
The Contribution of Epigenetics to Evolutionary Adaptation in Zingiber kawagoii Hayata (Zingiberaceae) Endemic to Taiwan


Abstract
:1. Introduction
2. Materials and Methods
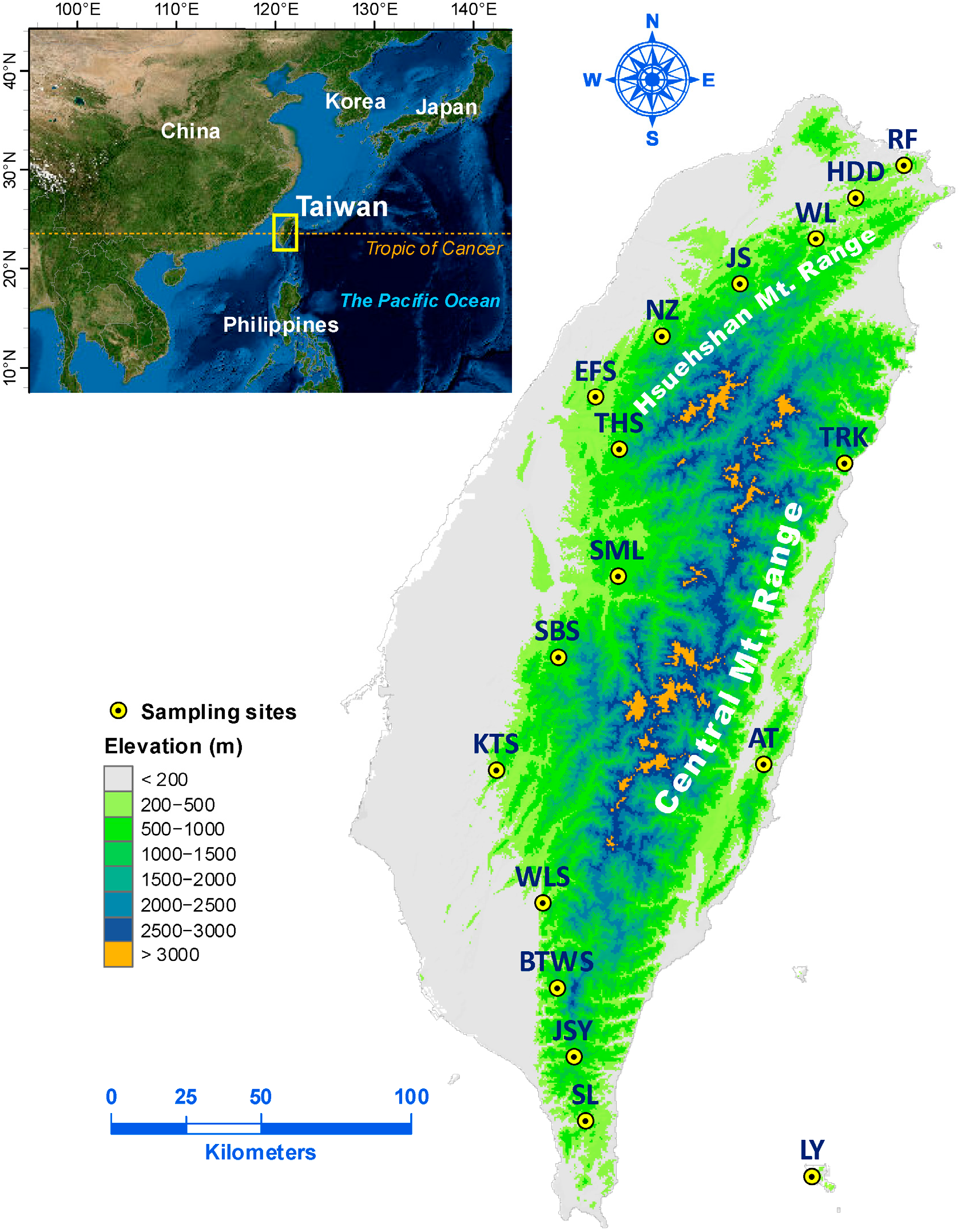
2.1. Sample Collection and Epigenotyping
2.2. Environmental Variables
2.3. Epigenetic Diversity, Differentiation, and Clustering
2.4. Test for FST Outliers
2.5. Test for Associations between Epigenetic Loci and Environmental Variables
2.6. Linear Relationships between Environmental Variables and the Ordination Axes of the Redundancy Analysis
2.7. Epigenetic Isolation-by-Environment Controlling for Geographic or Genetic Effects
3. Results
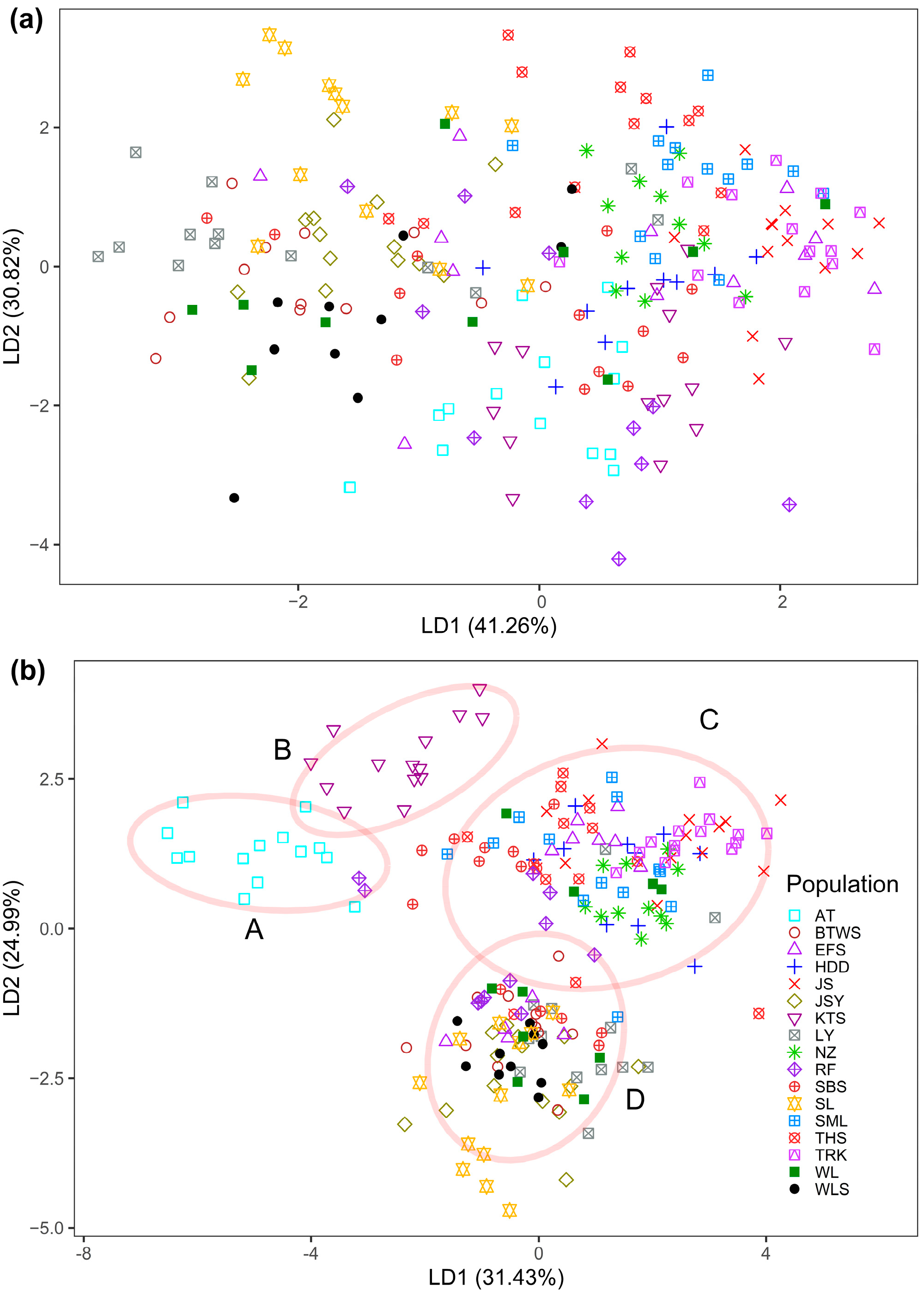
3.1. Epigenetic Diversity and Structure
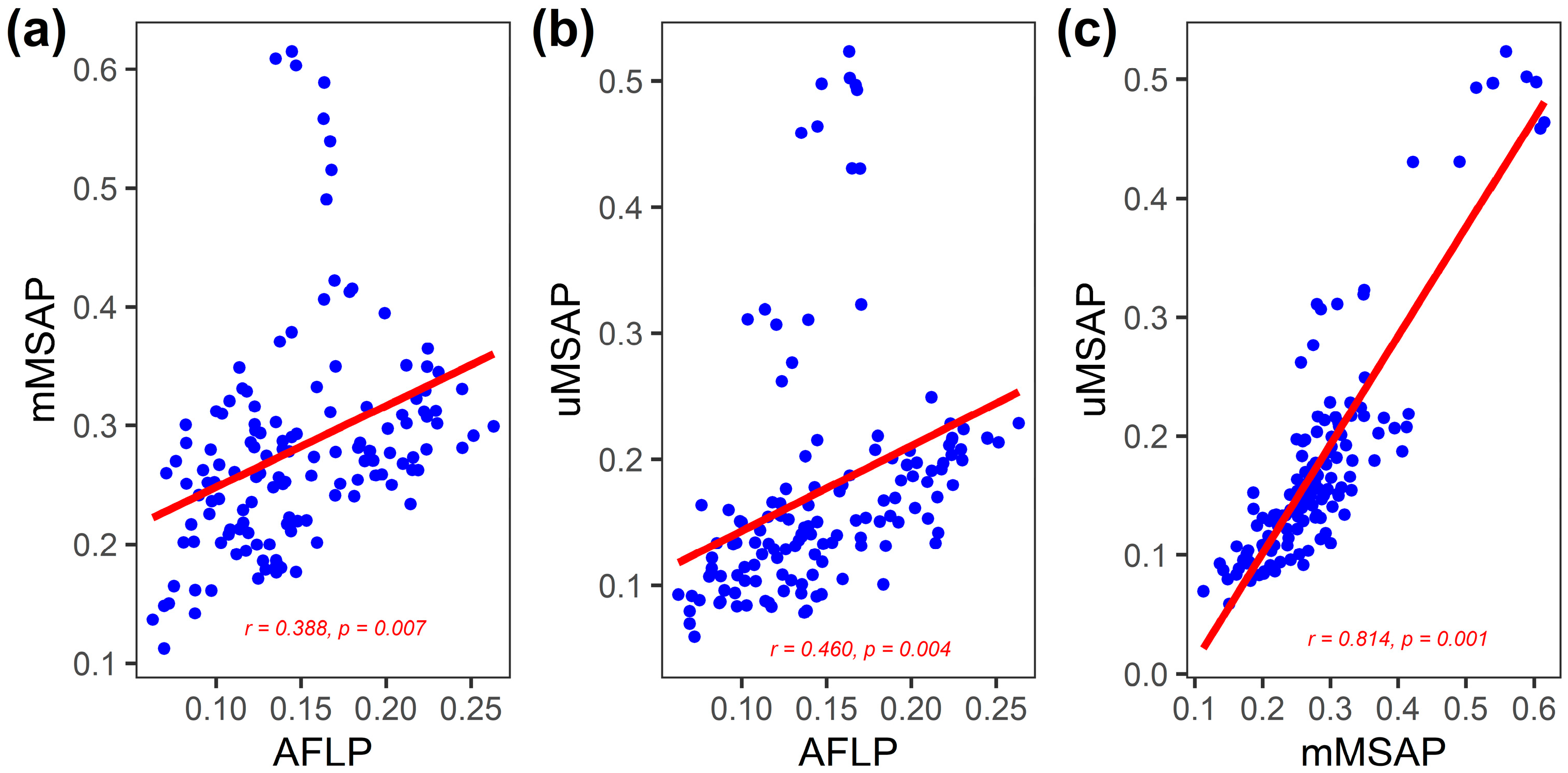
3.2. Inter-Population Relationship between Genetic and Epigenetic Variation
3.3. FST Outlier Identification and Association between Outliers and Environmental Variables
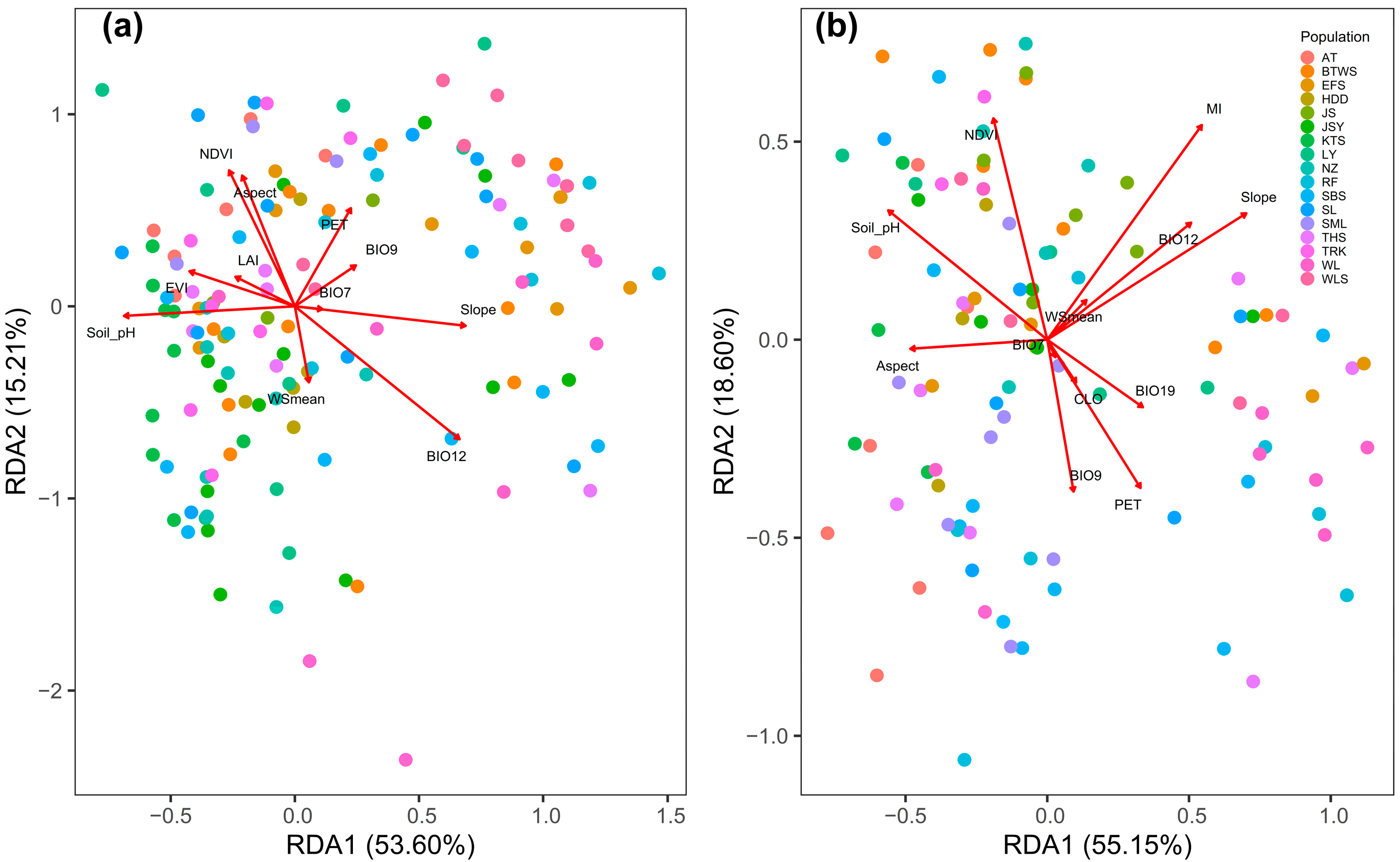
3.4. Environmental Effect on Outlier Epigenetic Variation
3.5. Contribution of Environment to Explaining the Total and Outlier Epigenetic Variation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ackerly, D.D. Community assembly, niche conservatism, and adaptive evolution in changing environments. Int. J. Plant Sci. 2003, 164, S165–S184. [Google Scholar] [CrossRef]
- Anderson, B.J.; Akçakaya, H.R.; Araújo, M.B.; Fordham, D.A.; Martinez-Meyer, E.; Thuiller, W.; Brook, B.W. Dynamics of range margins for metapopulations under climate change. Proc. R. Soc. B 2009, 276, 1415–1420. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.A.; Sgrὸ, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Evidence for ecological speciation and its alternative. Science 2009, 323, 737–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, C.L.; Bossdorf, O.; Pigliucci, M. What role does heritable epigenetic variation play in phenotypic evolution? BioScience 2010, 60, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Bossdorf, O.; Richards, C.L.; Pigliucci, M. Epigenetics for ecologists. Ecol. Lett. 2008, 11, 106–115. [Google Scholar] [CrossRef]
- Klironomos, F.D.; Berg, J.; Collins, S. How epigenetic mutations can affect genetic evolution: Model and mechanism. BioEssays 2013, 35, 571–578. [Google Scholar] [CrossRef]
- Jablonka, E.; Lachmann, M.; Lamb, M.J. Evidence, mechanisms and models for the inheritance of acquired characters. J. Theor. Biol. 1992, 158, 245–268. [Google Scholar] [CrossRef]
- Merilä, J.; Hendry, A.P. Climate change, adaptation, and phenotypic plasticity: The problem and the evidence. Evol. Appl. 2013, 7, 1–14. [Google Scholar] [CrossRef]
- Lande, R. Evolution of phenotypic plasticity in colonizing species. Mol. Ecol. 2015, 24, 2038–2045. [Google Scholar] [CrossRef]
- Furrow, R.E. Epigenetic inheritance, epimutation, and the response to selection. PLoS ONE 2014, 9, e101559. [Google Scholar] [CrossRef] [PubMed]
- Kronholm, I.; Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 2016, 25, 1856–1868. [Google Scholar] [CrossRef]
- Cervera, M.-T.; Ruiz-García, L.; Martínez-Zapater, J. Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genom. 2002, 268, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, R.A.; Wendel, J.F. Epigenetics and plant evolution. New Phytol. 2005, 168, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, M.W.; Tanurdžić, M.; Lippman, Z.; Jiang, H.; Carrasquillo, R.; Rabinowicz, P.D.; Dedhia, N.; McCombie, W.R.; Agier, N.; Bulski, A.; et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007, 5, e174. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress–induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010, 185, 1108–1118. [Google Scholar] [CrossRef]
- Richards, C.L.; Bossdorf, O.; Muth, N.Z.; Gurevitch, J.; Pigliucci, M. Jack of all trades, master of some? On the role of phenotypic plasticity in plant invasions. Ecol. Lett. 2006, 9, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Riddle, N.C.; Richards, E.J. The control of natural variation in cytosine methylation in Arabidopsis. Genetics 2002, 162, 355–363. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Genetic and epigenetic divergence between disturbed and undisturbed subpopulations of a Mediterranean shrub: A 20-year field experiment. Ecol. Evol. 2016, 6, 3832–3847. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.J.; Glor, R.E.; Losos, J.B. Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol. Lett. 2013, 16, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Sexton, J.P.; Hangartner, S.B.; Hoffmann, A.A. Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution 2014, 68, e12258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-S.; Liao, P.-C.; Chang, C.-T.; Hwang, S.-Y. Pattern of adaptive divergence in Zingiber kawagoii Hayata (Zingiberaceae) along a narrow latitudinal range. Plants 2022, 11, 2490. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-P. Contrasting pattern of population genetic structure and local adaptation of two Taiwan endemic ginger species. Master’s Thesis, National Taiwan Normal University, Taipei, Taiwan, 2020. [Google Scholar]
- Kirkpatrick, M.; Barton, N.H. Evolution of a species’ range. Am. Nat. 1997, 150, 286054. [Google Scholar] [CrossRef] [Green Version]
- Polechová, J.; Barton, N.H. Limits to adaptation along environmental gradients. Proc. Natl. Acad. Sci. USA 2015, 112, 6401–6406. [Google Scholar] [CrossRef] [Green Version]
- Herrera, C.M.; Bazaga, P. Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 2010, 187, 867–876. [Google Scholar] [CrossRef]
- Latzel, V.; Allan, E.; Silveira, A.B.; Colot, V.; Fischer, M.; Bossdorf, O. Epigenetic diversity increases the productivity and stability of plant populations. Nat. Commu. 2013, 4, 2875. [Google Scholar] [CrossRef] [Green Version]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjalmsson, B. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef]
- Foust, C.M.; Preite, V.; Schrey, A.W.; Alvarez, M.; Robertson, M.H.; Verhoeven, K.J.F.; Richards, C.L. Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol. Ecol. 2016, 25, 1639–1652. [Google Scholar] [CrossRef]
- Cao, J.-J.; Li, Y.-S.; Chang, C.-T.; Chung, J.-D.; Hwang, S.-Y. Adaptive divergence without distinct species relationships indicate early stage ecological speciation in species of the Rhododendron pseudochrysanthum complex endemic to Taiwan. Plants 2022, 11, 1226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.J.; Tricker, P.J. Epigenomic plasticity within populations: Its evolutionary significance and potential. Heredity 2010, 105, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-L.; Chen, J.-H.; Tsang, M.-H.; Chung, J.-D.; Chang, C.-T.; Hwang, S.-Y. Influences of environmental and spatial factors on genetic and epigenetic variations in Rhododendron oldhamii (Ericaceae). Tree Genet. Genom. 2015, 11, 823. [Google Scholar] [CrossRef]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic variation in mangrove plants occurring in contrasting natural environments. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef] [PubMed]
- Whipple, A.V.; Holeski, L.M. Epigenetic inheritance across the landscape. Front. Genet. 2016, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, G.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.Z.; Xu, C.G.; Saghai Maroof, M.A.; Zhang, Q. Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol. Gen. Genet. 1999, 261, 439–446. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Stucki, S.; Orozco-terWengel, P.; Forester, B.R.; Duruz, S.; Colli, L.; Masembe, C.; Negrini, R.; Landguth, E.; Jones, M.R.; The NEXTGEN Consortium; et al. High performance computation of landscape genomic models integrating local indices of spatial association. Mol. Ecol. Resour. 2017, 17, 1072–1089. [Google Scholar] [CrossRef] [Green Version]
- Foll, M.; Gaggiotti, O. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Goodrich, B.; Gabry, J.; Ali, I.; Brilleman, S. rstanarm: Bayesian Applied Regression Modeling via Stan. R Package Version 2.21.1. 2020. Available online: https://mc-stan.org/rstanarm (accessed on 3 April 2018).
- Borcard, D.; Legendre, P.; Drapeau, P. Partialling out the spatial component of ecological variation. Ecology 1992, 73, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure from small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Schulz, B.; Eckstein, R.L.; Durka, W. Scoring and analysis of methylation-sensitive amplification polymorphisms for epigenetic population studies. Mol. Ecol. Resour. 2013, 134, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing, (Version 3.6.3); R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 6 January 2022).
- Bonin, A.; Bellemain, E.; Bronken, E.P.; Pompanon, F.; Brochmann, C.; Taberlet, P. How to track and assess genotyping errors in population genetics studies. Mol. Ecol. 2004, 13, 3261–3273. [Google Scholar] [CrossRef]
- Vekemans, X.; Beauwens, T.; Lemaire, M.; Roldán-Ruiz, I. Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol. 2002, 11, 139–151. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; ISBN 9780231886710. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed effects models using lme4. J. Stat. Soft. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; Sage: Thousand Oaks, CA, USA, 2011; ISBN 978-141-297-518-8. Available online: http://socserv.socsci.mcmaster.ca/jfox/Books/Companion (accessed on 11 March 2019).
- Lenth, R.V. Emmeans: Estimated Marginal Means, Aka Least-Squares Means. R Package Version 1.8.0. 2022. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 6 January 2022).
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-2. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 15 January 2018).
- Dray, S.; Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Soft. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foll, M. Bayescan 2.1 User Manual. Available online: http://cmpg.unibe.ch/software/BayeScan/files/BayeScan2.1_manual.pdf (accessed on 23 June 2012).
- Storey, J.D.; Bass, A.J.; Dabney, A.; Robinson, D. qvalue: Q-Value Estimation for False Discovery Rate Control. R Package Version 2.28.0. 2022. Available online: http://github.com/jdstorey/qvalue (accessed on 8 January 2019).
- Schulz, B.; Eckstein, R.L.; Durka, W. Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 2014, 23, 3523–3537. [Google Scholar] [CrossRef]
- Liu, L.; Du, N.; Pei, C.; Guo, X.; Guo, W. Genetic and epigenetic variations associated with adaptation to heterogeneous habitat conditions in a deciduous shrub. Ecol. Evol. 2018, 8, 2594–2606. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-S.; Chang, C.-T.; Wang, C.-N.; Thomas, P.; Chung, J.-D.; Hwang, S.-Y. The contribution of neutral and environmentally dependent processes in driving population and lineage divergence in Taiwania (Taiwania cryptomerioides). Front. Plant Sci. 2018, 9, 1148. [Google Scholar] [CrossRef] [Green Version]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef]
- Jiang, C.; Mithani, A.; Belfield, E.J.; Mott, R.; Hurst, L.D.; Harberd, N.P. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 2014, 24, 1821–1829. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.L.; Schrey, A.W.; Pigliucci, M. Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with high epigenetic differentiation. Ecol. Lett. 2012, 15, 1016–1025. [Google Scholar] [CrossRef]
- Medrano, M.; Herrera, C.M.; Bazaga, P. Epigenetic variation predicts regional and local intraspecific functional diversity in a perennial herb. Mol. Ecol. 2014, 23, 4926–4938. [Google Scholar] [CrossRef]
- Trucchi, E.; Mazzarella, A.B.; Gilfillan, G.D.; Lorenzo, M.T.; Schönswetter, P.; Paun, O. BsRADseq: Screening DNA methylation in natural populations of non-model species. Mol. Ecol. 2016, 25, 1697–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Liu, X.; Li, J.; Zhao, H.; Li, Q.; Wang, Y.; Yuan, C.; Zhang, L.; Dong, Y. Genetic and epigenetic diversity of wild and cultivated soybean in local populations in Northern Huang Huai region of China. Plant Omics 2014, 7, 415–423. [Google Scholar]
- Richards, E.J. Population genetics. Curr. Opin. Genet. Dev. 2008, 18, 221–226. [Google Scholar] [CrossRef]
- Fraser, D.J.; Lippe, C.; Bernatchez, L. Consequences of unequal population size, asymmetric gene flow and sex-biased dispersal on population structure in brook charr (Salvelinus fontinalis). Mol. Ecol. 2004, 13, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Hänfling, B.; Weetman, D. Concordant genetic estimators of migration reveal anthropogenically enhanced source-sink population structure in the river sculpin, Cottus gobio. Genetics 2006, 173, 1487–1501. [Google Scholar] [CrossRef] [Green Version]
- Baulcombe, D.C.; Dean, C. Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 2014, 6, a019471. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P. Epigenetic variation and environmental change. J. Exp. Bot. 2015, 66, 3541–3548. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Sun, L.; Cheng, T.; Pan, H.; Wang, J.; Zhang, Q. Epigenetic variance, performing cooperative structure with genetics, is associated with leaf shape traits in widely distributed populations of ornamental tree Prunus mume. Front. Plant Sci. 2018, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Campoy, J.G.; Sobral, M.; Carro, B.; Lema, M.; Barreiro, R.; Retuerto, R. Epigenetic and phenotypic responses to experimental climate change of native and invasive Carpobrotus edulis. Front. Plant Sci. 2022, 13, 888391. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Z.; Yang, Y.; Liu, B. Epigenetic variation associated with responses to different habitats in the context of genetic divergence in Phragmites australlis. Ecol. Evol. 2021, 11, 11874–11889. [Google Scholar] [CrossRef] [PubMed]
- Zoldoš, V.; Biruš, I.; Muratović, E.; Šatović, Z.; Vojta, A.; Robin, O.; Pustahija, F.; Bogunić, F.; Bočkor, V.V.; Siljak-Yakovlev, S. Epigenetic differentiation of natural populations of Lilium bosniacum associated with contrasting habitat conditions. Genome Biol. Evol. 2018, 10, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammarco, I.; Münzbergová, Z.; Latzel, V. DNA methylation can mediate local adaptation and response to climate change in the clonal plant Fragaria vesca: Evidence from a European-scale reciprocal transplant experiment. Front. Plant Sci. 2022, 13, 827166. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; You, J.; Li, J.; Du, Y.; Zhao, W.; Chen, X. Genetic and epigenetic changes during the upward expansion of Deyeuxia angustifolia Kom. in the alpine tundra of the Changbai mountains, China. Plants 2021, 10, 291. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Latitude/Longitude | Altitude (m) | N | mMSAP | uMSAP | ||
|---|---|---|---|---|---|---|---|
| PPL (%) | uHE (SE) | PPL (%) | uHE (SE) | ||||
| Antong (AT) | 23.2847/121.3721 | 610 | 14 | 45.5 | 0.159 (0.007) | 31.9 | 0.112 (0.008) |
| Beitawushan (BTWS) | 22.6148/120.7022 | 1192 | 12 | 83.0 | 0.190 (0.005) | 34.7 | 0.140 (0.008) |
| Erfenshan (EFS) | 24.3919/120.8240 | 769 | 12 | 77.8 | 0.179 (0.006) | 30.5 | 0.133 (0.009) |
| Huangdidian (HDD) | 24.9894/121.6799 | 432 | 10 | 59.7 | 0.158 (0.007) | 38.1 | 0.109 (0.008) |
| Jianshi (JS) | 24.7307/121.2895 | 850 | 13 | 32.1 | 0.120 (0.007) | 26.3 | 0.093 (0.008) |
| Jinshuiying (JSY) | 22.4075/120.7564 | 1488 | 14 | 54.2 | 0.167 (0.006) | 37.3 | 0.150 (0.010) |
| Kantoushan (KTS) | 23.2671/120.5010 | 583 | 14 | 42.2 | 0.146 (0.008) | 31.1 | 0.096 (0.008) |
| Lanyu (LY) | 22.0496/121.5257 | 302 | 13 | 46.0 | 0.152 (0.006) | 33.1 | 0.136 (0.010) |
| Nanzhuang (NZ) | 24.5742/121.0436 | 467 | 11 | 49.1 | 0.130 (0.008) | 28.2 | 0.099 (0.008) |
| Ruifang (RF) | 25.0861/121.8385 | 349 | 11 | 76.9 | 0.185 (0.006) | 44.9 | 0.131 (0.008) |
| Shibishan (SBS) | 23.6077/120.7045 | 1347 | 13 | 82.3 | 0.179 (0.006) | 30.5 | 0.132 (0.010) |
| Shuangliu (SL) | 22.2140/120.7961 | 255 | 13 | 75.0 | 0.174 (0.006) | 30.2 | 0.128 (0.009) |
| Sunmoonlake (SML) | 23.8519/120.8982 | 816 | 13 | 33.0 | 0.119 (0.007) | 28.2 | 0.103 (0.008) |
| Tahsueshan (THS) | 24.2326/120.9003 | 937 | 14 | 47.4 | 0.158 (0.007) | 31.1 | 0.133 (0.010) |
| Taroko (TRK) | 24.1880/121.6382 | 929 | 14 | 40.1 | 0.136 (0.007) | 27.4 | 0.085 (0.007) |
| Wulai (WL) | 24.8663/121.5498 | 143 | 10 | 72.9 | 0.166 (0.005) | 45.2 | 0.134 (0.008) |
| Weiliaoshan (WLS) | 22.8695/120.6571 | 694 | 10 | 81.6 | 0.206 (0.005) | 42.4 | 0.099 (0.006) |
| Average | 12.4 | 58.8 | 0.160 | 33.6 | 0.119 | ||
| Source of Variation | df | Sum of Squares | Percent Variation | Φ Statistic (p) |
|---|---|---|---|---|
| mMSAP | ||||
| Total data | ||||
| Between populations | 16 | 1456.462 | 7.08 | ΦST = 0.071 (0.0001) |
| Within populations | 194 | 9083.036 | 92.92 | |
| Total | 210 | 10,539.498 | 100 | |
| Outlier data | ||||
| Between species | 16 | 131.396 | 28.39 | ΦST = 0.284 (0.0001) |
| Within populations | 194 | 269.249 | 71.61 | |
| Total | 210 | 400.645 | 100 | |
| uMSAP | ||||
| Total data | ||||
| Between populations | 16 | 1372.462 | 18.13 | ΦST = 0.181 (0.0001) |
| Within populations | 194 | 4442.694 | 81.87 | |
| Total | 210 | 5815.156 | 100 | |
| Outlier data | ||||
| Between species | 16 | 172.242 | 40.42 | ΦST = 0.404 (0.0001) |
| Within populations | 194 | 221.853 | 59.58 | |
| Total | 210 | 394.095 | 100 |
| Environmental Variable | mMSAP | uMSAP | ||
|---|---|---|---|---|
| R2 | Adjusted p | R2 | Adjusted p | |
| Aspect | 0.127 | 0.002 | 0.106 | 0.002 |
| Elevation | 0.005 | 0.655 | 0.001 | 0.968 |
| Slope | 0.065 | 0.004 | 0.104 | 0.002 |
| CLO | 0.009 | 0.455 | 0.083 | 0.002 |
| EVI | 0.079 | 0.002 | 0.016 | 0.247 |
| LAI | 0.055 | 0.005 | 0.013 | 0.303 |
| MI | 0.004 | 0.739 | 0.230 | 0.002 |
| NDVI | 0.091 | 0.002 | 0.076 | 0.002 |
| PET | 0.039 | 0.016 | 0.189 | 0.002 |
| RH | 0.021 | 0.159 | 0.009 | 0.409 |
| Soil pH | 0.100 | 0.002 | 0.042 | 0.015 |
| WSmean | 0.201 | 0.002 | 0.090 | 0.002 |
| BIO7 | 0.308 | 0.002 | 0.129 | 0.002 |
| BIO9 | 0.101 | 0.002 | 0.097 | 0.002 |
| BIO12 | 0.243 | 0.002 | 0.097 | 0.002 |
| BIO19 | 0.002 | 0.805 | 0.057 | 0.002 |
| Environmental Variable | mMSAP | uMSAP | ||
|---|---|---|---|---|
| RDA1 | RDA2 | RDA1 | RDA2 | |
| Aspect | −0.141 | 0.454 | −0.322 | −0.015 |
| Slope | 0.459 | −0.067 | 0.468 | 0.213 |
| CLO | 0.068 | −0.072 | ||
| EVI | −0.283 | 0.122 | ||
| LAI | −0.160 | 0.102 | ||
| MI | 0.364 | 0.361 | ||
| NDVI | −0.177 | 0.472 | −0.127 | 0.373 |
| PET | 0.151 | 0.341 | 0.219 | −0.249 |
| Soil pH | −0.459 | −0.033 | −0.374 | 0.218 |
| WSmean | 0.038 | −0.264 | 0.092 | 0.067 |
| BIO7 | 0.074 | −0.010 | 0.019 | −0.029 |
| BIO9 | 0.164 | 0.142 | 0.062 | −0.256 |
| BIO12 | 0.442 | −0.461 | 0.338 | 0.197 |
| BIO19 | 0.225 | −0.114 | ||
| Mantel Test | Partial Mantel Test | |||||||
|---|---|---|---|---|---|---|---|---|
| IBE | IBD | IBE controlling for geographic effect | IBE controlling for genetic effect | |||||
| r (p) | r (p) | r (p) | r (p) | |||||
| Total data | ||||||||
| mMSAP | 0.148 (0.002) | 0.126 (0.001) | 0.121 (0.013) | 0.091 (0.031) | ||||
| uMSAP | 0.195 (0.001) | 0.184 (0.001) | 0.150 (0.001) | 0.121 (0.001) | ||||
| Outlier data | ||||||||
| mMSAP | 0.243 (0.001) | 0.253 (0.001) | 0.191 (0.001) | 0.150 (0.001) | ||||
| uMSAP | 0.222 (0.001) | 0.188 (0.001) | 0.184 (0.001) | 0.191 (0.001) | ||||
| MMRR | ||||||||
| IBE | IBD | IBE controlling for geographic effect | IBE controlling for genetic effect | |||||
| r (p) | r (p) | R2 | βD (p) | βE (p) | R2 | βG (p) | βE (p) | |
| Total data | ||||||||
| mMSAP | 0.131 (0.002) | 0.112 (0.001) | 0.030 | 0.084 (0.002) | 0.110 (0.012) | 0.036 | 0.138 (0.009) | 0.086 (0.051) |
| uMSAP | 0.184 (0.001) | 0.174 (0.001) | 0.056 | 0.130 (0.001) | 0.145 (0.001) | 0.062 | 0.190 (0.001) | 0.121 (0.001) |
| Outlier data | ||||||||
| mMSAP | 0.163 (0.001) | 0.171 (0.001) | 0.098 | 0.138 (0.001) | 0.128 (0.001) | 0.102 | 0.182 (0.001) | 0.105 (0.002) |
| uMSAP | 0.157 (0.001) | 0.132 (0.001) | 0.068 | 0.100 (0.001) | 0.132 (0.001) | 0.070 | 0.122 (0.001) | 0.136 (0.001) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-S.; Liao, P.-C.; Chang, C.-T.; Hwang, S.-Y. The Contribution of Epigenetics to Evolutionary Adaptation in Zingiber kawagoii Hayata (Zingiberaceae) Endemic to Taiwan. Plants 2023, 12, 1558. https://doi.org/10.3390/plants12071558
Li Y-S, Liao P-C, Chang C-T, Hwang S-Y. The Contribution of Epigenetics to Evolutionary Adaptation in Zingiber kawagoii Hayata (Zingiberaceae) Endemic to Taiwan. Plants. 2023; 12(7):1558. https://doi.org/10.3390/plants12071558
Chicago/Turabian StyleLi, Yi-Shao, Pei-Chun Liao, Chung-Te Chang, and Shih-Ying Hwang. 2023. "The Contribution of Epigenetics to Evolutionary Adaptation in Zingiber kawagoii Hayata (Zingiberaceae) Endemic to Taiwan" Plants 12, no. 7: 1558. https://doi.org/10.3390/plants12071558