
Miniature Inverted-Repeat Transposable Elements: Small DNA Transposons That Have Contributed to Plant MICRORNA Gene Evolution

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
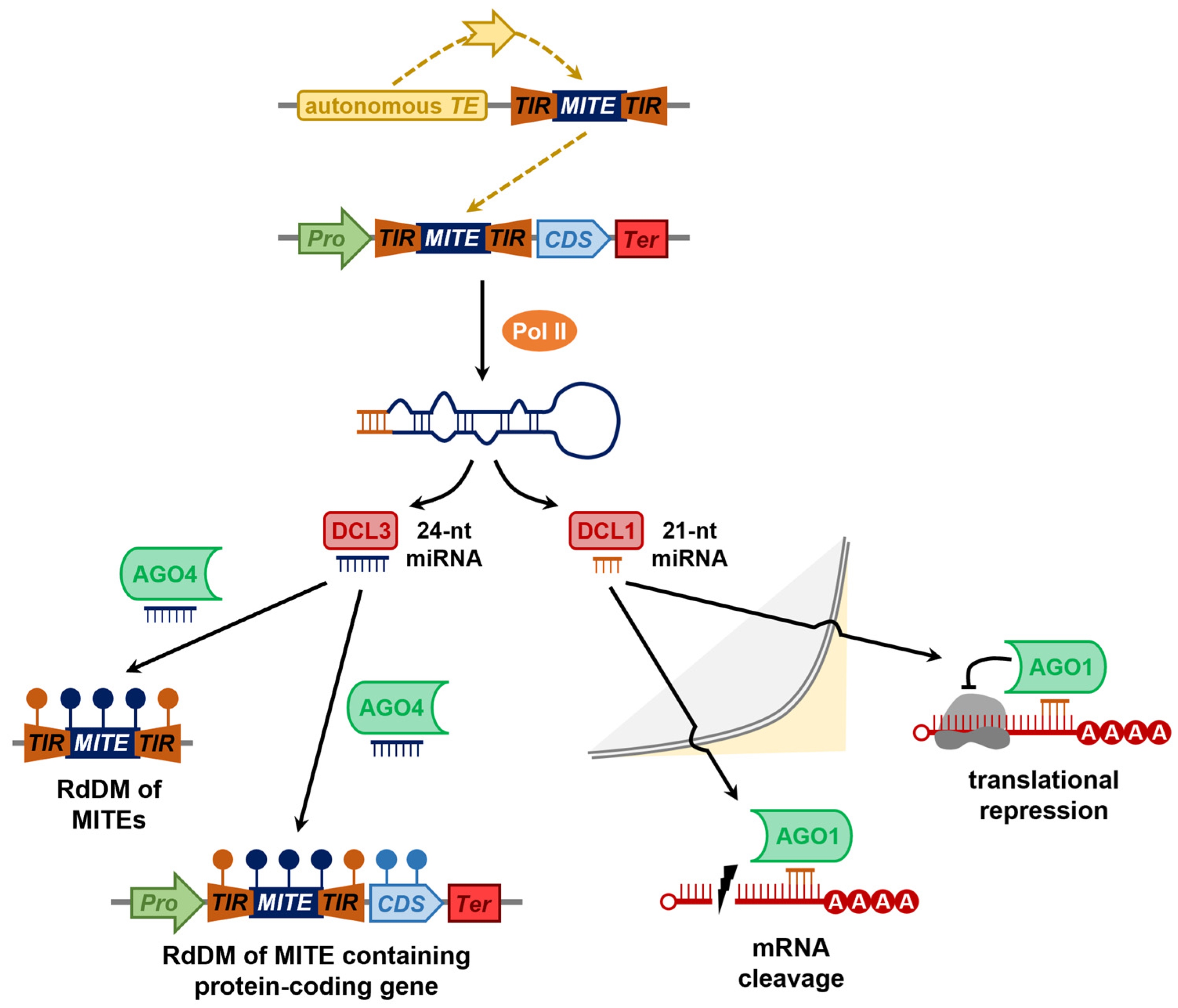{kind=link}
Abstract
:1. Introduction
2. Angiosperm Transposable Elements: Type I and Type II Transposons
3. Miniature Inverted-Repeat Transposable Elements (MITEs): Short Nonautonomous Type II Transposons
4. Repression of TE Activity by the RNA-Directed DNA Methylation (RdDM) Pathway, and TE Mechanisms to Avoid RdDM-Mediated Repression
5. The Plant microRNA Pathway
6. Plant MIR Gene Evolution Models
7. MITEs as a Source of MIR Genes in Angiosperms
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A Triptych of the Evolution of Plant Transposable Elements. Trends Plant Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Byng, J.W. The Number of Known Plants Species in the World and Its Annual Increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.D.; Leitch, I.J. Nuclear DNA Amounts in Angiosperms: Targets, Trends and Tomorrow. Ann. Bot. 2011, 107, 467–590. [Google Scholar] [CrossRef]
- Oliver, K.R.; McComb, J.A.; Greene, W.K. Transposable Elements: Powerful Contributors to Angiosperm Evolution and Diversity. Genome Biol. Evol. 2013, 5, 1886–1901. [Google Scholar] [CrossRef]
- Ito, H. Environmental Stress and Transposons in Plants. Genes Genet. Syst. 2022, 97, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and Evolution of a Minute Plant Genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramírez-González, R.H.; De Oliveira, R.; Mayer, K.F.X.; Paux, E.; Choulet, F. Impact of Transposable Elements on Genome Structure and Evolution in Bread Wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef]
- Mirouze, M.; Vitte, C. Transposable Elements, a Treasure Trove to Decipher Epigenetic Variation: Insights from Arabidopsis and Crop Epigenomes. J. Exp. Bot. 2014, 65, 2801–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Brachypodium Initiative, T. Genome Sequencing and Analysis of the Model Grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guo, K.; Zhu, X.; Chen, P.; Li, Y.; Xie, G.; Wang, L.; Wang, Y.; Persson, S.; Peng, L. Domestication of Rice Has Reduced the Occurrence of Transposable Elements within Gene Coding Regions. BMC Genom. 2017, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Hung, N.N.; Kim, D.G.; Lyu, J.I.; Park, K.C.; Kim, J.M.; Kim, J.B.; Ha, B.K.; Kwon, S.J. Detecting Genetic Mobility Using a Transposon-Based Marker System in Gamma-Ray Irradiated Soybean Mutants. Plants 2021, 10, 373. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of Allotetraploid Cotton (Gossypium Hirsutum L. Acc. TM-1) Provides a Resource for Fiber Improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Zimmermann, W.; Perovic, D.; Paterson, A.H.; Ganal, M.; Graner, A.; Stein, N. A Detailed Look at 7 Million Years of Genome Evolution in a 439 Kb Contiguous Sequence at the Barley Hv-ElF4E Locus: Recombination, Rearrangements and Repeats. Plant J. 2005, 41, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling Genome Size without Polyploidization: Dynamics of Retrotransposition-Driven Genomic Expansions in Oryza australiensis, a Wild Relative of Rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The Paleontology of Intergene Retrotransposons of Maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Kim, H.R.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential Lineage-Specific Amplification of Transposable Elements is Responsible for Genome Size Variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vielle-Calzada, J.P.; De La Vega, O.M.; Hernández-Guzmán, G.; Ibarra-LacLette, E.; Alvarez-Mejía, C.; Vega-Arreguín, J.C.; Jiménez-Moraila, B.; Fernández-Cortés, A.; Corona-Armenta, G.; Herrera-Estrella, L.; et al. The Palomero Genome Suggests Metal Effects on Domestication. Science 2009, 326, 1078. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Du, J. Young But Not Relatively Old Retrotransposons Are Preferentially Located in Gene-Rich Euchromatic Regions in Tomato (Solanum lycopersicum) Plants. Plant J. 2014, 80, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.D.; Springer, N.M. Transposable Element Influences on Gene Expression in Plants. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2017, 1860, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, B.; Walko, R.; Hake, S. Mutator Insertions in an Intron of the Maize Knotted1 Gene Result in Dominant Suppressible Mutations. Genetics 1994, 138, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected Consequences of a Sudden and Massive Transposon Amplification on Rice Gene Expression. Nature 2009, 461, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Asano, T.; Kanemitsu, Y.; Goto, T.; Yoshida, Y.; Yasuba, K.; Misawa, Y.; Nakatani, S.; Kobata, K. Positional Differences of Intronic Transposons in PAMT Affect the Pungency Level in Chili Pepper through Altered Splicing Efficiency. Plant J. 2019, 100, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Yoneda, Y.; Kuboyama, T. A Stowaway Transposon Disrupts the InWDR1 Gene Controlling Flower and Seed Coloration in a Medicinal Cultivar of the Japanese Morning Glory. Genes Genet. Syst. 2016, 91, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Hollister, J.D.; Gaut, B.S. Epigenetic Silencing of Transposable Elements: A Trade-off between Reduced Transposition and Deleterious Effects on Neighboring Gene Expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennetzen, J.L. Transposable Element Contributions to Plant Gene and Genome Evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A Unified Classification System for Eukaryotic Transposable Elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant Transposable Elements: Where Genetics Meets Genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef]
- Charles, M.; Belcram, H.; Just, J.; Huneau, C.; Viollet, A.; Couloux, A.; Segurens, B.; Carter, M.; Huteau, V.; Coriton, O.; et al. Dynamics and Differential Proliferation of Transposable Elements during the Evolution of the B and A Genomes of Wheat. Genetics 2008, 180, 1071–1086. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved Maize Reference Genome with Single-Molecule Technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Ohshima, K.; Okada, N. SINEs and LINEs: Symbionts of Eukaryotic Genomes With a Common Tail. Cytogenet. Genome Res. 2005, 110, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Yadav, B.S.; Ahmad, K.; Kumar, A. Mapping and Analysis of the LINE and SINE Type of Repetitive Elements in Rice. Bioinformation 2011, 7, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sugimoto, K.; Otsuki, H.; Hirochika, H. A 13-bp cis-Regulatory Element in the LTR Promoter of the Tobacco Retrotransposon Tto1 is Involved in Responsiveness to Tissue Culture, Wounding, Methyl Jasmonate and Fungal Elicitors. Plant J. 1999, 18, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; González, E.; Casaretto, J.A.; Casacuberta, J.M.; Ruiz-Lara, S. The Promoter of the TLC1.1 Retrotransposon from Solanum chilense is Activated by Multiple Stress-Related Signaling Molecules. Plant Cell Rep. 2007, 26, 1861–1868. [Google Scholar] [CrossRef]
- Casacuberta, J.M.; Santiago, N. Plant LTR-Retrotransposons and MITEs: Control of Transposition and Impact on the Evolution of Plant Genes and Genomes. Gene 2003, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.M.; Casacuberta, J.M. Additional ORFs in Plant LTR-Retrotransposons. Front. Plant Sci. 2020, 11, 555. [Google Scholar] [CrossRef]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Rice, D.; Rafii, M.Y.; Azizi, P.; Osman, M.; Taheri, S.; Bakar, M.F.A.; Isa, M.N.M.; et al. Contribution of Transposable Elements in the Plant’s Genome. Gene 2018, 665, 155–166. [Google Scholar] [CrossRef]
- Lisch, D. How Important Are Transposons for Plant Evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Lisch, D. Epigenetic Regulation of Transposable Elements in Plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef] [Green Version]
- Fattash, I.; Rooke, R.; Wong, A.; Hui, C.; Luu, T.; Bhardwaj, P.; Yang, G. Miniature Inverted-Repeat Transposable Elements: Discovery, Distribution, and Activity1. Genome 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Venkatesh; Nandini, B. Miniature Inverted-Repeat Transposable Elements (MITEs), Derived Insertional Polymorphism as a Tool of Marker Systems for Molecular Plant Breeding. Mol. Biol. Rep. 2020, 47, 3155–3167. [Google Scholar] [CrossRef]
- Bureau, T.E.; Wessler, S.R. Tourist: A Large Family of Small Inverted Repeat Elements Frequently Associated with Maize Genes. Plant Cell 1992, 4, 1283–1294. [Google Scholar] [PubMed] [Green Version]
- Bureau, T.E.; Wessler, S.R. Stowaway: A New Family of Inverted Repeat Elements Associated with the Genes of Both Monocotyledonous and Dicotyledonous Plants. Plant Cell 1994, 6, 907–916. [Google Scholar] [PubMed] [Green Version]
- Casacuberta, E.; Casacuberta, J.M.; Puigdomènech, P.; Monfort, A. Presence of Miniature Inverted-Repeat Transposable Elements (MITEs) in the Genome of Arabidopsis thaliana: Characterisation of the Emigrant Family of Elements. Plant J. 1998, 16, 79–85. [Google Scholar] [CrossRef]
- Hu, J.; Reddy, V.S.; Wessler, S.R. The Rice R Gene Family: Two Distinct Subfamilies Containing Several Miniature Inverted-Repeat Transposable Elements. Plant Mol. Biol. 2000, 42, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Jeong, C.S.; Song, M.T.; Kim, N.S. A New MITE Family, Pangrangja, in Gramineae Species. Mol. Cells 2003, 15, 373–380. [Google Scholar]
- Feschotte, C.; Mouchès, C. Recent Amplification of Miniature Inverted-Repeat Transposable Elements in the Vector Mosquito Culex Pipiens: Characterization of the Mimo Family. Gene 2000, 250, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Dong, J.; Chandrasekharan, M.; Hall, T. Kiddo, a New Transposable Element Family Closely Associated with Rice Genes. Mol. Genet. Genom. 2001, 266, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feschotte, C.; Zhang, Q.; Jiang, N.; Eggleston, W.B.; Wessler, S.R. P Instability Factor: An Active Maize Transposon System Associated with the Amplification of Tourist-like MITEs and a New Superfamily of Transposases. Proc. Natl. Acad. Sci. USA 2001, 98, 12572–12577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Bao, Z.; Zhang, X.; Hirochika, H.; Eddy, S.R.; McCouch, S.R.; Wessler, S.R. An Active DNA Transposon Family in Rice. Nature 2003, 421, 163–167. [Google Scholar] [CrossRef]
- Yang, G.; Fattash, I.; Lee, C.N.; Liu, K.; Cavinder, B. Birth of Three Stowaway-like MITE Families via Microhomology-Mediated Miniaturization of a Tc1/Mariner Element in the Yellow Fever Mosquito. Genome Biol. Evol. 2013, 5, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Lu, D.; Mason, A.S.; Li, B.; An, S.; Li, G.; Cai, D. Distribution of MITE Family Monkey King in Rapeseed (Brassica napus L) and Its Influence on Gene Expression. Genomics 2021, 113, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Spinelli, M.; Ye, C.; Li, Q.Q.; Liang, C. Genome-Wide Comparative Analysis of Miniature Inverted Repeat Transposable Elements in 19 Arabidopsis thaliana Ecotype Accessions. Sci. Rep. 2017, 7, 2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hu, Q.; Zhang, Y.; Lu, C.; Kuang, H. P-MITE: A Database for Plant Miniature Inverted-Repeat Transposable Elements. Nucleic Acids Res. 2014, 42, D1176–D1181. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Bao, Z.; Zhang, X.; Eddy, S.R.; Wessler, S.R. Pack-MULE Transposable Elements Mediate Gene Evolution in Plants. Nature 2004, 431, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Oki, N.; Yano, K.; Okumoto, Y.; Tsukiyama, T.; Teraishi, M.; Tanisaka, T. A Genome-Wide View of Miniature Inverted-Repeat Transposable Elements (MITEs) in Rice, Oryza sativa ssp. Japonica. Genes Genet. Syst. 2008, 83, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Sampath, P.; Lee, S.C.; Lee, J.; Izzah, N.K.; Choi, B.S.; Jin, M.; Park, B.S.; Yang, T.J. Characterization of a New High Copy Stowaway Family MITE, BRAMI-1 in Brassica Genome. BMC Plant Biol. 2013, 13, 56. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Hou, J.; Long, Y.; Wang, J.; Li, C.; Xiao, Q.; Jiang, X.; Zou, X.; Zou, J.; Meng, J. Widespread and Evolutionary Analysis of a MITE Family Monkey King in Brassicaceae. BMC Plant Biol. 2015, 15, 149. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, M.K.; Smith, A.M.; Ellis, T.H.N.; Hedley, C.; Martin, C. The Wrinkled-Seed Character of Pea Described by Mendel is Caused by a Transposon-like Insertion in a Gene Encoding Starch-Branching Enzyme. Cell 1990, 60, 115–122. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Martin, C.; Smith, A. The Importance of Starch Biosynthesis in the Wrinkled Seed Shape Character of Peas Studied by Mendel. Plant Mol. Biol. 1993, 22, 525–531. [Google Scholar] [CrossRef]
- Patel, M.; Jung, S.; Moore, K.; Powell, G.; Ainsworth, C.; Abbott, A. High-Oleate Peanut Mutants Result from a MITE Insertion into the FAD2 Gene. Theor. Appl. Genet. 2004, 108, 1492–1502. [Google Scholar] [CrossRef]
- Yang, M.; Xu, Y. Oleate Accumulation, Induced by Silencing of Microsomal Omega-6 Desaturase, Declines with Leaf Expansion in Transgenic Tobacco. J. Plant Physiol. 2007, 164, 23–30. [Google Scholar] [CrossRef]
- Momose, M.; Abe, Y.; Ozeki, Y. Miniature Inverted-Repeat Transposable Elements of Stowaway Are Active in Potato. Genetics 2010, 186, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.V.; Garvin, D.F.; Wang, Y.; Sorrells, M.E.; Klein, P.E.; Schaffert, R.E.; Li, L.; Kochian, L.V. Comparative Mapping of a Major Aluminum Tolerance Gene in Sorghum and Other Species in the Poaceae. Genetics 2004, 167, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.V.; Liu, J.; Guimarães, C.T.; Lana, U.G.P.; Alves, V.M.C.; Wang, Y.H.; Schaffert, R.E.; Hoekenga, O.A.; Piñeros, M.A.; Shaff, J.E.; et al. A Gene in the Multidrug and Toxic Compound Extrusion (MATE) Family Confers Aluminum Tolerance in Sorghum. Nat. Genet. 2007, 39, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Castelletti, S.; Tuberosa, R.; Pindo, M.; Salvi, S. A MITE Transposon Insertion is Associated with Differential Methylation at the Maize Flowering Time QTL Vgt1. G3 Genes Genomes Genet. 2014, 4, 805–812. [Google Scholar]
- Zerjal, T.; Rousselet, A.; Mhiri, C.; Combes, V.; Madur, D.; Grandbastien, M.A.; Charcosset, A.; Tenaillon, M.I. Maize Genetic Diversity and Association Mapping Using Transposable Element Insertion Polymorphisms. Theor. Appl. Genet. 2012, 124, 1521–1537. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Helguera, M.; Kato, K.; Fukuyama, S.; Sherman, J.; Dubcovsky, J. Allelic Variation at the VRN-1 Promoter Region in Polyploid Wheat. Theor. Appl. Genet. 2004, 109, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Loukoianov, A.; Tranquilli, G.; Helguera, M.; Fahima, T.; Dubcovsky, J. Positional Cloning of the Wheat Vernalization Gene VRN1. Proc. Natl. Acad. Sci. USA 2003, 100, 6263–6268. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Carver, B.F.; Yan, L. TamiR1123 Originated from a Family of Miniature Inverted-Repeat Transposable Elements (MITE) Including One Inserted in the Vrn-A1a Promoter in Wheat. Plant Sci. 2014, 215–216, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazaki, T.; Okumoto, Y.; Horibata, A.; Yamahira, S.; Teraishi, M.; Nishida, H.; Inoue, H.; Tanisaka, T. Mobilization of a Transposon in the Rice Genome. Nature 2003, 421, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Cao, X.; Song, X. Epigenetic Mutation of RAV6 Affects Leaf Angle and Seed Size in Rice. Plant Physiol. 2015, 169, 2118–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Gu, L.; Song, X.; Cui, X.; Lu, Z.; Zhou, M.; Wang, L.; Hu, F.; Zhai, J.; Meyers, B.C.; et al. Dicer-like 3 Produces Transposable Element-Associated 24-nt siRNAs That Control Agricultural Traits in Rice. Proc. Natl. Acad. Sci. USA 2014, 111, 3877–3882. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, R.K.; Martienssen, R. Transposable Elements and the Epigenetic Regulation of the Genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Fultz, D.; Choudury, S.G.; Slotkin, R.K. Silencing of Active Transposable Elements in Plants. Curr. Opin. Plant Biol. 2015, 27, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, P.; Zhai, J.; Zhou, M.; Ma, L.; Liu, B.; Jeong, D.H.; Nakano, M.; Cao, S.; Liu, C.; et al. Roles of DCL4 and DCL3b in Rice Phased Small RNA Biogenesis. Plant J. 2012, 69, 462–474. [Google Scholar] [CrossRef]
- Kasschau, K.D.; Fahlgren, N.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Carrington, J.C. Genome-Wide Profiling and Analysis of Arabidopsis siRNAs. PLoS Biol. 2007, 5, e57. [Google Scholar] [CrossRef] [Green Version]
- Pontier, D.; Yahubyan, G.; Vega, D.; Bulski, A.; Saez-Vasquez, J.; Hakimi, M.A.; Lerbs-Mache, S.; Colot, V.; Lagrange, T. Reinforcement of Silencing at Transposons and Highly Repeated Sequences Requires the Concerted Action of Two Distinct RNA Polymerases IV in Arabidopsis. Genes Dev. 2005, 19, 2030–2040. [Google Scholar] [CrossRef] [Green Version]
- Mosher, R.A.; Schwach, F.; Studholme, D.; Baulcombe, D.C. PolIVb Influences RNA-Directed DNA Methylation Independently of its Role in siRNA Biogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3145–3150. [Google Scholar] [CrossRef] [Green Version]
- Lucero, L.; Fonouni-Farde, C.; Crespi, M.; Ariel, F. Long Noncoding RNAs Shape Transcription in Plants. Transcription 2020, 11, 160–171. [Google Scholar] [CrossRef]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and Functional Diversification of Small RNA Pathways in Plants. PLoS Biol. 2004, 2, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasciolli, V.; Mallory, A.C.; Bartel, D.P.; Vaucheret, H. Partially Redundant Functions of Arabidopsis DICER-like Enzymes and a Role for DCL4 in Producing Trans-Acting siRNAs. Curr. Biol. 2005, 15, 1491–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontes, O.; Li, C.F.; Nunes, P.C.; Haag, J.; Ream, T.; Vitins, A.; Jacobsen, S.E.; Pikaard, C.S. The Arabidopsis Chromatin-Modifying Nuclear siRNA Pathway Involves a Nucleolar RNA Processing Center. Cell 2006, 126, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Axtell, M.J. AGO4 Is Specifically Required for Heterochromatic siRNA Accumulation at Pol V-Dependent Loci in Arabidopsis thaliana. Plant J. 2017, 90, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuthikattu, S.; McCue, A.D.; Panda, K.; Fultz, D.; DeFraia, C.; Thomas, E.N.; Keith Slotkin, R. The Initiation of Epigenetic Silencing of Active Transposable Elements is Triggered by RDR6 and 21-22 Nucleotide Small Interfering RNAs. Plant Physiol. 2013, 162, 116–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, K.; Ji, L.; Neumann, D.A.; Daron, J.; Schmitz, R.J.; Slotkin, R.K. Full-Length Autonomous Transposable Elements are Preferentially Targeted by Expression-Dependent Forms of RNA-Directed DNA Methylation. Genome Biol. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creasey, K.M.; Zhai, J.; Borges, F.; Van Ex, F.; Regulski, M.; Meyers, B.C.; Martienssen, R.A. MiRNAs Trigger Widespread Epigenetically Activated siRNAs from Transposons in Arabidopsis. Nature 2014, 508, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Panda, K.; McCue, A.D.; Slotkin, R.K. Arabidopsis RNA Polymerase IV Generates 21–22 Nucleotide Small RNAs That Can Participate in RNA-Directed DNA Methylation and May Regulate Genes. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190417. [Google Scholar] [CrossRef]
- Jia, Y.; Lisch, D.R.; Ohtsu, K.; Scanlon, M.J.; Nettleton, D.; Schnable, P.S. Loss of RNA-Dependent RNA Polymerase 2 (RDR2) Function Causes Widespread and Unexpected Changes in the Expression of Transposons, Genes, and 24-nt Small RNAs. PLoS Genet. 2009, 5, e1000737. [Google Scholar] [CrossRef] [Green Version]
- Hirochika, H.; Okamoto, H.; Kakutani, T. Silencing of Retrotransposons in Arabidopsis and Reactivation by the ddm1 Mutation. Plant Cell 2000, 12, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Miura, A.; Bender, J.; Jacobsen, S.E.; Kakutani, T. Role of CG and non-CG Methylation in Immobilization of Transposons in Arabidopsis. Curr. Biol. 2003, 13, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Li, Y.; Messing, J.; Dooner, H.K. Gene Movement by Helitron Transposons Contributes to the Haplotype Variability of Maize. Proc. Natl. Acad. Sci. USA 2005, 102, 9068–9073. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fefelova, N.; Caronna, J.; He, L.; Dooner, H.K. The Polychromatic Helitron Landscape of the Maize Genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19916–19921. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Stupar, R.M. Allelic Variation and Heterosis in Maize: How Do Two Halves Make More Than a Whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Vallejo, V.; Nobuta, K.; Slotkin, R.K.; Lisch, D.; Meyers, B.C.; Shiu, S.H.; Jiang, N. The Functional Role of Pack-MULEs in Rice Inferred From Purifying Selection and Expression Profile. Plant Cell 2009, 21, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Kuang, H.; Padmanabhan, C.; Li, F.; Kamei, A.; Bhaskar, P.B.; Ouyang, S.; Jiang, J.; Robin Buell, C.; Baker, B. Identification of Miniature Inverted-Repeat Transposable Elements (MITEs) and Biogenesis of Their siRNAs in the Solanaceae: New Functional Implications for MITEs. Genome Res. 2009, 19, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, Y.; Yang, K.; Sun, Z.; Fu, Y.; Chen, X.; Fang, R. Small RNAs from MITE-Derived Stem-Loop Precursors Regulate Abscisic Acid Signaling and Abiotic Stress Responses in Rice. Plant J. 2011, 65, 820–828. [Google Scholar] [CrossRef]
- Zhou, M.; Tao, G.; Pi, P.; Zhu, Y.; Bai, Y.; Meng, X. Genome-Wide Characterization and Evolution Analysis of Miniature Inverted-Repeat Transposable Elements (MITEs) in Moso Bamboo (Phyllostachys heterocycla). Planta 2016, 244, 775–787. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Jordan, I.K. Dual Coding of siRNAs and miRNAs by Plant Transposable Elements. RNA 2008, 14, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, C.; Xia, J.; Jin, Y. Domestication of Transposable Elements into Microrna Genes in Plants. PLoS ONE 2011, 6, e19212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.; Li, J.; Song, R.; Messing, J.; Chen, X. CARPEL FACTORY, a Dicer Homolog, and HEN1, a Novel Protein, Act in MicroRNA Metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Finnegan, E.J.; Margis, R.; Waterhouse, P.M. Posttranscriptional Gene Silencing Is Not Compromised in the Arabidopsis CARPEL FACTORY (DICER-LIKE1) Mutant, a Homolog of Dicer-1 from Drosophila. Curr. Biol. 2003, 13, 236–240. [Google Scholar] [CrossRef]
- Zhang, Q.; Arbuckle, J.; Wessler, S.R. Recent, Extensive, and Preferential Insertion of Members of the Miniature Inverted-Repeat Transposable Element Family Heartbreaker into Genic Regions of Maize. Proc. Natl. Acad. Sci. USA 2000, 97, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Wood, T.C.; Yu, Y.; Budiman, M.A.; Tomkins, J.; Woo, S.S.; Sasinowski, M.; Presting, G.; Frisch, D.; Goff, S.; et al. Rice Transposable Elements: A Survey of 73,000 Sequence-Tagged-Connectors. Genome Res. 2000, 10, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in Plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Poretti, M.; Praz, C.R.; Meile, L.; Kälin, C.; Schaefer, L.K.; Schläfli, M.; Widrig, V.; Sanchez-Vallet, A.; Wicker, T.; Bourras, S. Domestication of High-Copy Transposons Underlays the Wheat Small RNA Response to an Obligate Pathogen. Mol. Biol. Evol. 2020, 37, 839–848. [Google Scholar] [CrossRef]
- Crescente, J.M.; Zavallo, D.; del Vas, M.; Asurmendi, S.; Helguera, M.; Fernandez, E.; Vanzetti, L.S. Genome-Wide Identification of MITE-Derived MicroRNAs and Their Targets in Bread Wheat. BMC Genom. 2022, 23, 154. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Chen, T.; Zhu, X.; Fan, L.; Sun, J.; Llewellyn, D.J.; Wilson, I.; Zhu, Q.H. Expansion of MIR482/2118 by a Class-II Transposable Element in Cotton. Plant J. 2020, 103, 2084–2099. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like Genes, MYB33 and MYB65, Are MicroRNA-Regulated Genes That Redundantly Facilitate Anther Development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef] [Green Version]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C.P. MicroRNA-Mediated Repression of Rolled Leaf1 Specifies Maize Leaf Polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef]
- Subramanian, S.; Fu, Y.; Sunkar, R.; Barbazuk, W.B.; Zhu, J.K.; Yu, O. Novel and Nodulation-Regulated MicroRNAs in Soybean Roots. BMC Genom. 2008, 9, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanca, A.S.; Vicentini, R.; Ortiz-Morea, F.A.; Del Bem, L.E.V.; da Silva, M.J.; Vincentz, M.; Nogueira, F.T.S. Identification and Expression Analysis of MicroRNAs and Targets in the Biofuel Crop Sugarcane. BMC Plant Biol. 2010, 10, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Luo, Y.; Gong, X.; Zeng, W.; Li, S. Computational Identification of 48 Potato MicroRNAs and Their Targets. Comput. Biol. Chem. 2009, 33, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA Regulation of NAC-Domain Targets is Required for Proper Formation and Separation of Adjacent Embryonic, Vegetative, and Floral Organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Achard, P.; Herr, A.; Baulcombe, D.C.; Harberd, N.P. Modulation of Floral Development by a Gibberellin-Regulated MicroRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [Green Version]
- Aukerman, M.J.; Sakai, H. Regulation of Flowering Time and Floral Organ Identity by a MicroRNA and its APETALA2-Like Target Genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Pegler, J.L.; Oultram, J.M.J.; Nguyen, D.Q.; Grof, C.P.L.; Eamens, A.L. Microrna-Mediated Responses to Cadmium Stress in Arabidopsis thaliana. Plants 2021, 10, 130. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.M.J.; Grof, C.P.L.; Eamens, A.L. Molecular Manipulation of the MiR399/PHO2 Expression Module Alters the Salt Stress Response of Arabidopsis thaliana. Plants 2021, 10, 73. [Google Scholar] [CrossRef]
- Nguyen, D.Q.; Brown, C.W.; Pegler, J.L.; Eamens, A.L.; Grof, C.P.L. Molecular Manipulation of MicroRNA397 Abundance Influences the Development and Salt Stress Response of Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 7879. [Google Scholar] [CrossRef]
- He, X.F.; Fang, Y.Y.; Feng, L.; Guo, H.S. Characterization of Conserved and Novel MicroRNAs and Their Targets, Including a TuMV-Induced TIR-NBS-LRR Class R Gene-Derived Novel miRNA in Brassica. FEBS Lett. 2008, 582, 2445–2452. [Google Scholar] [CrossRef] [Green Version]
- López-Márquez, D.; Del-Espino, Á.; López-Pagán, N.; Rodríguez-Negrete, E.A.; Rubio-Somoza, I.; Ruiz-Albert, J.; Bejarano, E.R.; Beuzón, C.R. miR825-5p Targets the TIR-NBS-LRR Gene MIST1 and down-Regulates Basal Immunity against Pseudomonas syringae in Arabidopsis. J. Exp. Bot. 2021, 72, 7316–7334. [Google Scholar] [CrossRef] [PubMed]
- Radwan, O.; Liu, Y.; Clough, S.J. Transcriptional Analysis of Soybean Root Response to Fusarium virguliforme, the Causal Agent of Sudden Death Syndrome. Mol. Plant-Microbe Interact. 2011, 24, 958–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeeswaran, G.; Zheng, Y.; Sumathipala, N.; Jiang, H.; Arrese, E.L.; Soulages, J.L.; Zhang, W.; Sunkar, R. Deep Sequencing of Small RNA Libraries Reveals Dynamic Regulation of Conserved and Novel MicroRNAs and MicroRNA-Stars during Silkworm Development. BMC Genom. 2010, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, B.; Green, P.; Lu, C. MiRNAs in the Plant Genome: All Things Great and Small. Genome Dyn. 2008, 4, 108–118. [Google Scholar]
- Xie, Z.; Khanna, K.; Ruan, S. Expression of MicroRNAs and its Regulation in Plants. Semin. Cell Dev. Biol. 2010, 21, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Han, M.H.; Goud, S.; Song, L.; Fedoroff, N. The Arabidopsis Double-Stranded RNA-Binding Protein HYL1 Plays a Role in MicroRNA-Mediated Gene Regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Z.; Lu, F.; Dong, A.; Huang, H. SERRATE is a Novel Nuclear Regulator in Primary MicroRNA Processing in Arabidopsis. Plant J. 2006, 47, 841–850. [Google Scholar] [CrossRef]
- Lobbes, D.; Rallapalli, G.; Schmidt, D.D.; Martin, C.; Clarke, J. SERRATE: A New Player on the Plant MicroRNA Scene. EMBO Rep. 2006, 7, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Gasciolli, V.; Crété, P.; Vaucheret, H. The Nuclear dsRNA Binding Protein HYL1 is Required for MicroRNA Accumulation and Plant Development, but Not Posttranscriptional Transgene Silencing. Curr. Biol. 2004, 14, 346–351. [Google Scholar] [CrossRef]
- Dong, Z.; Han, M.H.; Fedoroff, N. The RNA-Binding Proteins HYL1 and SE Promote Accurate in vitro Processing of Pri-MiRNA by DCL1. Proc. Natl. Acad. Sci. USA 2008, 105, 9970–9975. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, Y.; Takashi, Y.; Watanabe, Y. The Interaction Between DCL1 and HYL1 is Important for Efficient and Precise Processing of Pri-MiRNA in Plant MicroRNA Biogenesis. RNA 2006, 12, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, J.; Cheng, Y.; Jia, D. HEN1 Functions Pleiotropically in Arabidopsis Development and Acts in C Function in the Flower. Development 2002, 129, 1085–1094. [Google Scholar] [CrossRef]
- Baumberger, N.; Baulcombe, D.C. Arabidopsis ARGONAUTE1 is an RNA Slicer That Selectively Recruits MicroRNAs and Short Interfering RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 11928–11933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaucheret, H.; Vazquez, F.; Crété, P.; Bartel, D.P. The Action of ARGONAUTE1 in the miRNA Pathway and its Regulation by the miRNA Pathway are Crucial for Plant Development. Genes Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Bollman, K.M.; Aukerman, M.J.; Park, M.Y.; Hunter, C.; Berardini, T.Z.; Poethig, S.R. HASTY, the Arabidopsis Ortholog of Exportin 5/MSN5, Regulates Phase Change and Morphogenesis. Development 2003, 130, 1493–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liu, L.; Zhuang, X.; Yu, Y.; Liu, X.; Cui, X.; Ji, L.; Pan, Z.; Cao, X.; Mo, B.; et al. MicroRNAs Inhibit the Translation of Target mRNAs on the Endoplasmic Reticulum in Arabidopsis. Cell 2013, 153, P562–P574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, R.S.; Hart-Smith, G.; Eamens, A.L.; Wilkins, M.R.; Waterhouse, P.M. Gene Regulation by Translational Inhibition is Determined by Dicer Partnering Proteins. Nat. Plants 2015, 1, 14027. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A Diverse and Evolutionarily Fluid Set of MicroRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, Y.; Hiraguri, A.; Moriyama, H.; Fukuhara, T. The dsRNA-Binding Protein DRB4 Interacts with the Dicer-like Protein DCL4 in vivo and Functions in the trans-acting siRNA Pathway. Plant Mol. Biol. 2007, 63, 777–785. [Google Scholar] [CrossRef]
- Pélissier, T.; Clavel, M.; Chaparro, C.; Pouch-PéLissier, M.N.; Vaucheret, H.; Deragon, J.M. Double-Stranded RNA Binding Proteins DRB2 and DRB4 Have an Antagonistic Impact on Polymerase IV-Dependent siRNA Levels in Arabidopsis. RNA 2011, 17, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Eamens, A.L.; Kim, K.W.; Curtin, S.J.; Waterhouse, P.M. DRB2 Is Required for MicroRNA Biogenesis in Arabidopsis thaliana. PLoS ONE 2012, 7, e35933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eamens, A.L.; Kim, K.W.; Waterhouse, P.M. DRB2, DRB3 and DRB5 Function in a Non-Canonical MicroRNA Pathway in Arabidopsis thaliana. Plant Signal. Behav. 2012, 7, 1224–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhu, Q.H.; Guo, X.; Gui, Y.; Bao, J.; Helliwell, C.; Fan, L. Molecular Evolution and Selection of a Gene Encoding Two Tandem MicroRNAs in Rice. FEBS Lett. 2007, 581, 4789–4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guddeti, S.; Zhang, D.C.; Li, A.L.; Leseberg, C.H.; Kang, H.; Li, X.G.; Zhai, W.X.; Johns, M.A.; Mao, L. Molecular Evolution of the Rice MIR395 Gene Family. Cell Res. 2005, 15, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, K.H.; Gouy, M.; Yang, Y.W.; Sharp, P.M.; Li, W.H. Date of the Monocot-Dicot Divergence Estimated from Chloroplast DNA Sequence Data. Proc. Natl. Acad. Sci. USA 1989, 86, 6201–6205. [Google Scholar] [CrossRef] [Green Version]
- Ganie, S.A.; Debnath, A.B.; Gumi, A.M.; Mondal, T.K. Comprehensive Survey and Evolutionary Analysis of Genome-Wide miRNA Genes From Ten Diploid Oryza Species. BMC Genom. 2017, 18, 711. [Google Scholar] [CrossRef] [Green Version]
- Lu, S. de novo Origination of MIRNAs through Generation of Short Inverted Repeats in Target Genes. RNA Biol. 2019, 16, 846–859. [Google Scholar] [CrossRef]
- Guo, Z.; Kuang, Z.; Tao, Y.; Wang, H.; Wan, M.; Hao, C.; Shen, F.; Yang, X.; Li, L. Miniature Inverted-Repeat Transposable Elements Drive Rapid MicroRNA Diversification in Angiosperms. Mol. Biol. Evol. 2022, 39, msac224. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Sung, G.H.; Spatafora, J.W.; Carrington, J.C. Evolution of MicroRNA Genes by Inverted Duplication of Target Gene Sequences in Arabidopsis thaliana. Nat. Genet. 2004, 36, 1282–1290. [Google Scholar] [CrossRef]
- Fahlgren, N.; Jogdeo, S.; Kasschau, K.D.; Sullivan, C.M.; Chapman, E.J.; Laubinger, S.; Smith, L.M.; Dasenko, M.; Givan, S.A.; Weigel, D.; et al. Microrna Gene Evolution in Arabidopsis Lyrata and Arabidopsis thaliana. Plant Cell 2010, 22, 1074–1089. [Google Scholar] [CrossRef] [Green Version]
- De Meaux, J.; Hu, J.Y.; Tartler, U.; Goebel, U. Structurally Different Alleles of the Ath-MIR824 MicroRNA Precursor Are Maintained at High Frequency in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2008, 105, 8994–8999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Mao, L. Evolution of Plant MicroRNA Gene Families. Cell Res. 2007, 17, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; D’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel Long Non-Protein Coding RNAs Involved in Arabidopsis Differentiation and Stress Responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; You, C.; Chen, X. The Evolution of MicroRNAs in Plants. Curr. Opin. Plant Biol. 2017, 35, 61–67. [Google Scholar] [CrossRef] [Green Version]
- De Felippes, F.F.; Schneeberger, K.; Dezulian, T.; Huson, D.H.; Weigel, D. Evolution of Arabidopsis thaliana MicroRNAs from Random Sequences. RNA 2008, 14, 2455–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, M.; Miura, S.; Nei, M. Origins and Evolution of MicroRNA Genes in Plant Species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread Translational Inhibition by Plant miRNAs and siRNAs. Science 2008, 320, 1185. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational Identification of Plant MicroRNAs and Their Targets, Including a Stress-Induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Zhang, S.D.; Ling, L.Z.; Zhang, Q.F.; Xu, J.D.; Cheng, L. Evolutionary Comparison of Two Combinatorial Regulators of SBP-Box Genes, miR156 and miR529, in Plants. PLoS ONE 2015, 10, e0124621. [Google Scholar] [CrossRef] [Green Version]
- Lorenzetti, A.P.; de Antonio, G.Y.; Paschoal, A.R.; Domingues, D.S. PlanTE-MIR DB: A Database for Transposable Element-Related MicroRNAs in Plant Genomes. Funct. Integr. Genom. 2016, 16, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Pedro, D.L.F.; Lorenzetti, A.P.R.; Domingues, D.S.; Paschoal, A.R. PlaNC-TE: A Comprehensive Knowledgebase of Non-Coding RNAs and Transposable Elements in Plants. Database 2018, 2018, bay078. [Google Scholar] [CrossRef]
- Arteaga-Vázquez, M.; Caballero-Pérez, J.; Vielle-Calzada, J.P. A Family of MicroRNAs Present in Plants and Animals. Plant Cell 2006, 18, 3355–3369. [Google Scholar] [CrossRef] [Green Version]
- De Vries, S.; Kloesges, T.; Rose, L.E. Evolutionarily Dynamic, But Robust, Targeting of Resistance Genes by the MIR482/2118 Gene Family in the Solanaceae. Genome Biol. Evol. 2015, 7, 3307–3321. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive Families of MIRNAs and PHAS Loci in Norway Spruce Demonstrate the Origins of Complex phasiRNA Networks in Seed Plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.C.M.; Baulcombe, D.C. A MicroRNA Superfamily Regulates Nucleotide Binding Site-Leucine-Rich Repeats and Other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarilar, V.; Marmagne, A.; Brabant, P.; Joets, J.; Alix, K. BraSto, a Stowaway MITE from Brassica: Recently Active Copies Preferentially Accumulate in the Gene Space. Plant Mol. Biol. 2011, 77, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Wang, B.; Zhu, X.; Mu, Q.; Wang, C.; Tao, R.; Fang, J. Cloning, Expression, and Characterization of MIR058 and Its Target PPO during the Development of Grapevine Berry Stone. Gene 2014, 548, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Figueroa, B.E.; Gao, L.; Wu, Z.; Zhou, X.; Zhu, J.; Jin, H.; Liu, R.; Zhu, J.K. High Throughput Sequencing Reveals Novel and Abiotic Stress-Regulated MicroRNAs in the Inflorescences of Rice. BMC Plant Biol. 2012, 12, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou-Yang, F.; Luo, Q.J.; Zhang, Y.; Richardson, C.R.; Jiang, Y.; Rock, C.D. Transposable Element-Associated MicroRNA Hairpins Produce 21-nt sRNAs Integrated Into Typical MicroRNA Pathways in Rice. Funct. Integr. Genom. 2013, 13, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, S.; Sánchez-Sanuy, F.; Camargo-Ramírez, R.; Gómez-Ariza, J.; Baldrich, P.; Campos-Soriano, L.; Soto-Suárez, M.; San Segundo, B. A Novel Transposable Element-Derived MicroRNA Participates in Plant Immunity to Rice Blast Disease. Plant Biotechnol. J. 2021, 19, 1798–1811. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Q.; Zhou, H.; Ni, F.; Wu, X.; Qi, Y. Rice MicroRNA Effector Complexes and Targets. Plant Cell 2009, 21, 3421–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA Methylation Mediated by a MicroRNA Pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pegler, J.L.; Oultram, J.M.J.; Mann, C.W.G.; Carroll, B.J.; Grof, C.P.L.; Eamens, A.L. Miniature Inverted-Repeat Transposable Elements: Small DNA Transposons That Have Contributed to Plant MICRORNA Gene Evolution. Plants 2023, 12, 1101. https://doi.org/10.3390/plants12051101
Pegler JL, Oultram JMJ, Mann CWG, Carroll BJ, Grof CPL, Eamens AL. Miniature Inverted-Repeat Transposable Elements: Small DNA Transposons That Have Contributed to Plant MICRORNA Gene Evolution. Plants. 2023; 12(5):1101. https://doi.org/10.3390/plants12051101
Chicago/Turabian StylePegler, Joseph L., Jackson M. J. Oultram, Christopher W. G. Mann, Bernard J. Carroll, Christopher P. L. Grof, and Andrew L. Eamens. 2023. "Miniature Inverted-Repeat Transposable Elements: Small DNA Transposons That Have Contributed to Plant MICRORNA Gene Evolution" Plants 12, no. 5: 1101. https://doi.org/10.3390/plants12051101