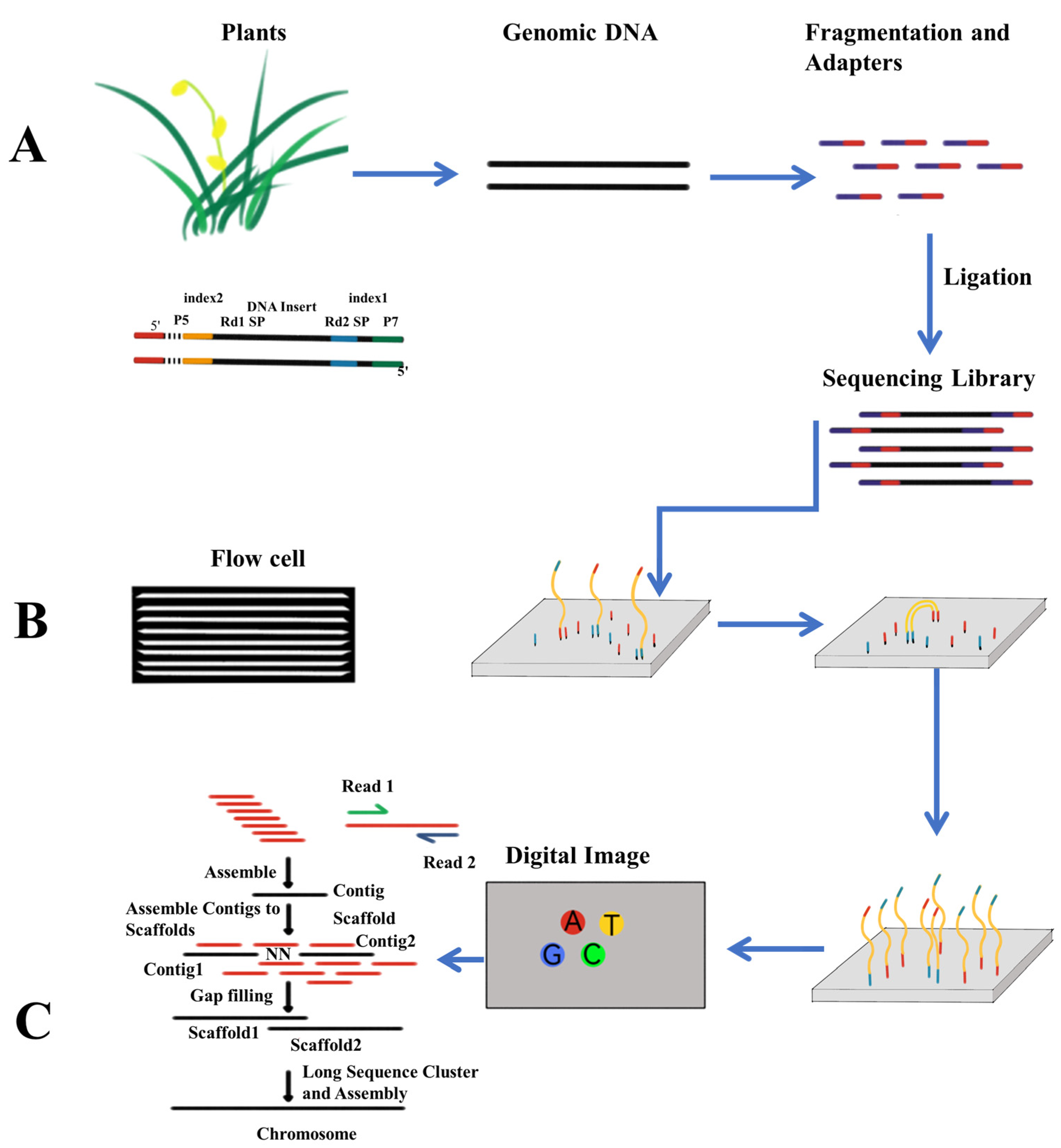
2.2. Next Generation Sequencing
2.2.1. Roche 454 Sequencing Technology
2.2.2. Illumina Sequencing and Solexa Technology
2.2.3. ABI SOLiD Sequencing Technology
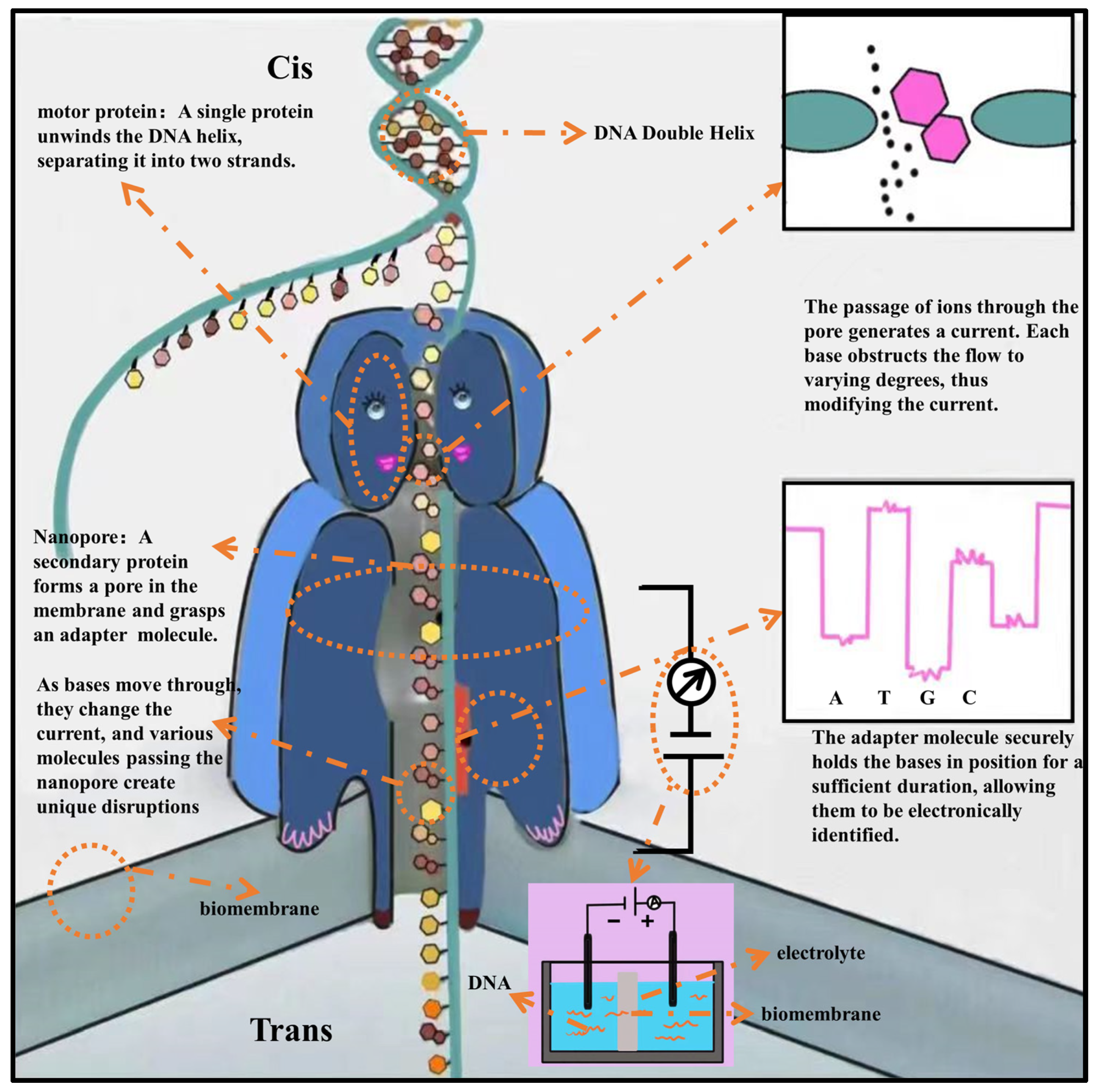
2.3. Third−Generation Genome Sequencing Technology (TGS)
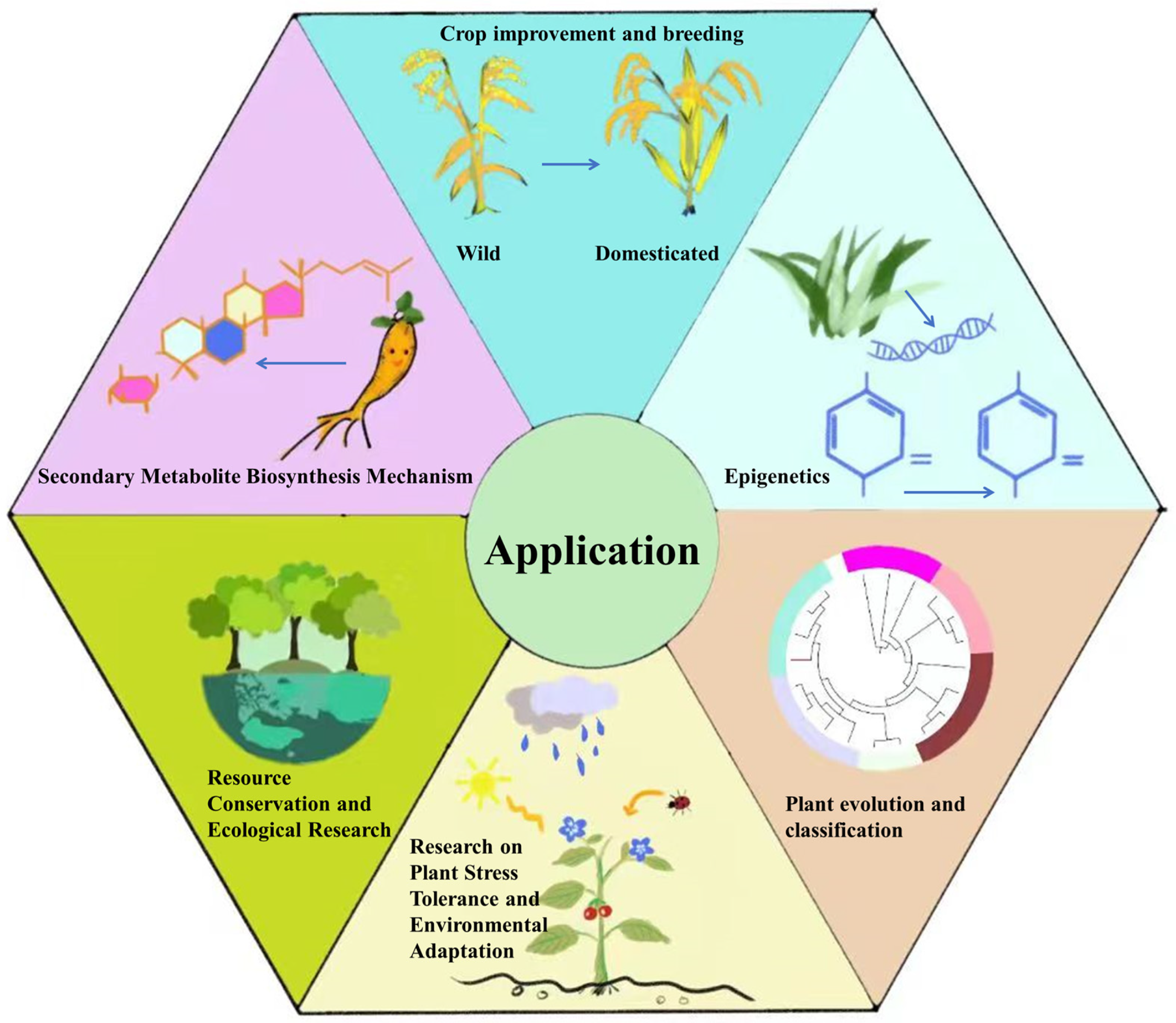
3. Applications of Genome Sequencing Technology
3.1. Enhancing Crop Quality through Molecular Breeding
3.2. Exploration of Epigenetic Regulatory Mechanisms
3.3. Evolutionary Analysis of the Origin of Species
3.4. Biodiversity
3.5. Abiotic Stress and Biotic Stress
3.5.1. Genes/QTL and Plant Stress
3.5.2. DNA Methylation and Plant Stress
3.6. The Synthesis of Secondary Metabolites in Plants
3.7. Conservation of Endangered Gymnosperms
3.7.1. Current Status of Gene Sequencing in Gymnosperms
3.7.2. Gene Sequencing Technology and Endangered Gymnosperms
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, K.; Wen, S.; Yang, D.; Gao, J.; Wang, Z.; Zhu, P.; Bie, Z.; Cheng, J. Phloem unloading in cultivated melon fruits follows an apoplasmic pathway during enlargement and ripening. Hortic. Res. 2023, 10, uhad123. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xu, Z.; Fan, R.; Wang, G.; Wang, F.; Qin, X.; Yan, L.; Ji, X.; Meng, M.; Sim, S. The complex genome and adaptive evolution of polyploid Chinese pepper (Zanthoxylum armatum and Zanthoxylum bungeanum). Plant Biotechnol. J. 2023, 21, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Jin, M.; Li, Z.; Li, H.; Zhang, L.; Yu, S.; Zhang, Z.; Fan, R.; Liu, J.; Xu, Q. Population Genomics Provide Insights into the Evolution and Adaptation of the Asia Corn Borer. Mol. Biol. Evol. 2023, 40, msad112. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; He, Q.; Wang, J.; Wang, B.; Zhao, J.; Huang, S.; Yang, T.; Tang, Y.; Yang, S.; Aisimutuola, P. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 2023, 55, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Soltis, D.E.; Kersey, P.J.; Wegrzyn, J.L.; Leebens-Mack, J.H.; Gostel, M.R.; Liu, X.; Soltis, P.S. Green plant genomes: What we know in an era of rapidly expanding opportunities. Proc. Natl. Acad. Sci. USA 2022, 119, e2115640118. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Gong, Y.; Liu, Z.; Zhou, Y.; Dai, C.; Wang, Q. Evolution of complex genome architecture in gymnosperms. GigaScience 2022, 11, giac078. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Song, L.; Li, X.; Zi, H.; Chen, W.; Gao, Y.; Zheng, S.; Fei, Z.; Sun, X.; Wu, J. The Torreya grandis genome illuminates the origin and evolution of gymnosperm-specific sciadonic acid biosynthesis. Nat. Commun. 2023, 14, 1315. [Google Scholar] [CrossRef]
- Sheldon, N.D.; Smith, S.Y.; Stein, R.; Ng, M. Carbon isotope ecology of gymnosperms and implications for paleoclimatic and paleoecological studies. Glob. Planet. Chang. 2020, 184, 103060. [Google Scholar] [CrossRef]
- Dror, D.; Klein, T. The effect of elevated CO2 on aboveground and belowground carbon allocation and eco-physiology of four species of angiosperm and gymnosperm forest trees. Tree Physiol. 2022, 42, 831–847. [Google Scholar] [CrossRef]
- Van Konijnenburg-van Cittert, J.H.; Pott, C.; Schmeißner, S.; Dütsch, G.; Kustatscher, E. The Rhaetian flora of Wüstenwelsberg, Bavaria, Germany: Description of selected gymnosperms (Ginkgoales, Cycadales, Coniferales) together with an ecological assessment of the locally prevailing vegetation. Rev. Palaeobot. Palynol. 2021, 288, 104398. [Google Scholar] [CrossRef]
- Subedi, S.C.; Bhattarai, K.R.; Perez, T.M.; Sah, J.P. Gymnosperm species richness patterns along the elevational gradient and its comparison with other plant taxonomic groups in the Himalayas. Front. Biogeogr. 2020, 12, e44232. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Bae, E.-K.; Tran, T.N.A.; Lee, H.; Ko, J.-H. Exploring the Seasonal Dynamics and Molecular Mechanism of Wood Formation in Gymnosperm Trees. Int. J. Mol. Sci. 2023, 24, 8624. [Google Scholar] [CrossRef] [PubMed]
- Swor, K.; Satyal, P.; Poudel, A.; Setzer, W.N. Gymnosperms of Idaho: Chemical Compositions and Enantiomeric Distributions of Essential Oils of Abies lasiocarpa, Picea engelmannii, Pinus contorta, Pseudotsuga menziesii, and Thuja plicata. Molecules 2023, 28, 2477. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Kirkness, E.F. Whole genome sequencing. In Genetic Variation: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2010; pp. 215–226. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- McElhinney, L.M.; Marston, D.A.; Ellis, R.J.; Freuling, C.M.; Müller, T.F.; Fooks, A.R. Sanger Sequencing of Lyssaviruses. In Current Laboratory Techniques in Rabies Diagnosis, Research and Prevention; Academic Press: Cambridge, MA, USA, 2014; pp. 159–170. [Google Scholar] [CrossRef]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger sequencing and molecular assay monitoring. J. Vet. Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef]
- Metzker, M.L. Emerging technologies in DNA sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef]
- Blazej, R.G.; Kumaresan, P.; Mathies, R.A. Microfabricated bioprocessor for integrated nanoliter-scale Sanger DNA sequencing. Proc. Natl. Acad. Sci. USA 2006, 103, 7240–7245. [Google Scholar] [CrossRef]
- Muzzey, D.; Kash, S.; Johnson, J.I.; Melroy, L.M.; Kaleta, P.; Pierce, K.A.; Ready, K.; Kang, H.P.; Haas, K.R. Software-assisted manual review of clinical next-generation sequencing data: An alternative to routine Sanger sequencing confirmation with equivalent results in >15,000 germline DNA screens. J. Mol. Diagn. 2019, 21, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Menon, S. Comparison of High-Throughput Next generation sequencing data processing pipelines. Int. Res. J. Mod. Eng. Technol. Sci. (IRJMETS) 2021, 3, 125–136. [Google Scholar]
- Saeed, M.; Jamil, Z.; Shehzad, T.; ul Hasan, S.Z.; Bibi, R.; Malik, S.N.; Matee-ur-Rehman, H.; Ahmed, R. Role of Next Generation Sequencing (NGS) in Plant Disease Management: A Review. J. Appl. Res. Plant Sci. 2023, 4, 512–517. [Google Scholar] [CrossRef]
- Cheng, C.; Fei, Z.; Xiao, P. Methods to improve the accuracy of next-generation sequencing. Front. Bioeng. Biotechnol. 2023, 11, 982111. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Yeh, Y.-C.; Wang, L.-C.; Lin, Y.-Y.; Lin, S.-Y.; Wang, S.-Y.; Chu, P.-Y.; Liu, Z.-Y.; Su, Y.-C.; Ho, H.-L. Genomic Profiling With Large-Scale Next-Generation Sequencing Panels Distinguishes Separate Primary Lung Adenocarcinomas From Intrapulmonary Metastases. Mod. Pathol. 2023, 36, 100047. [Google Scholar] [CrossRef] [PubMed]
- Heydari, A.A.; Sindi, S.S. Deep learning in spatial transcriptomics: Learning from the next next-generation sequencing. Biophys. Rev. 2023, 4, 11306. [Google Scholar] [CrossRef]
- Xu, Z.; Yan, S.; Yuan, S.; Wu, C.; Chen, S.; Guo, Z.; Li, Y. Efficient Two-Stage Analysis for Complex Trait Association with Arbitrary Depth Sequencing Data. Stats 2023, 6, 468–481. [Google Scholar] [CrossRef]
- Hassan, S.; Bahar, R.; Johan, M.F.; Mohamed Hashim, E.K.; Abdullah, W.Z.; Esa, E.; Abdul Hamid, F.S.; Zulkafli, Z. Next-generation sequencing (NGS) and third-generation sequencing (TGS) for the diagnosis of thalassemia. Diagnostics 2023, 13, 373. [Google Scholar] [CrossRef]
- Thun, G.A.; Gueuning, M.; Mattle-Greminger, M. Long-read sequencing in blood group genetics. Transfus. Med. Hemother. 2023, 50, 184–197. [Google Scholar] [CrossRef]
- Wang, D.; Cheng, J.; Peng, L.; Li, Y. Comparison of Metagenomic Second-and Third-Generation Sequencing by Diagnostic Sensitivity and Specificity in Tuberculosis Patients. Clin. Lab. 2023, 69, 1831–1837. [Google Scholar] [CrossRef]
- Zhuang, J.; Chen, C.; Fu, W.; Wang, Y.; Zhuang, Q.; Lu, Y.; Xie, T.; Xu, R.; Zeng, S.; Jiang, Y. Third-Generation Sequencing as a New Comprehensive Technology for Identifying Rare α-and β-Globin Gene Variants in Thalassemia Alleles in the Chinese Population. Arch. Pathol. Lab. Med. 2023, 147, 208–214. [Google Scholar] [CrossRef]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genom. 2012, 11, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-generation sequencing: The spearhead towards the radical transformation of modern genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Razali, S.A.; Deris, Z.M.; Danish-Daniel, M.; Tan, M.P.; Nor, S.A.M.; Ma, H.; Min, W.; Yantao, L.; Asaduzzaman, M. Application of second-generation sequencing (SGS) and third generation sequencing (TGS) in aquaculture breeding program. Aquaculture 2022, 548, 737633. [Google Scholar] [CrossRef]
- Saini, M.K.; Gaurav, H.B.; Kumar, J.; Sanu, K. DNA Sequencing techniques: Sanger to Next Generation Sequencing. DNA 2023, 3, 2378–2393. [Google Scholar] [CrossRef]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. 2010, 12, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S. Sequencing nucleic acids: From chemistry to medicine. Chem. Commun. 2011, 47, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- McKernan, K.J.; Peckham, H.E.; Costa, G.L.; McLaughlin, S.F.; Fu, Y.; Tsung, E.F.; Clouser, C.R.; Duncan, C.; Ichikawa, J.K.; Lee, C.C. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009, 19, 1527–1541. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-generation sequencing in the clinical laboratory: Exploring the advantages and challenges of nanopore sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.; Gu, L.; Reddy, A.S. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2018, 19, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quinlan, A.R. Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics 2014, 30, 3399–3401. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, H.; Punchihewa, S.; Senanayake, A.; Hammond, J.M.; Stevanovski, I.; Ferguson, J.M.; Ragel, R.; Gamaarachchi, H.; Deveson, I.W. Genopo: A nanopore sequencing analysis toolkit for portable Android devices. Commun. Biol. 2020, 3, 538. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.M.; Smith, M.A. SquiggleKit: A toolkit for manipulating nanopore signal data. Bioinformatics 2019, 35, 5372–5373. [Google Scholar] [CrossRef] [PubMed]
- Pryszcz, L.P.; Novoa, E.M. ModPhred: An integrative toolkit for the analysis and storage of nanopore sequencing DNA and RNA modification data. Bioinformatics 2022, 38, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Tham, C.-Y.; Poon, L.; Yan, T.; Koh, J.Y.P.; Ramlee, M.K.; Teoh, V.S.I.; Zhang, S.; Cai, Y.; Hong, Z.; Lee, G.S. High-throughput telomere length measurement at nucleotide resolution using the PacBio high fidelity sequencing platform. Nat. Commun. 2023, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef]
- Coupland, P.; Chandra, T.; Quail, M.; Reik, W.; Swerdlow, H. Direct sequencing of small genomes on the Pacific Biosciences RS without library preparation. Biotechniques 2012, 53, 365–372. [Google Scholar] [CrossRef]
- Sikic, M. Facilitating genome structural variation analysis. Nat. Methods 2023, 20, 491–492. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef]
- Haque, F.; Li, J.; Wu, H.-C.; Liang, X.-J.; Guo, P. Solid-state and biological nanopore for real-time sensing of single chemical and sequencing of DNA. Nano Today 2013, 8, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Ying, Y.-L.; Cao, C.; He, P.; Long, Y.-T. Accurate data process for nanopore analysis. Anal. Chem. 2015, 87, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Schlotter, T.; Kloter, T.; Nakatsuka, N.; Aramesh, M.; Voros, J.; Zambelli, T. Interface nanopores as a flexible technology for next-generation single-molecule protein sensing. Biophys. J. 2022, 121, 541a. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Jain, C.; Cheng, H.; Au, K.F.; Li, H.; Aluru, S. Real-time mapping of nanopore raw signals. Bioinformatics 2021, 37, i477–i483. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Nanopore, O. Oxford Nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 2012, 30, 295. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Wyres, K.L.; Holt, K.E. Recovery of small plasmid sequences via Oxford Nanopore sequencing. Microb. Genom. 2021, 7, 000631. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, X.; Liu, H.; Tian, Y.; Lian, J.; Yang, R.; Hao, S.; Wang, X.; Yang, S.; Li, Q. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 2015, 8, 922–934. [Google Scholar] [CrossRef]
- Xu, Z.; Pu, X.; Gao, R.; Demurtas, O.C.; Fleck, S.J.; Richter, M.; He, C.; Ji, A.; Sun, W.; Kong, J. Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol. 2020, 18, 63. [Google Scholar] [CrossRef]
- Shang, J.; Tian, J.; Cheng, H.; Yan, Q.; Li, L.; Jamal, A.; Xu, Z.; Xiang, L.; Saski, C.A.; Jin, S. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020, 21, 1–28. [Google Scholar] [CrossRef]
- Song, B.; Ning, W.; Wei, D.; Jiang, M.; Zhu, K.; Wang, X.; Edwards, D.; Odeny, D.A.; Cheng, S. Plant genome resequencing and population genomics: Current status and future prospects. Mol. Plant 2023, 16, 1252–1268. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.A.; Mascher, M.; Buluç, A.; Barry, K.; Georganas, E.; Session, A.; Strnadova, V.; Jenkins, J.; Sehgal, S.; Oliker, L. A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 2015, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaegashi, H.; Obara, T.; Oikawa, K.; Utsushi, H.; Kanzaki, E. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Wang, W.; Jiang, X.; Wu, M.; Bai, H.; Wu, C.; Shen, L. Transcriptome analysis to identify candidate genes related to chlorogenic acid biosynthesis during development of Korla fragrant pear in Xinjiang. Food Sci. Hum. Wellness 2022, 11, 854–864. [Google Scholar] [CrossRef]
- Yan, H.; Sun, M.; Zhang, Z.; Jin, Y.; Zhang, A.; Lin, C.; Wu, B.; He, M.; Xu, B.; Wang, J. Pangenomic analysis identifies structural variation associated with heat tolerance in pearl millet. Nat. Genet. 2023, 55, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, S.; Yang, S.; Yang, Z.; Liu, S.; Wang, Y.; Gao, H.; Zhang, S.; Yang, X.; Jiang, C. Genome assembly and genetic dissection of a prominent drought-resistant maize germplasm. Nat. Genet. 2023, 55, 496–506. [Google Scholar] [CrossRef]
- Kumar, S.; Mohapatra, T. Dynamics of DNA methylation and its functions in plant growth and development. Front. Plant Sci. 2021, 12, 596236. [Google Scholar] [CrossRef]
- Bai, D.; Yi, C. Advances in the Profiling of Single-Cell DNA Modifications. Small Methods 2019, 3, 1900137. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Nie, F.; Zhang, J.; Zhang, Z.; Wu, B.; Bai, L.; Liu, W.; Xiao, C.-L.; Luo, F. Genome-wide detection of cytosine methylations in plant from Nanopore data using deep learning. Nat. Commun. 2021, 12, 5976. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Perez, R.; Bazaga, P.; Medrano, M.; Herrera, C.M. MSAP markers and global cytosine methylation in plants: A literature survey and comparative analysis for a wild-growing species. Mol. Ecol. Resour. 2016, 16, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, J.; Xu, T.; Kang, H. Epitranscriptomic mRNA modifications governing plant stress responses: Underlying mechanism and potential application. Plant Biotechnol. J. 2022, 20, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Zhang, Q.; Yu, X.; Sun, Z.; He, Y.; Guo, W. Role of main RNA methylation in hepatocellular carcinoma: N6-methyladenosine, 5-methylcytosine, and N1-methyladenosine. Front. Cell Dev. Biol. 2021, 9, 767668. [Google Scholar] [CrossRef]
- Zhang, L.; Rong, W.; Ma, J.; Li, H.; Tang, X.; Xu, S.; Wang, L.; Wan, L.; Zhu, Q.; Jiang, B. Comprehensive analysis of DNA 5-methylcytosine and N6-adenine methylation by nanopore sequencing in hepatocellular carcinoma. Front. Cell Dev. Biol. 2022, 10, 827391. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.-D.; Xie, Y.-Y.; Chen, H.-X.; Lan, Y.-L.; Liu, X.-H.; Ji, J.-Y.; Wu, F.; Jin, L.; Chen, J.; Mak, D.W. Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat. Commun. 2023, 14, 1906. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Xi, F.; Wang, H.; Han, X.; Wei, W.; Zhang, H.; Zhang, Q.; Zheng, Y.; Zhu, Q. Profiling of circular RNA N6-methyladenosine in moso bamboo (Phyllostachys edulis) using nanopore-based direct RNA sequencing. J. Integr. Plant Biol. 2020, 62, 1823–1838. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M. N6-methyladenine DNA methylation in Japonica and Indica rice genomes and its association with gene expression, plant development, and stress responses. Mol. Plant 2018, 11, 1492–1508. [Google Scholar] [CrossRef]
- Yang, W.; Meng, J.; Liu, J.; Ding, B.; Tan, T.; Wei, Q.; Yu, Y. The N1-methyladenosine methylome of petunia mRNA. Plant Physiol. 2020, 183, 1710–1724. [Google Scholar] [CrossRef]
- Zhang, J.; Sheng, H.; Hu, C.; Li, F.; Cai, B.; Ma, Y.; Wang, Y.; Ma, Y. Effects of DNA methylation on gene expression and phenotypic traits in cattle: A review. Int. J. Mol. Sci. 2023, 24, 11882. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ng, H.K.; Drautz-Moses, D.I.; Schuster, S.C.; Beck, S.; Kim, C.; Chambers, J.C.; Loh, M. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci. Rep. 2019, 9, 10383. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics 2022, 17, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 2020, 10, 8013. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Deng, Y.; Wei, H.; Lu, S. Characteristics of Salvia miltiorrhiza methylome and the regulatory mechanism of DNA methylation in tanshinone biosynthesis. Hortic. Res. 2023, 10, uhad114. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Hu, S.; Cheng, S.; Wang, L.; Kan, L.; Wang, Z.; Xu, Q.; Liu, Z.; Kang, C. Factor of DNA methylation 1 affects woodland strawberry plant stature and organ size via DNA methylation. Plant Physiol. 2023, 191, 335–351. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.-L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Serre, D.; Lee, B.H.; Ting, A.H. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010, 38, 391–399. [Google Scholar] [CrossRef]
- Rothkegel, K.; Sánchez, E.; Montes, C.; Greve, M.; Tapia, S.; Bravo, S.; Prieto, H.; Almeida, A.M. DNA methylation and small interference RNAs participate in the regulation of MADS-box genes involved in dormancy in sweet cherry (Prunus avium L.). Tree Physiol. 2017, 37, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jia, X.; Wei, H.; Fan, S.; Wang, H.; Guo, Y.; Duan, S.; Pang, C.; Yu, S. Global analysis of DNA methylation in young (J1) and senescent (J2) Gossypium hirsutum L. cotyledons by MeDIP-Seq. PLoS ONE 2017, 12, e0179141. [Google Scholar] [CrossRef] [PubMed]
- Paun, O.; Verhoeven, K.J.; Richards, C.L. Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. New Phytol. 2019, 221, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.; Richards, C. Opportunities and challenges of next-generation sequencing applications in ecological epigenetics. Mol. Ecol. 2015, 24, 3799–3801. [Google Scholar] [CrossRef] [PubMed]
- Yadav, C.B.; Pandey, G.; Muthamilarasan, M.; Prasad, M. Epigenetics and epigenomics of plants. Plant Genet. Mol. Biol. 2018, 164, 237–261. [Google Scholar] [CrossRef]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Haghani, A.; Li, C.Z.; Robeck, T.R.; Zhang, J.; Lu, A.T.; Ablaeva, J.; Acosta-Rodríguez, V.A.; Adams, D.M.; Alagaili, A.N.; Almunia, J. DNA methylation networks underlying mammalian traits. Science 2023, 381, eabq5693. [Google Scholar] [CrossRef]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Lowe, R.; Yan, Q.; Zhang, J.; Vu, H. Universal DNA methylation age across mammalian tissues. Innov. Aging 2021, 5, 410. [Google Scholar] [CrossRef]
- Fang, Q.; Yuan, Z.; Hu, H.; Zhang, W.; Wang, G.; Wang, X. Genome-wide discovery of circulating cell-free DNA methylation biomarkers for colorectal cancer detection. Clin. Epigenet. 2023, 15, 119. [Google Scholar] [CrossRef]
- Klughammer, J.; Romanovskaia, D.; Nemc, A.; Posautz, A.; Seid, C.A.; Schuster, L.C.; Keinath, M.C.; Lugo Ramos, J.S.; Kosack, L.; Evankow, A. Comparative analysis of genome-scale, base-resolution DNA methylation profiles across 580 animal species. Nat. Commun. 2023, 14, 232. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.C.; Harrison, C.J.; Paps, J.; Schneider, H. The evolutionary emergence of land plants. Curr. Biol. 2021, 31, R1281–R1298. [Google Scholar] [CrossRef] [PubMed]
- Batalova, A.Y.; Putintseva, Y.A.; Sadovsky, M.G.; Krutovsky, K.V. Comparative genomics of seasonal senescence in forest trees. Int. J. Mol. Sci. 2022, 23, 3761. [Google Scholar] [CrossRef]
- Haas, M.; Schreiber, M.; Mascher, M. Domestication and crop evolution of wheat and barley: Genes, genomics, and future directions. J. Integr. Plant Biol. 2019, 61, 204–225. [Google Scholar] [CrossRef]
- Song, A.; Su, J.; Wang, H.; Zhang, Z.; Zhang, X.; Van de Peer, Y.; Chen, F.; Fang, W.; Guan, Z.; Zhang, F. Analyses of a chromosome-scale genome assembly reveal the origin and evolution of cultivated chrysanthemum. Nat. Commun. 2023, 14, 2021. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Li, W.; Li, Y.; Liu, M.; Cao, H.; Provart, N.; Ding, X.; Sun, M.; Tang, Z.; Yue, C. The red flower wintersweet genome provides insights into the evolution of magnoliids and the molecular mechanism for tepal color development. Plant J. 2021, 108, 1662–1678. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Duan, S.; Xia, Q.; Liang, Z.; Dong, X.; Margaryan, K.; Musayev, M.; Goryslavets, S.; Zdunić, G.; Bert, P.-F. Dual domestications and origin of traits in grapevine evolution. Science 2023, 379, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.I.; Heuberger, M.; Schoen, A.; Koo, D.-H.; Quiroz-Chavez, J.; Adhikari, L.; Raupp, J.; Cauet, S.; Rodde, N.; Cravero, C. Einkorn genomics sheds light on history of the oldest domesticated wheat. Nature 2023, 620, 830–838. [Google Scholar] [CrossRef]
- Li, A.; Yang, Q.; Li, R.; Dai, X.; Cai, K.; Lei, Y.; Jia, K.; Jiang, Y.; Zan, L. Chromosome-level genome assembly for takin (Budorcas taxicolor) provides insights into its taxonomic status and genetic diversity. Mol. Ecol. 2023, 32, 1323–1334. [Google Scholar] [CrossRef]
- Schneider, H. Integrating genomics and conservation to safeguard plant diversity. Integr. Conserv. 2023, 2, 10–18. [Google Scholar] [CrossRef]
- Barbour, M.A.; Kliebenstein, D.J.; Bascompte, J. A keystone gene underlies the persistence of an experimental food web. Science 2022, 376, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yu, Z.; Gao, X.; Liu, G.; Zhang, Y.; Šmarda, P.; Guo, Q. Genetic diversity, population structure, and genome-wide association analysis of ginkgo cultivars. Hortic. Res. 2023, 10, uhad136. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.P.; Tang, J.Y.; Yu, J.G.; Smith, D.R.; Zhu, Y.M.; Wang, Y.R.; Kang, J.S.; Yang, J.; Zhang, X.C. The evolution of extremely diverged plastomes in Selaginellaceae (lycophyte) is driven by repeat patterns and the underlying DNA maintenance machinery. Plant J. 2022, 111, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Fang, D.; Dong, S.; Guo, X.; Li, N.; Campos, L.; Chen, X.; Fang, D.; Dong, S.; et al. Genomes shed light on the evolution of Begonia, a mega-diverse genus. New Phytol. 2022, 234, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Stansell, Z.; Björkman, T. From landrace to modern hybrid broccoli: The genomic and morphological domestication syndrome within a diverse B. oleracea collection. Hortic. Res. 2020, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; So, T.; Thammavong, B.; Chamchumroon, V.; Theilade, I.; Phourin, C.; Bouamanivong, S.; Hartvig, I.; Gaisberger, H.; Jalonen, R. Range-wide differential adaptation and genomic vulnerability in critically endangered Asian rosewoods. Proc. Natl. Acad. Sci. USA 2023, 120, e2301603120. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Deng, T.; Zhang, A.; Moore, M.J.; Landis, J.B.; Lin, N.; Zhang, H.; Zhang, X.; Huang, J.; Zhang, X. Genome sequencing of the endangered Kingdonia uniflora (Circaeasteraceae, Ranunculales) reveals potential mechanisms of evolutionary specialization. IScience 2020, 23, 101124. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, T.; Zhang, L.; Kang, M.; Zhang, Z.; Zheng, Z.; Sun, P.; Shrestha, N.; Liu, J.; Yang, Y. Genomic analyses of a “living fossil”: The endangered dove-tree. Mol. Ecol. Resour. 2020, 20, 756–769. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Y.; Liu, D.; Sun, W.; Liu, X.; Wan, Y.; Zhang, X.; Zhang, R.; Yun, Q.; Wang, J. Chromosome-level genome assembly and population genetic analysis of a critically endangered rhododendron provide insights into its conservation. Plant J. 2021, 107, 1533–1545. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, T.; Wang, Z.; Lu, Z.; Li, Y.; Fu, C.; Chen, X.; Zhao, M.; Olson, M.S.; Liu, J. Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nat. Commun. 2018, 9, 5449. [Google Scholar] [CrossRef]
- Liang, Z.; Myers, Z.A.; Petrella, D.; Engelhorn, J.; Hartwig, T.; Springer, N.M. Mapping responsive genomic elements to heat stress in a maize diversity panel. Genome Biol. 2022, 23, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.M.; Oladzad, A.; Koh, C.; Ramsay, L.; Hart, J.P.; Mamidi, S.; Hoopes, G.; Sreedasyam, A.; Wiersma, A.; Zhao, D. The tepary bean genome provides insight into evolution and domestication under heat stress. Nat. Commun. 2021, 12, 2638. [Google Scholar] [CrossRef]
- Mekonnen, T.; Haileselassie, T.; Tesfaye, K. Identification, mapping and pyramiding of genes/quantitative trait loci (qtls) for durable resistance of crops to biotic stresses. J. Plant Pathol. Microbiol. 2017, 8, 1000412. [Google Scholar] [CrossRef]
- Sallam, A.; Eltaher, S.; Alqudah, A.M.; Belamkar, V.; Baenziger, P.S. Combined GWAS and QTL mapping revealed candidate genes and SNP network controlling recovery and tolerance traits associated with drought tolerance in seedling winter wheat. Genomics 2022, 114, 110358. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Rao, G.J.; Varier, M.; Prakash, A.; Prasad, D. Improved Tapaswini having four BB resistance genes pyramided with six genes/QTLs, resistance/tolerance to biotic and abiotic stresses in rice. Sci. Rep. 2018, 8, 2413. [Google Scholar] [CrossRef] [PubMed]
- Parihar, A.K.; Kumar, J.; Gupta, D.S.; Lamichaney, A.; Naik Sj, S.; Singh, A.K.; Dixit, G.P.; Gupta, S.; Toklu, F. Genomics enabled breeding strategies for major biotic stresses in pea (Pisum sativum L.). Front. Plant Sci. 2022, 13, 861191. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.J.; Reddy, J.N.; Variar, M.; Mahender, A. Molecular breeding to improve plant resistance to abiotic stresses. In Advances in Plant Breeding Strategies: Agronomic, Abiotic and Biotic Stress Traits; Springer: Cham, Swizerland, 2016; pp. 283–326. [Google Scholar] [CrossRef]
- Laosatit, K.; Somta, P.; Chen, X.; Srinives, P. Genomic approaches to biotic stresses. In The Mungbean Genome; Springer: Cham, Swizerland, 2020; pp. 133–167. [Google Scholar] [CrossRef]
- Liu, P.; Liu, R.; Xu, Y.; Zhang, C.; Niu, Q.; Lang, Z. DNA cytosine methylation dynamics and functional roles in horticultural crops. Hortic. Res. 2023, 10, uhad170. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Cao, S.; Chen, K.; Lu, K.; Chen, S.; Zhang, X.; Shen, C.; Zhu, S.; Niu, Y.; Fan, L.; Chen, Z.J. Asymmetric variation in DNA methylation during domestication and de-domestication of rice. Plant Cell 2023, 35, koad160. [Google Scholar] [CrossRef]
- Cao, Q.; Huang, L.; Li, J.; Qu, P.; Tao, P.; Crabbe, M.J.C.; Zhang, T.; Qiao, Q. Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis). BMC Plant Biol. 2022, 22, 613. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.-h.; Wang, J.-b. DNA-methylation changes induced by salt stress in wheat Triticum aestivum. Afr. J. Biotechnol. 2009, 8, 22. [Google Scholar] [CrossRef]
- Shan, X.; Wang, X.; Yang, G.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Yuan, Y. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J. Plant Biol. 2013, 56, 32–38. [Google Scholar] [CrossRef]
- Wan, T.; Liu, Z.; Leitch, I.J.; Xin, H.; Maggs-Kölling, G.; Gong, Y.; Li, Z.; Marais, E.; Liao, Y.; Dai, C. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat. Commun. 2021, 12, 4247. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, F.; Yuan, T.; Li, W.; Li, Y.; Wu, H.X.; Wei, H.; Niu, S. The methylation landscape of giga-genome and the epigenetic timer of age in Chinese pine. Nat. Commun. 2023, 14, 1947. [Google Scholar] [CrossRef]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Deng, M.; Yang, X.; Yu, W.; Cai, J.; Shi, Y.; Zhu, Z.; Zhou, T.; Xue, L.; Cao, F. Genome-Wide Identification and Coexpression Network Analysis of DNA Methylation Pathway Genes and Their Differentiated Functions in Ginkgo biloba L. Forests 2020, 11, 1076. [Google Scholar] [CrossRef]
- Castander-Olarieta, A.; Pereira, C.; Sales, E.; Meijón, M.; Arrillaga, I.; Cañal, M.J.; Goicoa, T.; Ugarte, M.D.; Moncaleán, P.; Montalbán, I.A. Induction of radiata pine somatic embryogenesis at high temperatures provokes a long-term decrease in DNA methylation/hydroxymethylation and differential expression of stress-related genes. Plants 2020, 9, 1762. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z. Unlocking plant metabolic diversity: A (pan)-genomic view. Plant Commun. 2022, 3, 100300. [Google Scholar] [CrossRef]
- Shang, X.; Yi, X.; Xiao, L.; Zhang, Y.; Huang, D.; Xia, Z.; Ou, K.; Ming, R.; Zeng, W.; Wu, D. Chromosomal-level genome and multi-omics dataset of Pueraria lobata var. thomsonii provide new insights into legume family and the isoflavone and puerarin biosynthesis pathways. Hortic. Res. 2022, 9, uhab035. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Z.; Yang, Y.; Wang, Z.; Mu, W.; Liu, J. Multi-omics reveal differentiation and maintenance of dimorphic flowers in an alpine plant on the Qinghai-Tibet Plateau. Mol. Ecol. 2023, 32, 1411–1424. [Google Scholar] [CrossRef]
- Li, C.; Wood, J.C.; Vu, A.H.; Hamilton, J.P.; Rodriguez Lopez, C.E.; Payne, R.M.; Serna Guerrero, D.A.; Gase, K.; Yamamoto, K.; Vaillancourt, B. Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nat. Chem. Biol. 2023, 19, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; Langley, C.H.; Salzberg, S.L.; Wegrzyn, J.L. Open access to tree genomes: The path to a better forest. Genome Biol. 2013, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E. Sequence of the sugar pine megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U. The Douglas-fir genome sequence reveals specialization of the photosynthetic apparatus in Pinaceae. G3: Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Mosca, E.; Cruz, F.; Gómez-Garrido, J.; Bianco, L.; Rellstab, C.; Brodbeck, S.; Csilléry, K.; Fady, B.; Fladung, M.; Fussi, B. A reference genome sequence for the European silver fir (Abies alba Mill.): A community-generated genomic resource. G3 Genes Genomes Genet. 2019, 9, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.A.; Feranchuk, S.I.; Sharov, V.V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20, 35–46. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, X.; Liu, X.; Zhu, X.; Li, Z.; Chu, H.; Wang, Q.; Lou, Q.; Cai, B.; Yang, Y. Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol. Plant 2021, 14, 1199–1209. [Google Scholar] [CrossRef]
- Xiong, X.; Gou, J.; Liao, Q.; Li, Y.; Zhou, Q.; Bi, G.; Li, C.; Du, R.; Wang, X.; Sun, T. The Taxu s genome provides insights into paclitaxel biosynthesis. Nat. Plants 2021, 7, 1026–1036. [Google Scholar] [CrossRef]
- Song, C.; Fu, F.; Yang, L.; Niu, Y.; Tian, Z.; He, X.; Yang, X.; Chen, J.; Sun, W.; Wan, T. Taxus yunnanensis genome offers insights into gymnosperm phylogeny and taxol production. Commun. Biol. 2021, 4, 1203. [Google Scholar] [CrossRef]
- Scott, A.D.; Zimin, A.V.; Puiu, D.; Workman, R.; Britton, M.; Zaman, S.; Caballero, M.; Read, A.C.; Bogdanove, A.J.; Burns, E. A reference genome sequence for giant sequoia. G3 Genes Genomes Genet. 2020, 10, 3907–3919. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xie, Y.H.; Li, Z.; Liu, Y.J.; Sun, X.M.; Li, J.J.; Quan, W.P.; Zeng, Q.Y.; Van de Peer, Y.; Zhang, S.G. The Larix kaempferi genome reveals new insights into wood properties. J. Integr. Plant Biol. 2022, 64, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, X.; Wang, G.; Cui, P.; Wu, S.; Ai, C.; Hu, N.; Li, A.; He, B.; Shao, X. The nearly complete genome of Ginkgo biloba illuminates gymnosperm evolution. Nat. Plants 2021, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Liu, Z.-M.; Li, L.-F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.-J.; Xin, H.-P.; Gong, Y.-B.; Liu, Y. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, S.; Li, L.; Yang, T.; Dong, S.; Wei, T.; Wu, S.; Liu, Y.; Gong, Y.; Feng, X. The Cycas genome and the early evolution of seed plants. Nat. Plants 2022, 8, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Song, C.; Wen, C.; Yang, L.; Guo, Y.; Yang, X.; Shu, Z.; Li, X.; Feng, Y.; Liu, B. The Metasequoia genome and evolutionary relationships among redwoods. Plant Commun. 2023, 4, 100643. [Google Scholar] [CrossRef]
- Scott, A.D.; Stenz, N.W.; Ingvarsson, P.K.; Baum, D.A. Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers. New Phytol. 2016, 211, 186–193. [Google Scholar] [CrossRef]
- Yue, M.; Chen, H.; Xuan, L.; Yang, Y.; Chong, X.; Li, M.; Yu, C.; Lu, X.; Zhang, F. Novel molecular markers for Taxodium breeding from the chloroplast genomes of four artificial Taxodium hybrids. Front. Genet. 2023, 14. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Yang, G.; Qian, H.; Li, M. Characterization of the complete chloroplast genome sequence of Tsuga longibracteata WC Cheng (Pinaceae). Conserv. Genet. Resour. 2019, 11, 117–120. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Zhang, L.; Yang, X. The complete chloroplast genome of Cycas Szechuanensis, an extremely endangered species. Mitochondrial DNA 2018, 3, 974–975. [Google Scholar] [CrossRef]
- Yang, X.; Deng, T.; Tang, W.; Wu, T. Characterization of the complete chloroplast genome of Cycas ferruginea, a vulnerable species. Mitochondrial DNA 2022, 7, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Wu, C.-S.; Huang, Y.-Y.; Chaw, S.-M. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol. Evol. 2012, 4, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Li, J. The complete chloroplast genome sequence of Podocarpus imbricatus (Podocarpaceae) and its phylogenetic analysis. Mitochondrial DNA 2019, 4, 368–369. [Google Scholar] [CrossRef]
- Li, J.; Xu, B.; Yang, Q.; Liu, Z.-L. The complete chloroplast genome sequence of Picea schrenkiana (Pinaceae). Mitochondrial DNA 2020, 5, 2191–2192. [Google Scholar] [CrossRef]
- Parmar, R.; Cattonaro, F.; Phillips, C.; Vassiliev, S.; Morgante, M.; Rajora, O.P. Assembly and annotation of Red Spruce (Picea rubens) chloroplast genome, identification of simple sequence repeats, and phylogenetic analysis in Picea. Int. J. Mol. Sci. 2022, 23, 15243. [Google Scholar] [CrossRef]
- Yang, M.-Q.; Du, Y.; Ling, L.-Z. Characterization of the complete chloroplast genome of Pinus wangii (Pinaceae), an endangered and endemic species in China. Mitochondrial DNA 2018, 3, 1195–1197. [Google Scholar] [CrossRef]
- Wu, L.-X.; Wang, Y.-H.; Gong, X. Characterization of the complete chloroplast genome of Cycas hongheensis (Cycadaceae), an endemic species in the red river region of China. Mitochondrial DNA 2021, 6, 3513–3514. [Google Scholar] [CrossRef]
- Kersten, B.; Rellstab, C.; Schroeder, H.; Brodbeck, S.; Fladung, M.; Krutovsky, K.V.; Gugerli, F. The mitochondrial genome sequence of Abies alba Mill. reveals a high structural and combinatorial variation. BMC Genom. 2022, 23, 776. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.I.; Ru, D.; Miao, J.; Tao, W.; Zhang, L.; Xu, J.; Liu, J.; Mao, K. Allopatric divergence and hybridization within Cupressus chengiana (Cupressaceae), a threatened conifer in the northern Hengduan Mountains of western China. Mol. Ecol. 2020, 29, 1250–1266. [Google Scholar] [CrossRef]
- WANG, H.W.; Ge, S. Phylogeography of the endangered Cathaya argyrophylla (Pinaceae) inferred from sequence variation of mitochondrial and nuclear DNA. Mol. Ecol. 2006, 15, 4109–4122. [Google Scholar] [CrossRef] [PubMed]
- Meena, B.; Singh, N.; Mahar, K.S.; Sharma, Y.K.; Rana, T.S. Molecular analysis of genetic diversity and population genetic structure in Ephedra foliata: An endemic and threatened plant species of arid and semi-arid regions of India. Physiol. Mol. Biol. Plants 2019, 25, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Cannon, A.; Lisch, D. A Molecular Cloning and Sanger Sequencing-based Protocol for Detecting Site-specific DNA Methylation. Bio-Protocol 2022, 12, e4408. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kang, M.-Y.; Shim, E.-J.; Oh, J.; Seo, K.-I.; Kim, K.S.; Sim, S.-C.; Chung, S.-M.; Park, Y.; Lee, G.P. Genome-wide core sets of SNP markers and Fluidigm assays for rapid and effective genotypic identification of Korean cultivars of lettuce (Lactuca sativa L.). Hortic. Res. 2022, 9, uhac119. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, K.; Li, Z.; Li, Y.; He, J.; Li, H.; Wang, B.; Xin, T.; Tian, H.; Tian, J. Architecture design of cucurbit crops for enhanced productivity by a natural allele. Nat. Plants 2022, 8, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Xu, W.; Xin, T.; Song, J. Application of third-generation sequencing to herbal genomics. Front. Plant Sci. 2023, 14, 1124536. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ding, C.; Zhang, W.; Zhang, T.; Li, Z.; Zhang, J.; Chu, Y.; Su, X. The Populus koreana genome provides insights into the biosynthesis of plant aroma. Ind. Crops Prod. 2023, 197, 116453. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, J.; Zhao, X.; Jiang, H.; Ma, X.; Pan, W. CIDP: A multi-functional platform for designing CRISPR sgRNAs. Hortic. Res. 2022, 10, uhad092. [Google Scholar] [CrossRef]
- Xu, D.; Jin, K.; Jiang, H.; Gong, D.; Yang, J.; Yu, W.; Yang, Y.; Li, J.; Pan, W. GFAP: Ultra-fast and accurate gene functional annotation software for plants. Plant Physiol. 2023, 193, 1745–1748. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, L.; Zhu, Q.-H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Pioneering Generation Genome Sequencing Technology (Sanger) | Offers reliable data for small-scale projects. | Slower, for small-scale projects. | [19,20] |
| Fits shorter DNA sequencing like gene or Sanger sequencing. | Costly reagents and equipment expenses. | [21,22] | |
| Mature workflows and analysis tools after years of development. | Can’t meet high-throughput sequencing needs. | [23,24] | |
| NGS | Fast sequencing of many DNA fragments for high-throughput projects. | Produces shorter reads, limiting applications like complex genome assembly. | [25,26] |
| Relatively inexpensive for large-scale sequencing. | Generates abundant data, and needs intricate processing and analysis. | [27,28] | |
| Suitable for multi-sample sequencing for population and ecology studies. | Some techniques introduce sequencing biases or errors. | [29,30] | |
| Third-generation genome sequencing technology (TGS) | Long-read sequencing aids complex genome assembly and detection. | Longer data generation time is not ideal for high-throughput sequencing. | [30,31] |
| No PCR, fewer errors. | Demands more computing and complex data analysis. | [32,33] | |
| Useful for genome structure research: Reveals repetitive sequences and chromosomal rearrangements with long reads. | High cost. | [34,35] | |
| Provides real-time data for monitoring DNA synthesis and modifications. | [36] |
| Name | Advantages | Disadvantages | Reference |
|---|---|---|---|
| PacBio HiFi | Long reads aid in complex genome assembly and detection. | Lower throughput extends data generation time. | [58] |
| High accuracy lowers errors. | High cost. | [48,59] | |
| Direct DNA sequencing, no PCR, fewer errors. | [60] | ||
| Useful for genome structure research: Reveals repetitive sequences and chromosomal rearrangements with long reads. | [61] | ||
| Nanopore ONT | Offers real-time data for monitoring DNA synthesis and modifications. | Low accuracy, requires multiple runs for data quality. | [62,63] |
| Compact and lightweight device. | Complex data processing needs more computing resources and tools. | [64,65] | |
| Enables real-time data analysis, accelerating the research process. | [66,67] | ||
| Lower sequencing costs. | Relatively shorter read lengths are unsuitable for some long-read studies. | [68,69] |
| Name | Methods | Size | Reference |
|---|---|---|---|
| Pinus taeda | Sanger + Illumina | 23 G | [156] |
| Picea glauca | Illumina | 23.6 G | [157] |
| Pinus lambertiana | Illumina | 27.6 G | [158] |
| Pseudotsuga menziesii | Illumina | 15.7 G | [159] |
| Abies alba | Illumina | 18.2 G | [160] |
| Larix sibirica | Illumina | 12.3 G | [161] |
| Taxus wallichiana | Illumina | 10.9 Gb | [162] |
| Taxus chinensis | PacBio + Hi-C + Illumina | 10.23 Gb | [163] |
| Taxus yunnanensis | Illumina + Nanopore | 10.7 Gb | [164] |
| Sequoiadendron giganteum | Illumina +Hi-C + Nanopore | 8 GB | [165] |
| Picea abies | Shortgun | 20 GB | [166] |
| Pinus tabuliformis | PacBio + Hi-C + Illumina | 25.4 Gb | [149] |
| Larix kaempferi | PacBio + Bionano + Illumina | 10.97 GB | [167] |
| Ginkgo biloba | PacBio + Hi-C | 9.88 Gb | [168] |
| Gnetum montanum | Illumina | 4.5 G | [169] |
| Welwitschia mirabilis | Illumina + Nanopore | 6.86 Gb | [147] |
| Cycas panzhihuaensis | PacBio + Illumina | 10.5 Gb | [170] |
| Torreya grandis | PacBio + HiFi + Illumina | 19 Gb | [8] |
| Metasequoia glyptostroboides | ONT + Illumina + Hi-C | 8.07 Gb | [171] |
| Sequoia sempervirens | PacBio HiFi | 27 Gb | [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, K.; Radian, Y.; Manda, T.; Xu, H.; Luo, Y. The Development of Plant Genome Sequencing Technology and Its Conservation and Application in Endangered Gymnosperms. Plants 2023, 12, 4006. https://doi.org/10.3390/plants12234006
Hong K, Radian Y, Manda T, Xu H, Luo Y. The Development of Plant Genome Sequencing Technology and Its Conservation and Application in Endangered Gymnosperms. Plants. 2023; 12(23):4006. https://doi.org/10.3390/plants12234006
Chicago/Turabian StyleHong, Kaiyue, Yasmina Radian, Teja Manda, Haibin Xu, and Yuming Luo. 2023. "The Development of Plant Genome Sequencing Technology and Its Conservation and Application in Endangered Gymnosperms" Plants 12, no. 23: 4006. https://doi.org/10.3390/plants12234006