
Wheat Omics: Advancements and Opportunities

 ,
,  and
and
Abstract
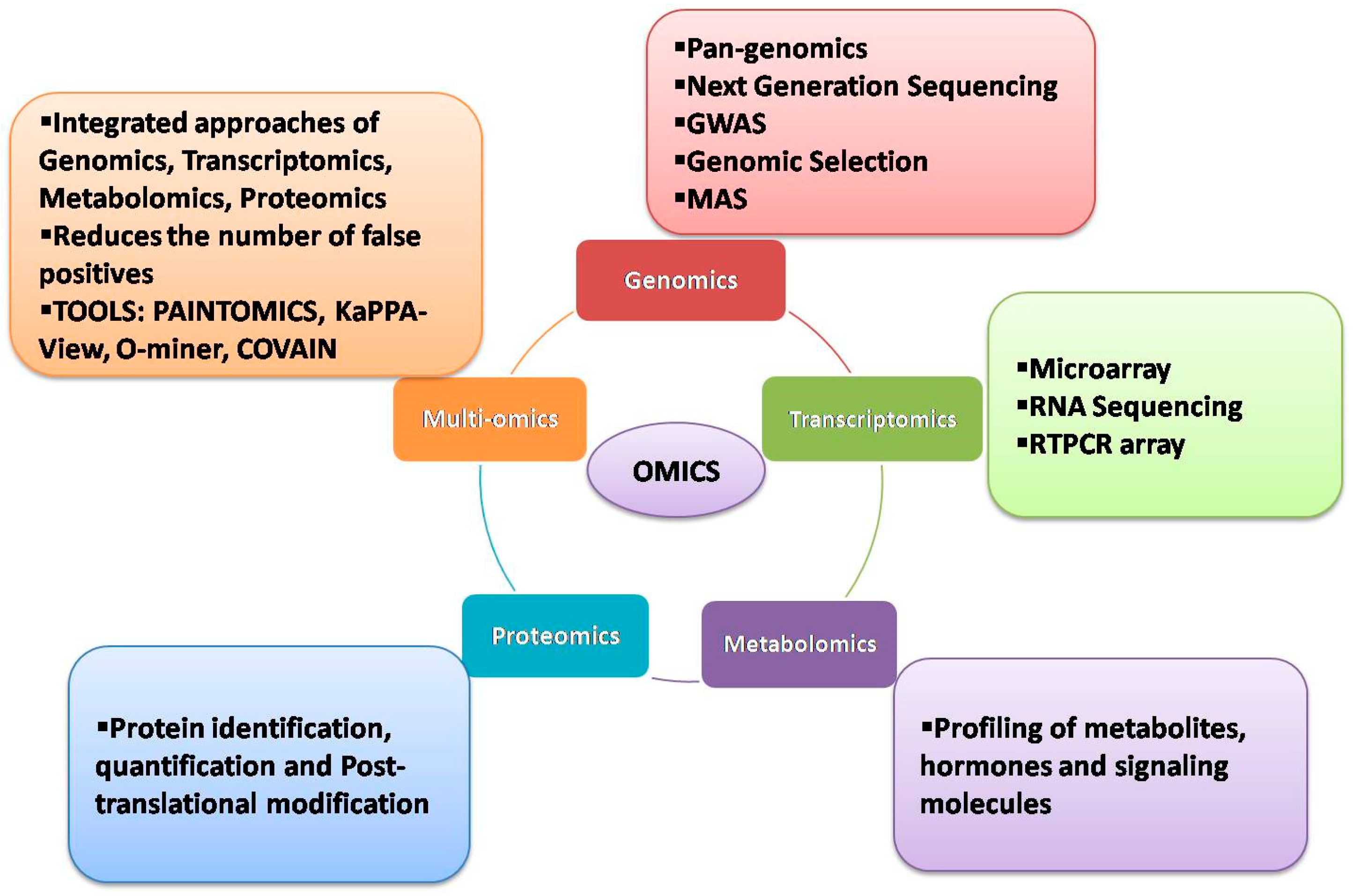
:1. Introduction
2. Genomic Approaches
3. Transcriptomic Approaches
4. Metabolomic Approaches
5. Proteomics Approaches
6. Multiomics Approaches
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAOSTAT. (FAO, 2022). Available online: https://www.fao.org/faostat/en/#data (accessed on 10 October 2022).
- Anwaar, H.A.; Perveen, R.; Mansha, M.Z.; Abid, M.; Sarwar, Z.M.; Aatif, H.M.; Alam, M.M. Assessment of grain yield indices in response to drought stress in wheat (Triticum aestivum L.). Saudi J. Biol. Sci. 2019, 27, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Xu, J.; Zou, X.; Cheng, Y.; Nasir, M.; Zhang, X. Omics approaches for engineering wheat production under abiotic stresses. Int. J. Mol. Sci. 2018, 19, 2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorilli, V.; Vannini, C.; Ortolani, F.; Garcia-Seco, D.; Chiapello, M.; Novero, M.; Domingo, G.; Terzi, V.; Morcia, C.; Bagnaresi, P.; et al. Omics approaches revealed how arbuscular mycorrhizal symbiosis enhances yield and resistance to leaf pathogen in wheat. Sci. Rep. 2018, 8, 9625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, M.; Lv, W.; Tang, X.; Zhao, D.; Wang, L.; Li, C.; Jiang, L. Overexpression of TaSNAC4-3D in common wheat (Triticum aestivum L.) negatively regulates drought tolerance. Front. Plant Sci. 2022, 13, 945272. [Google Scholar] [CrossRef]
- Yuan, Y.; Scheben, A.; Chan, C.K.K.; Edwards, D. Databases for wheatgenomics and crop improvement. In Wheat Biotechnology. Methods in Molecular Biology; Bhalla, P., Singh, M., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1679, pp. 277–291. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, L.; Rimbert, H.; Rodriguez, J.C.; Deal, K.R.; De Oliveira, R.; Choulet, F.; Keeble-Gagnere, G.; Tibbits, J.; Rogers, J.; et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 2021, 107, 303–314. [Google Scholar] [CrossRef]
- Ma, S.; Wang, M.; Wu, J.; Guo, W.; Chen, Y.; Li, G.; Wang, Y.; Shi, W.; Xia, G.; Fu, D.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant. 2021, 14, 1965–1968. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, C.; Chen, Z.; Gui, L.; Chen, C.; Li, D.; Xie, Z.; Zhang, Q.; Zhang, X.; Xia, C.; et al. WheatGmap: A comprehensive platform for wheat gene mapping and genomic studies. Mol. Plant. 2021, 14, 187–190. [Google Scholar] [CrossRef]
- Deshmukh, R.; Sonah, H.; Patil, G.; Echen, W.; Eprince, S.; Emutava, R.; Evuong, T.; Valliyodan, B.; Nguyen, H.T. Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 2014, 25, 244. [Google Scholar] [CrossRef]
- Yang, G.; Pan, W.; Zhang, R.; Pan, Y.; Guo, Q.; Song, W.; Zheng, W.; Nie, X. Genome-wide identification and characterization of caffeoyl-coenzyme A O-methyltransferase genes related to the Fusarium head blight response in wheat. BMC Genom. 2021, 22, 504. [Google Scholar] [CrossRef]
- Hussain, B.; Akpınar, B.A.; Alaux, M.; Algharib, A.M.; Sehgal, D.; Ali, Z.; Aradottir, G.I.; Batley, J.; Bellec, A.; Bentley, A.R.; et al. Capturing wheat phenotypes at the genome level. Front. Plant Sci. 2022, 13, 851079. [Google Scholar] [CrossRef]
- Rasheed, A.; Hao, Y.; Xia, X.; Khan, A.; Xu, Y.; Varshney, R.K.; He, Z. Crop breeding chips and genotyping platforms: Progress, challenges, and perspectives. Mol. Plant. 2017, 10, 1047–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrill, P.; Harrington, S.A.; Uauy, C. Applying the latest advances in genomics and phenomics for trait discovery in polyploid wheat. Plant J. 2019, 97, 56–72. [Google Scholar] [CrossRef]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef]
- Cavanagh, C.R.; Chao, S.; Wang, S.; Huang, B.E.; Stephen, S.; Kiani, S.; Forrest, K.; Saintenac, C.; Brown-Guedira, G.L.; Akhunova, A.; et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 8057–8062. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, B.; Lehnert, H.; Keilwagen, J.; Plieske, J.; Ordon, F.; Naseri Rad, S.; Ganal, M.; Beier, S.; Perovic, D. Comparison between core set selection methods using different Illumina marker platforms: A case study of assessment of diversity in wheat. Front. Plant Sci. 2020, 11, 1040. [Google Scholar] [CrossRef]
- Winfield, M.O.; Allen, A.M.; Burridge, A.J.; Barker, G.L.; Benbow, H.R.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; Scopes, G.; et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 2016, 14, 1195–1206. [Google Scholar] [CrossRef]
- Allen, A.M.; Winfield, M.O.; Burridge, A.J.; Downie, R.C.; Benbow, H.R.; Barker, G.L.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; et al. Characterization of a wheat breeder’ Array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum). Plant Biotechnol. J. 2017, 15, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Rimbert, H.; Darrier, B.; Navarro, J.; Kitt, J.; Choulet, F.; Leveugle, M.; Duarte, J.; Rivière, N.; Eversole, K.; Le Gouis, J.; et al. International wheat genome sequencing consortium, Le Gouis J. high throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 2018, 13, e0186329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeble-Gagnère, G.; Pasam, R.; Forrest, K.L.; Wong, D.; Robinson, H.; Godoy, J.; Rattey, A.; Moody, D.; Mullan, D.; Walmsley, T.; et al. Novel design of imputation-enabled snp arrays for breeding and research applications supporting multi-species hybridization. Front. Plant Sci. 2021, 12, 756877. [Google Scholar] [CrossRef]
- Sehgal, D.; Vikram, P.; Sansaloni, C.P.; Ortiz, C.; Pierre, C.S.; Payne, T.; Ellis, M.; Amri, A.; Petroli, C.D.; Wenzl, P.; et al. Exploring and mobilizing the gene bank biodiversity for wheat improvement. PLoS ONE 2015, 10, e0132112. [Google Scholar] [CrossRef]
- Ando, K.; Krishnan, V.; Rynearson, S.; Rouse, M.N.; Danilova, T.; Friebe, B.; See, D.; Pumphrey, M.O. Introgression of a novel ug99-effective stem rust resistance gene into wheat and development of Dasypyrum villosum chromosome-specific markers via genotyping-by-sequencing (GBS). Plant Dis. 2019, 103, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Alipour, H.; Bai, G.; Zhang, G.; Bihamta, M.R.; Mohammadi, V.; Peyghambari, S.A. Imputation accuracy of wheat genotyping-by-sequencing (GBS) data using barley and wheat genome references. PLoS ONE 2019, 14, e0208614. [Google Scholar] [CrossRef]
- Ladejobi, O.; Mackay, I.J.; Poland, J.; Praud, S.; Hibberd, J.M.; Bentley, A.R. Reference genome anchoring of high-density markers for association mapping and genomic prediction in European winter wheat. Front. Plant Sci. 2019, 10, 1278. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Abdelsalam, N.R.; Khalaf, L.; Chuang, W.P.; Zhao, L.; Smith, C.M.; Carver, B.; Bai, G. Development of single nucleotide polymorphism markers for the wheat curl mite resistance gene Cmc4. Crop Sci. 2019, 59, 1567–1575. [Google Scholar] [CrossRef]
- Edae, E.A.; Rouse, M.N. Association mapping of resistance to emerging stem rust pathogen races in spring wheat using genotyping-by-sequencing. Plant Genome 2020, 13, e20050. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, S.; Abdelsalam, N.R.; Carver, B.F.; Bai, G. Characterization of wheat curl mite resistance gene Cmc4 in OK05312. Theor. Appl. Genet. 2021, 134, 993–1005. [Google Scholar] [CrossRef]
- Juliana, P.; He, X.; Poland, J.; Roy, K.K.; Malaker, P.K.; Mishra, V.K.; Chand, R.; Shrestha, S.; Kumar, U.; Roy, C.; et al. Genomic selection for spot blotch in bread wheat breeding panels, full-sibs and half-sibs and index-based selection for spot blotch, heading and plant height. Theor. Appl. Genet. 2022, 13, 1965–1983. [Google Scholar] [CrossRef]
- Li, H.; Zhang, F.; Zhao, J.; Bai, G.; Amand, P.S.; Bernardo, A.; Ni, Z.; Sun, Q.; Su, Z. Identification of a novel major QTL from Chinese wheat cultivar Ji5265 for Fusarium head blight resistance in greenhouse. Theor. Appl. Genet. 2022, 135, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, X.; Wang, Z.; Ren, M.; An, C.; Zhu, S.; Xu, R. Genetic mapping of powdery mildew resistance genes in wheat landrace Guizi 1 via genotyping by sequencing. Mol. Biol. Rep. 2022, 49, 4461–4468. [Google Scholar] [CrossRef] [PubMed]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Puiu, D.; Hall, R.; Sarah Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 2017, 6, giw016. [Google Scholar] [CrossRef] [Green Version]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 7, 885–896. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef]
- Guo, W.; Xin, M.; Wang, Z.; Yao, Y.; Hu, Z.; Song, W.; Yu, K.; Chen, Y.; Wang, X.; Guan, P.; et al. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat. Commun. 2020, 11, 5085. [Google Scholar] [CrossRef]
- Shi, M.; Wang, F.; Lan, P.; Zhang, Y.; Zhang, M.; Yan, Y.; Liu, Y. Effect of ultrasonic intensity on structure and properties of wheat starch-monoglyceride complex and its influence on quality of norther-style Chinese steamed bread. LWT 2021, 138, 110677. [Google Scholar] [CrossRef]
- Sato, K.; Abe, F.; Mascher, M.; Haberer, G.; Gundlach, H.; Spannagl, M.; Shirasawa, K.; Isobe, S. Chromosome-scale genome assembly of the transformation-amenable common wheat cultivar ‘Fielder’. DNA Res. 2021, 28, dsab008. [Google Scholar] [CrossRef] [PubMed]
- Aury, J.M.; Engelen, S.; Istace, B.; Monat, C.; Lasserre-Zuber, P.; Belser, C.; Cruaud, C.; Rimbert, H.; Leroy, P.; Arribat, S.; et al. Long-read and chromosome-scale assembly of the hexaploid wheat genome achieves high resolution for research and breeding. GigaScience 2022, 11, giac034. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Teng, W.; Fang, S.; Li, H.; Li, B.; Chu, J.; Li, Z.; Zheng, Q. Transcriptome analysis of salt-stress response in three seedling tissues of commonwheat. Crop J. 2019, 7, 378–392. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Huang, M.; Yao, Y.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.G.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Ling, H.Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Jayakodi, M.; Schreiber, M.; Stein, N.; Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 2021, 28, dsaa030. [Google Scholar] [CrossRef]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.K.K.; Visendi, P.; Lai, K.; Dolezel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Xiao, Y.; Wen, Y.; Li, K.; Ma, Z.; Yang, L.; Zhu, Y.; Yin, J. Genome-wide characterization and function analysis uncovered roles of wheat LIMs in responding to adverse stresses and TaLIM8-4D function as a susceptible gene. Plant Genome 2022, 15, e20246. [Google Scholar] [CrossRef]
- Wang, L.; Xiang, L.; Hong, J.; Xie, Z.; Li, B. Genome-wide analysis of bHLH transcription factor family reveals their involvement in biotic and abiotic stress responses in wheat (Triticum aestivum L.). 3 Biotech 2019, 9, 236. [Google Scholar] [CrossRef]
- Benbow, H.R.; Jermiin, L.S.; Doohan, F.M. Serpins: Genome-wide characterisation and expression analysis of the serine protease inhibitor family in Triticum aestivum. G3-Genes Genomes Genet. 2019, 9, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, Y.; Li, Z.; Gao, X.; Dong, J.; Zhang, J.; Zhang, L.; Thomashow, L.S.; Weller, D.M.; Yang, M. Genome-wide identification and expression profile analysis of the phospholipase C gene family in wheat (Triticum aestivum L.). Plant 2020, 7, 885. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Z.Y.; Kumar, V.; Xu, X.F.; Zhu, X.F.; Li, T.Y.; Jia, B.; Xuan, Y.H. Genome-wide identification of the SWEET gene family in wheat. Gene 2018, 642, 284–292. [Google Scholar] [CrossRef]
- Guo, J.; Islam, M.A.; Lin, H.; Ji, C.; Duan, Y.; Liu, P.; Zeng, Q.; Day, B.; Kang, Z.; Guo, J. Genome-wide identification of cyclic nucleotide-gated ion channel gene family in wheat and functional analyses of TaCNGC14 and TaCNGC16. Front. Plant Sci. 2018, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Feng, D. Genome-wide identification of the aspartic protease gene family and their response under powdery mildew stress in wheat. Mol. Biol. Rep. 2020, 47, 8949–8961. [Google Scholar] [CrossRef]
- Niu, H.; Xia, P.; Hu, Y.; Zhan, C.; Li, Y.; Gong, S.; Li, Y.; Ma, D. Genome-wide identification of ZF-HD gene family in Triticum aestivum: Molecular evolution mechanism and function analysis. PLoS ONE 2021, 16, e0256579. [Google Scholar] [CrossRef]
- Liu, Y.; Han, N.; Wang, S.; Chen, C.; Lu, J.; Riaz, M.W.; Si, H.; Sun, G.; Ma, C. Genome-wide identification of Triticum aestivum xylanase inhibitor gene family and inhibitory effects of XI-2 subfamily proteins on Fusarium graminearum GH11 xylanase. Front. Plant Sci. 2021, 12, 665501. [Google Scholar] [CrossRef]
- Ru, J.N.; Hou, Z.H.; Zheng, L.; Zhao, Q.; Wang, F.Z.; Chen, J.; Zhou, Y.B.; Chen, M.; Ma, Y.Z.; Xi, Y.J.; et al. Genome-wide analysis of DEAD-box RNA helicase family in wheat (Triticum aestivum) and functional identification of TaDEAD-box57 in abiotic stress responses. Front. Plant Sci. 2021, 12, 797276. [Google Scholar] [CrossRef]
- Ye, H.; Qiao, L.; Guo, H.; Guo, L.; Ren, F.; Bai, J.; Wang, Y. Genome-wide identification of wheat WRKY gene family reveals that TaWRKY75-A is referred to drought and salt resistances. Front. Plant Sci. 2021, 12, 663118. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, X.; Shao, W.; Song, J.; Jiang, W.; He, Y.; Yin, J.; Ma, D.; Qiao, Y. Genome-wide mining of wheat DUF966 gene family provides new insights into salt stress responses. Front. Plant Sci. 2020, 11, 569838. [Google Scholar] [CrossRef]
- Xiao, J.; Hu, R.; Gu, T.; Han, J.; Qiu, D.; Su, P.; Feng, J.; Chang, J.; Yang, G.; He, G. Genome-wide identification and expression profiling of trihelix gene family under abiotic stresses in wheat. BMC Genom. 2019, 20, 287. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, S.; Chunduri, V.; Kaur, A.; Kaur, S.; Malhotra, N.; Kumar, A.; Kapoor, P.; Kumari, A.; Kaur, J.; et al. Genome-wide identification and characterization of heat shock protein family reveals role in development and stress conditions in Triticum aestivum L. Sci. Rep. 2020, 10, 7858. [Google Scholar] [CrossRef]
- Guerin, C.; Roche, J.; Allard, V.; Ravel, C.; Mouzeyar, S.; Bouzidi, M.F. Genome-wide analysis, expansion and expression of the NAC family under drought and heat stresses in bread wheat (T. aestivum L.). PLoS ONE 2019, 14, e0213390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, P.; Baranwal, V.K.; Khurana, P. Genome-wide analysis of bZIP transcription factors in wheat and functional characterization of a TabZIP under abiotic stress. Sci. Rep. 2019, 9, 4608. [Google Scholar] [CrossRef] [Green Version]
- Zan, T.; Li, L.; Xie, T.; Zhang, L.; Li, X. Genome-wide identification and abiotic stress response patterns of abscisic acid stress ripening protein family members in Triticum aestivum L. Genomics 2020, 112, 3794–3802. [Google Scholar] [CrossRef]
- Jiang, W.; Geng, Y.; Liu, Y.; Chen, S.; Cao, S.; Li, W.; Chen, H.; Ma, D.; Yin, J. Genome-wide identification and characterization of SRO gene family in wheat: Molecular evolution and expression profiles during different stresses. Plant Physiol. Biochem. 2020, 154, 590–611. [Google Scholar] [CrossRef]
- Han, Z.; Liu, Y.; Deng, X.; Liu, D.; Liu, Y.; Hu, Y.; Yan, Y. Genome-wide identification and expression analysis of expansin gene family in common wheat (Triticum aestivum L.). BMC Genom. 2019, 20, 101. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Han, Y.; Feng, X.; Gao, H.; Cao, B.; Song, L. Genome-wide identification of BAM (β-amylase) gene family in jujube (Ziziphus jujuba Mill.) and expression in response to abiotic stress. BMC Genom. 2022, 23, 438. [Google Scholar] [CrossRef]
- Zan, T.; Zhang, L.; Xie, T.; Li, L. Genome-wide identification and analysis of the growth-regulating factor (GRF) gene family and GRF-interacting factor family in Triticum aestivum L. Biochem. Genet. 2020, 58, 705–724. [Google Scholar] [CrossRef]
- Tian, R.; Yang, Y.; Chen, M. Genome-wide survey of the amino acid transporter gene family in wheat (Triticum aestivum L.): Identification, expression analysis and response to abiotic stress. Int. J. Biol. Macromol. 2020, 162, 1372–1387. [Google Scholar] [CrossRef]
- Sehgal, D.; Dreisigacker, S. GWAS case studies in wheat. In Genome-Wide Association Studies. Methods in Molecular Biology; Torkamaneh, D., Belzile, F., Eds.; Humana: New York, NY, USA, 2022; Volume 2481, pp. 341–351. [Google Scholar] [CrossRef]
- Saini, D.K.; Srivastava, P.; Pal, N.; Gupta, P.K. Meta-QTLs, ortho-meta-QTLs and candidate genes for grain yield and associated traits in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2022, 135, 1049–1081. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, D.; Autrique, E.; Singh, R.; Ellis, M.; Singh, S.; Dreisigacker, S. Identification of genomic regions for grain yield and yield stability and their epistatic interactions. Sci. Rep. 2017, 7, 41578. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, D.; Mondal, S.; Guzman, C.; Barrios, G.G.; Franco, C.; Singh, R.; Dreisigacker, S. Validation of candidate gene-based markers and identification of novel loci for thousand-grain weight in spring bread wheat. Front. Plant Sci. 2019, 10, 1189. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, D.; Mondal, S.; Crespo-Herrera, L.; Velu, G.; Juliana, P.; Huerta-Espino, J.; Shrestha, S.; Poland, J.; Singh, R.; Dreisigacker, S. Haplotype-based, genome-wide association study reveals stable genomic regions for grain yield in CIMMYT spring bread wheat. Front. Genet. 2020, 11, 589490. [Google Scholar] [CrossRef]
- Sehgal, D.; Rosyara, U.; Mondal, S.; Singh, R.; Poland, J.; Dreisigacker, S. Incorporating genome-wide association mapping results into genomic prediction models for grain yield and yield stability in CIMMYT spring bread wheat. Front. Plant Sci. 2020, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Ledesma-Ramirez, L.; Solis-Moya, E.; Iturriaga, G.; Sehgal, D.; Reyes-Valdes, M.H.; Montero-Tavera, V.; Sansaloni, C.P.; Burgueno, J.; Ortiz, C.; Aguirre-Mancilla, C.L.; et al. GWAS to identify genetic loci for resistance to yellow rust in wheat pre-breeding lines derived from diverse exotic crosses. Front. Plant Sci. 2019, 10, 1390. [Google Scholar] [CrossRef]
- Li, F.; Wen, W.; Liu, J.; Zhang, Y.; Cao, S.; He, Z.; Rasheed, A.; Jin, H.; Zhang, C.; Yan, J.; et al. Genetic architecture of grain yield in bread wheat based on genome-wide association studies. BMC Plant Biol. 2019, 19, 168. [Google Scholar] [CrossRef]
- Kokhmetova, A.; Sehgal, D.; Ali, S.; Atishova, M.; Kumatbayeva, M.; Leonova, I.; Dreisigacker, S. Genome-wide association study of tan spot resistance in a hexaploid wheat collection from Kazakhstan. Front. Genet. 2021, 11, 581214. [Google Scholar] [CrossRef]
- Singh, S.; Vikram, P.; Sehgal, D.; Burgueño, J.; Sharma, A.; Singh, S.K.; Sansaloni, C.P.; Joynson, R.; Brabbs, T.; Ortiz, C.; et al. Harnessing genetic potential of wheat germplasm banks through impact oriented-prebreeding for future food and nutritional security. Nat. Sci. Rep. 2018, 8, 5640. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Jighly, A.; Sehgal, D.; Burgueño, J.; Joukhadar, R.; Sharma, A.; Vikram, P.; Sansaloni, C.P.; Govindan, V.; Bhavani, S.; et al. Direct introgression of untapped diversity into elite wheat lines. Nat. Food 2021, 2, 819–827. [Google Scholar] [CrossRef]
- Shokat, S.; Sehgal, D.; Fulai, L.; Sukhwinder, S. GWAS analysis of wheat pre-breeding germplasm for terminal drought stress using next generation sequencing technology. Int. J. Mol. Sci. 2020, 21, 3156. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, D.; Mondal, S.; Burgeno, J.; Rosyara, U.; Bentley, A.R.; Dreisigacker, S. Genomic selection in wheat: Progress, opportunities and challenges. In Genomic Selection in Plants: A Guide for Breeders, 1st ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2022; pp. 51–67. ISBN 9781003214991. [Google Scholar]
- Afzal, F.; Li, H.; Gul, A.; Subhani, A.; Ali, A.; Mujeeb-Kazi, A.; Ogbonnaya, F.; Trethowan, R.; Xia, X.; He, Z.; et al. Genome-wide analyses reveal footprints of divergent selection and drought adaptive traits in synthetic-derived wheats. G3-Genes Genomes Genet. 2019, 9, 1957–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, U.; Alipour, H.; Gul, A.; Jing, L.; Darvishzadeh, R.; Amir, R.; Munir, F.; Ilyas, M.K.; Ghafoor, A.; Siddiqui, S.U.; et al. Characterization of the genetic basis of local adaptation of wheat landraces from Iran and Pakistan using genome-wide association study. Plant Genome 2021, 14, e20096. [Google Scholar] [CrossRef] [PubMed]
- Mondaini, A.; Rosyara, U.; Sehgal, D.; Dreisigacker, S. Selection signatures in the CIMMYT International Elite Spring and Semi-arid Wheat Yield Trials. Plant Genome 2021, 15, e20165. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, N.M.; Mago, R.; Panwar, V.; Hewitt, T.; Luo, M.; Chen, J.; Sperschneider, J.; Nguyen-Phuc, H.; Wang, A.; Ortiz, D.; et al. Genomics accelerated isolation of a new stem rust avirulence gene–wheat resistance gene pair. Nat. Plants 2021, 7, 1220–1228. [Google Scholar] [CrossRef]
- Rutkoski, J.; Poland, J.; Mondal, S.; Autrique, E.; Perez, L.G.; Crossa, J.; Reynolds, M.; Singh, R. Canopy temperature and vegetation indices from high-throughput phenotyping improve accuracy of pedigree and genomic selection for grain yield in wheat. G3-Genes Genomes Genet. 2016, 6, 2799–2808. [Google Scholar] [CrossRef] [Green Version]
- Dunckel, S.; Crossa, J.; Wu, S.; Bonnett, D.; Poland, J. Genomic selection for increased yield in synthetic-derived wheat. J. Crop Sci. 2017, 57, 713–725. [Google Scholar] [CrossRef]
- Sun, J.; Rutkoski, J.E.; Poland, J.A.; Crossa, J.; Jannink, J.L.; Sorrells, M.E. Multitrait, random regression, or simple repeatability model in high-throughput phenotyping data improve genomic prediction for wheat grain yield. Plant Genome 2017, 10, plantgenome2016-11. [Google Scholar] [CrossRef]
- Crain, J.; Mondal, S.; Rutkoski, J.; Singh, R.P.; Poland, J. Combining high-throughput phenotyping and genomic information to increase prediction and selection accuracy in wheat breeding. Plant Genome 2018, 11, 170043. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Jarquin, D.; Crossa, J.; Reynolds, M. Genomic-enabled prediction accuracies increased by modeling genotype× environment interaction in durum wheat. Plant Genome 2018, 11, 170112. [Google Scholar] [CrossRef]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef]
- Merida-Garcia, R.; Liu, G.; He, S.; Gonzalez-Dugo, V.; Dorado, G.; Galvez, S.; Solis, I.; Zarco-Tejada, P.J.; Reif, J.C.; Hernandez, P. Genetic dissection of agronomic and quality traits based on association mapping and genomic selection approaches in durum wheat grown in Southern Spain. PLoS ONE 2019, 14, e0211718. [Google Scholar] [CrossRef] [Green Version]
- Sarinelli, J.M.; Murphy, J.P.; Tyagi, P.; Holland, J.B.; Johnson, J.W.; Mergoum, M.; Mason, R.E.; Babar, A.; Harrison, S.; Sutton, R.; et al. Training population selection and use of fixed effects to optimize genomic predictions in a historical USA winter wheat panel. Theor. Appl. Genet. 2019, 132, 1247–1261. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.E.; Poland, J.A.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.L.; Sorrells, M.E. Genomic selection for quantitative adult plant stem rust resistance in wheat. Plant Genome 2014, 7, plantgenome2014-02. [Google Scholar] [CrossRef] [Green Version]
- Herter, C.P.; Ebmeyer, E.; Kollers, S.; Korzun, V.; Würschum, T.; Miedaner, T. Accuracy of within-and among-family genomic prediction for Fusarium head blight and Septoria tritici blotch in winter wheat. Theor. Appl. Genet. 2019, 132, 1121–1135. [Google Scholar] [CrossRef]
- Verges, V.L.; Lyerly, J.; Dong, Y.; Van Sanford, D.A. Training Population Design With the Use of Regional Fusarium Head Blight Nurseries to Predict Independent Breeding Lines for FHB Traits. Front. Plant Sci. 2020, 11, 1083. [Google Scholar] [CrossRef] [PubMed]
- Larkin, D.L.; Holder, A.L.; Mason, R.E.; Moon, D.E.; Brown-Guedira, G.; Price, P.P.; Harrison, S.A.; Dong, Y. Genome-wide analysis and prediction of Fusarium head blight resistance in soft red winter wheat. Crop Sci. 2020, 60, 2882–2900. [Google Scholar] [CrossRef]
- Juliana, P.; Singh, R.P.; Singh, P.K.; Crossa, J.; Rutkoski, J.E.; Poland, J.A.; Bergstrom, G.C.; Sorrells, M.E. Comparison of models and whole-genome profiling approaches for genomic-enabled prediction of septoria tritici blotch, stagonosporanodorum blotch, and tan spot resistance in wheat. Plant Genome 2017, 10, plantgenome2016-08. [Google Scholar] [CrossRef]
- Alemu, A.; Brazauskas, G.; Gaikpa, D.S.; Henriksson, T.; Islamov, B.; Jørgensen, L.N.; Koppel, M.; Koppel, R.; Liatukas, Z.; Svensson, J.T.; et al. Genome-wide association analysis and genomic prediction for adult-plant resistance to Septoria tritici blotch and powdery mildew in winter wheat. Front. Genet. 2021, 12, 661742. [Google Scholar] [CrossRef]
- Juliana, P.; Singh, R.P.; Singh, P.K.; Crossa, J.; Huerta-Espino, J.; Lan, C.; Bhavani, S.; Rutkoski, J.E.; Poland, J.A.; Bergstrom, G.C.; et al. Genomic and pedigree-based prediction for leaf, stem, and stripe rust resistance in wheat. Theor. Appl. Genet. 2017, 130, 1415–1430. [Google Scholar] [CrossRef] [Green Version]
- Shahinnia, F.; Geyer, M.; Schurmann, F.; Rudolphi, S.; Holzapfel, J.; Kempf, H.; Stadlmeier, M.; Loschenberger, F.; Morales, L.; Buerstmayr, H.; et al. Genome-wide association study and genomic prediction of resistance to stripe rust in current Central and Northern European winter wheat germplasm. Theor. Appl. Genet. 2022, 135, 3583–3595. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.P.; Tzou, W.S. Computational methods for discovering gene networks from expression data. Brief. Bioinform. 2009, 10, 408–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, P.K.; Cao, Y.; Jailani, A.K.; Gupta, P.; Venglat, P.; Xiang, D.; Rai, R.; Sharma, R.; Thirunavukkarasu, N.; Abdin, M.Z.; et al. Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). GM Crops Food 2014, 5, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Dash, P.K. Transcriptome analysis for abiotic stresses in rice (Oryza sativa L.). In Transcriptome Analysis; Blumenberg, M., Ed.; IntechOpen: London, UK, 2019; pp. 61–75. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 11, pdb-top084970. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef] [Green Version]
- Goyal, E.; Amit, S.K.; Singh, R.S.; Mahato, A.K.; Chand, S.; Kanika, K. Transcriptome profiling of the salt-stress response in Triticum aestivum cv. Kharchia Local. Sci. Rep. 2016, 6, 27752. [Google Scholar] [CrossRef]
- He, G.H.; Xu, J.Y.; Wang, Y.X.; Liu, J.M.; Li, P.S.; Chen, M.; Ma, Y.Z.; Xu, Z.S. Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33fromwheat confer drought and/or heatresistance in Arabidopsis. BMC Plant Biol. 2016, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, C.; Sheng, H.; Wang, Y.; Zeng, J.; Kang, H.; Fan, X.; Sha, L.; Zhang, H.; Zhou, Y. Transcriptome-wide identification and expression analyses of ABC transporters in dwarf polish wheatundermetal stresses. Biol. Plant. 2017, 61, 293–304. [Google Scholar] [CrossRef]
- Dalal, M.; Sahu, S.; Tiwari, S.; Rao, A.R.; Gaikwad, K. Transcriptome analysis reveals interplay between hormones, ROS metabolism and cellwall biosynthesis for drought-induced root growth in wheat. Plant Physiol. Biochem. 2018, 130, 482–492. [Google Scholar] [CrossRef]
- Sharma, C.; Saripalli, G.; Kumar, S.; Gautam, T.; Kumar, A.; Rani, S.; Jain, N.; Prasad, P.; Raghuvanshi, S.; Jain, M.; et al. A study of transcriptome in leaf rust infected bread wheat involving seedling resistance gene Lr28. Funct. Plant Biol. 2018, 45, 1046–1064. [Google Scholar] [CrossRef]
- Ye, W.; Liu, T.; Zhang, W.; Li, S.; Zhu, M.; Li, H.; Kong, Y.; Xu, L. Disclosure of the molecular mechanism of wheat leaf spot disease caused by Bipolaris sorokiniana through comparative transcriptome and metabolomics analysis. Int. J. Mol. Sci. 2019, 20, 6090. [Google Scholar] [CrossRef] [Green Version]
- Vicente, R.; Bolger, A.M.; Martinez-Carrasco, R.; Perez, P.; Gutierrez, E.; Usadel, B.; Morcuende, R. De novo transcriptome analysis of durum wheat flag leaves provides new insights into the regulatory response to elevated CO2 and high temperature. Front. Plant Sci. 2019, 10, 1605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Fu, Y.; Guo, H.; Zhang, L.; Wang, C.; Song, W.; Yan, Z.; Wang, Y.; Ji, W. Transcriptome and proteome-based network analysis reveals a model of gene activation in wheat resistance to stripe rust. Int. J. Mol. Sci. 2019, 20, 1106. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, B.; Ayalew, H.; Mishina, K.; Tanaka, T.; Kawahara, Y.; Jafary, H.; Oono, Y. Comparative analysis of root transcriptome reveals candidate genes and expression divergence of homoeologous genes in response to water stress in wheat. Plants 2020, 9, 596. [Google Scholar] [CrossRef]
- Rangan, P.; Furtado, A.; Henry, R. Transcriptome profiling of wheat genotypes under heat stress during grain-filling. J. Cereal Sci. 2020, 91, 102895. [Google Scholar] [CrossRef]
- Dugasa, M.T.; Feng, X.; Wang, N.H.; Wang, J.; Wu, F. Comparative transcriptome and tolerance mechanism analysis in the two contrasting wheat (Triticum aestivum L.) cultivars in response to drought and salinity stresses. J. Plant Growth Regul. 2021, 94, 101–114. [Google Scholar] [CrossRef]
- Chaichi, M.; Badii, F.; Mohammadi, A.; Hashemi, M. Water resistance and mechanical properties of low methoxy-pectin nanocomposite film responses to interactions of Ca2+ ions and glycerol concentrations as crosslinking agents. Food Chem. 2019, 293, 429–437. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, Y.; Li, W.; Wei, M.; Dai, T.; Li, Z.; Wang, B. Physiological and transcriptomic analyses reveal exogenous trehalose is involved in the responses of wheat roots to high temperature stress. Plants 2021, 10, 2644. [Google Scholar] [CrossRef]
- Duarte-Delgado, D.; Dadshani, S.; Schoof, H.; Oyiga, B.C.; Schneider, M.; Mathew, B.; Leon, J.; Ballvora, A. Transcriptome profiling at osmotic and ionic phases of salt stress response in bread wheat uncovers trait-specific candidate genes. BMC Plant Biol. 2020, 20, 428. [Google Scholar] [CrossRef]
- Batyrshina, Z.S.; Shavit, R.; Yaakov, B.; Bocobza, S.; Tzin, V. The transcription factor TaMYB31 regulates the benzoxazinoid biosynthetic pathway in wheat. J. Exp. Bot. 2022, 73, 5634–5649. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, Y.; Henderson, I.R.; Guo, J. Artificial sweeteners stimulate horizontal transfer of extracellular antibiotic resistance genes through natural transformation. ISME J. 2022, 16, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bai, X.; Qiao, Y.; Si, L.; Yu, Z.; Ni, C.; Li, T.; Guo, C.; Xiao, K. tae-miR9674a, a microRNA member of wheat, confers plant drought and salt tolerance through modulating the stomata movement and ROS homeostasis. Plant Biotechnol. Rep. 2022, 1–18. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Firouzabadi, F.; Shobbar, Z.S. Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS ONE 2019, 14, e0213305. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2022, 15, 587–598. [Google Scholar] [CrossRef]
- Eren, H.; Pekmezci, M.Y.; Okay, S.; Turktas, M.; Inal, B.; Ilhan, E.; Atak, M.; Erayman, M.; Unver, T. Hexaploid wheat (Triticum aestivum) root miRNome analysis in response to salt stress. Ann. Appl. Biol. 2015, 167, 208–216. [Google Scholar] [CrossRef]
- Seifikalhor, M.; Aliniaeifard, S.; Shomali, A.; Azad, N.; Hassani, B.; Lastochkina, O.; Li, T. Calcium signaling and salt tolerance are diversely entwined in plants. Plant Signal. Behav. 2019, 14, 1665455. [Google Scholar] [CrossRef]
- Vaid, N.; Pandey, P.; Srivastava, V.K.; Tuteja, N. Pea lectin receptor-like kinase functions in salinity adaptation without yield penalty, by alleviating osmotic and ionic stresses and upregulating stress-responsive genes. Plant Mol. Biol. 2015, 88, 193–206. [Google Scholar] [CrossRef]
- Shahid, M.; Khalid, S.; Abbas, G.; Shahid, N.; Nadeem, M.; Sabir, M.; Aslam, M.; Dumat, C. Heavy Metal Stress and Crop Productivity. In Crop Production and Global Environmental Issues; Hakeem, K., Ed.; Springer: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- Zhang, T.; Xiao, J.; Zhao, Y.; Zhang, Y.; Jie, Y.; Shen, D.; Yue, C.; Huang, J.; Hua, Y.; Zhou, T. Comparative physiological and transcriptomic analyses reveal ascorbate and glutathione coregulation of cadmium toxicity resistance in wheat genotypes. BMC Plant Biol. 2021, 21, 459. [Google Scholar] [CrossRef]
- Francki, M.G.; Hayton, S.; Gummer, J.P.A.; Rawlinson, C.; Trengove, R.D. Metabolomic profiling and genomic analysis of wheat aneuploid lines to identify genes controlling biochemical pathways in mature grain. Plant Biotechnol. J. 2016, 14, 649–660. [Google Scholar] [CrossRef]
- Shewry, P.R.; Corol, D.I.; Jones, H.D.; Beale, M.H.; Ward, J.L. Defining genetic and chemical diversity in wheat grain by 1H-NMR spectroscopy of polar metabolites. Mol. Nutr. Food Res. 2017, 61, 1600807. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.M.; Hawkins, N.D.; Ward, J.L.; Lovegrove, A.; Napier, J.A.; Shewry, P.R.; Beale, M.H.A. Metabolomic study of substantial equivalence of field-grown genetically modified wheat. Plant Biotechnol. J. 2006, 4, 381–392. [Google Scholar] [CrossRef]
- Graham, S.F.; Amigues, E.; Migaud, M.; Browne, R.A. Application of NMR based metabolomics for mapping metabolite variation in european wheat. Metabolomics 2009, 5, 302–306. [Google Scholar] [CrossRef]
- Curtis, T.Y.; Muttucumaru, N.; Shewry, P.R.; Parry, M.A.J.; Powers, S.J.; Elmore, J.S.; Mottram, D.S.; Hook, S.; Halford, N.G. Effects of genotype and environment on free amino acid levels in wheat grain: Implications for acrylamide formation during processing. J. Agric. Food Chem. 2009, 57, 1013–1021. [Google Scholar] [CrossRef]
- Howarth, J.R.; Parmar, S.; Jones, J.; Shepherd, C.E.; Corol, D.I.; Galster, A.M.; Hawkins, N.D.; Miller, S.J.; Baker, J.M.; Verrier, P.J.; et al. Co-ordinated expression of amino acid metabolism in response to N and S deficiency during wheat grain filling. J. Exp. Bot. 2008, 59, 3675–3689. [Google Scholar] [CrossRef] [Green Version]
- Gunnaiah, R.; Kushalappa, A.C. Metabolomics deciphers the host resistance mechanisms in wheat cultivar Sumai-3, against trichothecene producing and non-producing isolates of Fusarium graminearum. Plant Physiol. Biochem. 2014, 83, 40–50. [Google Scholar] [CrossRef]
- Aranjuelo, I.; Erice, G.; Sanz-Sáez, A.; Abadie, C.; Gilard, F.; Gil-Quintana, E.; Avice, J.C.; Staudinger, C.; Wienkoop, S.; Araus, J.L.; et al. Differential CO2 effect on primary carbon metabolism of flag leaves in durum wheat (Triticum durum Desf.). Plant Cell Environ. 2015, 38, 2780–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogy, P.; Keck, M.; Niehaus, K.; Franzaring, J.; Fangmeier, A. Effects of atmospheric CO2 enrichment on biomass, yield and low molecular weight metabolites in wheat grain. J. Cereal Sci. 2010, 52, 215–220. [Google Scholar] [CrossRef]
- Hill, C.B.; Taylor, J.D.; Edwards, J.; Mather, D.; Langridge, P.; Bacic, A.; Roessner, U. Detection of QTL for metabolic and agronomic traits in wheat with adjustments for variation at genetic loci that affect plant phenology. Plant Science 2015, 233, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, M.G.; Ciarmiello, L.F.; Woodrow, P.; Maximova, E.; Fuggi, A.; Carillo, P. Durum wheat roots adapt to salinity remodeling the cellular content of nitrogen metabolites and sucrose. Front. Plant Sci. 2017, 7, 2035. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Babar, M.A.; Khan, N.; Guo, J.; Khan, J.; Islam, S.; Shrestha, S.; Shahi, D. Comparative metabolomic profiling in the roots and leaves in contrasting genotypes reveals complex mechanisms involved in post-anthesis drought tolerance in wheat. PLoS ONE 2019, 14, e0213502. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yu, S.; Lian, J.; Wang, Q.; He, Z.; Feng, Y.; Yang, X. Physiological and metabolomics responses of two wheat (Triticum aestivum L.) genotypes differing in grain cadmium accumulation. Sci. Total Environ. 2021, 769, 145345. [Google Scholar] [CrossRef]
- Qin, S.; Xu, Y.; Nie, Z.; Liu, H.; Gao, W.; Li, C.; Zhao, P. Metabolomic and antioxidant enzyme activity changes in response to cadmium stress under boron application of wheat (Triticum aestivum). Environ. Sci. Pollut. Res. 2022, 29, 34701–34713. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Griffiths, T.A.M.; Bleackley, M.R.; MacGillivray, R.T.A. Proteomics: Applications relevant to transfusion medicine. Transfus. Med. Rev. 2006, 20, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Faghani, E.; Gharechahi, J.; Komatsu, S.; Mirzaei, M.; Khavarinejad, R.A.; Najafi, F.; Farsad, L.K.; Salekdeh, G.H. Comparative physiology and proteomic analysis of two wheat genotypes contrasting in drought tolerance. J. Proteom. 2015, 114, 1–15. [Google Scholar] [CrossRef]
- Kang, G.; Li, G.; Xu, W.; Peng, X.; Han, Q.; Zhu, Y.; Guo, T. Proteomics reveals the effects of salicylic acid on growth and tolerance to subsequent drought stress in wheat. J. Proteome Res. 2012, 11, 6066–6079. [Google Scholar] [CrossRef]
- Alvarez, S.; Choudhury, S.R.; Pandey, S. Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 2014, 13, 1688–1701. [Google Scholar] [CrossRef]
- Michaletti, A.; Naghavi, M.R.; Toorchi, M.; Zolla, L.; Rinalducci, S. Metabolomics and proteomics reveal drought-stress responses of leaf tissues from spring-wheat. Sci. Rep. 2018, 8, 5710. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Ma, C.; Wang, S.; Gao, L.; Li, X.; Guo, G.; Ma, W.; Yan, Y. Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Anal. Bioanal. Chem. 2012, 402, 1297–1313. [Google Scholar] [CrossRef]
- Qin, N.; Xu, W.; Hu, L.; Li, Y.; Wang, H.; Qi, X.; Fang, Y.; Hua, X. Drought tolerance and proteomics studies of transgenic wheat containing the maize C4 phosphoenolpyruvate carboxylase (PEPC) gene. Protoplasma 2016, 253, 1503–1512. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; He, Q.; Li, H.; Zhang, X.; Zhang, F. Comparative proteomics illustrates the complexity of drought resistance mechanisms in two wheat (Triticum aestivum L.) cultivars under dehydration and rehydration. BMC Plant Biol. 2016, 16, 188. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Duan, W.; Cui, Y.; Zhang, J.; Zhu, D.; Zhang, M.; Yan, Y. 2D-DIGE based proteome analysis of wheat-Thinopyrum intermedium 7XL/7DS translocation line under drought stress. BMC Genom. 2022, 23, 369. [Google Scholar] [CrossRef]
- Ding, H.; Han, Q.; Ma, D.; Hou, J.; Huang, X.; Wang, C.; Xie, Y.; Kang, G.; Guo, T. Characterizing physiological and proteomic analysis of the action of H2S to mitigate drought stress in young seedling of wheat. Plant Mol. Biol. Rep. 2018, 36, 45–57. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Li, J.; Ren, Y.; Wang, Z.; Xin, Z.; Lin, T. Identification of two novel wheat drought tolerance-related proteins by comparative proteomic analysis combined with virus-induced gene silencing. Int. J. Mol. Sci. 2018, 19, 4020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lou, H.; Guo, D.; Zhang, R.; Su, M.; Hou, Z.; Zhou, H.; Liang, R.; Xie, C.; You, M.; et al. Identifying changes in the wheat kernel proteome under heat stress using iTRAQ. Crop J. 2018, 6, 600–610. [Google Scholar] [CrossRef]
- Kumar, R.R.; Singh, K.; Ahuja, S.; Tasleem, M.; Singh, I.; Kumar, S.; Grover, M.; Mishra, D.; Rai, G.K.; Goswami, S.; et al. Quantitative proteomic analysis reveals novel stress-associated active proteins (SAAPs) and pathways involved in modulating tolerance of wheat under terminal heat. Funct. Integr. Genom. 2019, 19, 329–348. [Google Scholar] [CrossRef]
- Chunduri, V.; Kaur, A.; Kaur, S.; Kumar, A.; Sharma, S.; Sharma, N.; Singh, P.; Kapoor, P.; Kaur, S.; Kumari, A.; et al. Gene expression and proteomics studies suggest an involvement of multiple pathways under day and day–night combined heat stresses during grain filling in wheat. Front. Plant Sci. 2021, 12, 973. [Google Scholar] [CrossRef]
- Wang, X.; Hou, L.; Lu, Y.; Wu, B.; Gong, X.; Liu, M.; Wang, J.; Sun, Q.; Vierling, E.; Xu, S. Metabolic adaptation of wheat grain contributes to a stable filling rate under heat stress. J. Exp. Bot. 2018, 69, 5531–5545. [Google Scholar] [CrossRef] [Green Version]
- Fercha, A.; Capriotti, A.L.; Caruso, G.; Cavaliere, C.; Gherroucha, H.; Samperi, R.; Stampachiacchiere, S.; Lagana, A. Gel-free proteomics reveal potential biomarkers of priming-induced salt tolerance in durum wheat. J. Proteom. 2013, 91, 486–499. [Google Scholar] [CrossRef]
- Guo, G.; Ge, P.; Ma, C.; Li, X.; Lv, D.; Wang, S.; Ma, W.; Yan, Y. Comparative proteomic analysis of salt response proteins in seedling roots of two wheat varieties. J. Proteom. 2012, 75, 1867–1885. [Google Scholar] [CrossRef]
- Yadav, R.; Santal, A.R.; Singh, N.P. Comparative root proteome analysis of two contrasting wheat genotypes Kharchia-65 (highly salt-tolerant) and PBW-373 (salt-sensitive) for salinity tolerance using LC–MS/MS approach. Vegetos 2022, 35, 133–139. [Google Scholar] [CrossRef]
- Gharechahi, J.; Alizadeh, H.; Naghavi, M.R.; Sharifi, G. A proteomic analysis to identify cold acclimation associated proteins in wild wheat (Triticum urartu L.). Mol. Biol. Rep. 2014, 41, 3897–3905. [Google Scholar] [CrossRef]
- Singh, R.P.; Runthala, A.; Khan, S.; Jha, P.N. Quantitative proteomics analysis reveals the tolerance of wheat to salt stress in response to Enterobacter cloacae SBP-8. PLoS ONE 2017, 12, e0183513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.; Xu, T.; Muhae-Ud-din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. ITRAQ-based proteomic analysis of wheat (Triticum aestivum) spikes in response to Tilletia controversa Kuhn and Tilletia foetida Kühn Infection, causal organisms of dwarf bunt and common bunt of wheat. Biology 2022, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Yang, X.; Xu, F.; Huang, Y.; Zhang, J.; Song, M.; Zhou, S.; Zhang, M.; He, D. TMT-based quantitative proteomic analysis reveals defense mechanism of wheat against the crown rot pathogen Fusarium pseudograminearum. BMC Plant Biol. 2021, 21, 82. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.W.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinf. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.C.; Tian, T.F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alcalde, F.; García-Lopez, F.; Dopazo, J.; Conesa, A. Paintomics: A web-based tool for the joint visualization of transcriptomics and metabolomics data. J. Bioinform. 2011, 27, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Tokimatsu, T.; Sakurai, N.; Suzuki, H.; Ohta, H.; Nishitani, K.; Koyama, T.; Umezawa, T.; Misawa, N.; Saito, K.; Shibata, D. KaPPA-View. A web-based analysis tool for integration of transcript and metabolite data on plant metabolic pathway maps. Plant Physiol. 2005, 138, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Weckwerth, W. COVAIN: A toolbox for uni- and multivariate statistics, time-series and correlation network analysis and inverse estimation of the differential Jacobian from metabolomics covariance data. Metabolomics 2012, 8, 81–93. [Google Scholar] [CrossRef]
- Sangaralingam, A.; Dayem Ullah, A.Z.; Marzec, J.; Gadaleta, E.; Nagano, A.; Ross-Adams, H.; Wang, J.; Lemoine, N.R.; Chelala, C. ‘Multi-omic’data analysis using O-miner. Brief. Bioinform. 2019, 20, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Biyiklioglu, S.; Alptekin, B.; Akpinar, B.A.; Varella, A.C.; Hofland, M.L.; Weaver, D.K.; Bothner, B.; Budak, H. A large-scale multiomics analysis of wheat stem solidness and the wheat stem sawfly feeding response, and syntenic associations in barley, Brachypodium, and rice. Funct. Integr. Genom. 2018, 18, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Velez-Bermúdez, I.C.; Salazar-Henao, J.E.; Fornalé, S.; López-Vidriero, I.; Franco-Zorrilla, J.M.; Grotewold, E.; Gray, J.; Solano, R.; Schmidt, W.; Pagés, M.; et al. A MYB/ZML complex regulates wound-induced lignin genes in maize. Plant Cell 2015, 27, 3245–3259. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, R.; Liu, H.; Liu, X.; Xu, K.; Xiao, K. Multi-omics analyses reveal the molecular mechanisms underlying the adaptation of wheat (Triticum aestivum L.) to potassium deprivation. Front. Plant Sci. 2020, 11, 588994. [Google Scholar] [CrossRef]
- Heang, D.; Sassa, H. An atypical bHLH protein encoded by positive regulator of grain length 2 is involved in controlling grain length and weight of rice through interaction with a typical bHLH protein APG. Breed Sci. 2012, 62, 133–141. [Google Scholar] [CrossRef]
- Thrash, A.; Tang, J.D.; DeOrnellis, M.; Peterson, D.G.; Warburton, M.L. PAST: The pathway association studies tool to infer biological meaning from GWAS datasets. Plants 2020, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Weckwerth, W.; Ghatak, A.; Bellaire, A.; Chaturvedi, P.; Varshney, R.K. PANOMICS meets germplasm. Plant Biotechnol. J. 2020, 18, 1507–1525. [Google Scholar] [CrossRef] [Green Version]
- Alqudah, A.M.; Haile, J.K.; Alomari, D.Z.; Pozniak, C.J.; Kobiljski, B.; Borner, A. Genome-wide and SNP network analyses reveal genetic control of spikelet sterility and yield-related traits in wheat. Sci. Rep. 2020, 10, 2098. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Y.; Tan, Y.; Zhang, M.; Zhu, L.; Xu, G.; Fan, X. Agronomic nitrogen-use efficiency of rice can be increased by driving Os NRT 2.1 expression with the Os NAR 2.1 promoter. Plant Biotechnol. J. 2016, 14, 1705–1715. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.G.; Lagudah, E.S.; Spielmeyer, W.; Dodds, P.N. The past, present and future of breeding rust resistant wheat. Front. Plant Sci. 2014, 5, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Espino, J.; Singh, R.P.; Germán, S.; McCallum, B.D.; Park, R.; Chen, W.Q.; Bhardwaj, S.C.; Goyeau, H. Global status of wheat leaf rust caused by Puccinia triticina. Euphytica 2011, 179, 143–160. [Google Scholar] [CrossRef]
- Singh, R.P.; Hodson, D.P.; Jin, Y.; Lagudah, E.S.; Ayliffe, M.A.; Bhavani, S.; Rouse, M.N.; Pretorius, Z.A.; Szabo, L.J.; Huerta-Espino, J.; et al. Emergence and spread of new races of wheat stem rust fungus: Continued threat to food security and prospects of genetic control. Phytopathology 2015, 105, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haile, T.A.; Heidecker, T.; Wright, D.; Neupane, S.; Ramsay, L.; Vandenberg, A.; Bett, K.E. Genomic selection for lentil breeding: Empirical evidence. Plant Genome 2020, 13, e20002. [Google Scholar] [CrossRef] [Green Version]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; GTEx Consortium; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef]


{kind=link}
| Gene Family | Putative Annotation | Stress | Reference |
|---|---|---|---|
| Biotic stress | |||
| LIM (Lin-11, IsI-1 and Mec-3) | Transcription factors | Fusarium head blight | [51] |
| bHLH (basic helix-loop-helix) | Transcription factors | Fusarium head blight and Septoria tritici blotch | [52] |
| Serpin (serine protease inhibitor) | Protease inhibitors | Fusarium head blight | [53] |
| SNARE | Transport proteins | Powdery mildew | [54] |
| SWEET | Sugar transporter | Stem rust | [55] |
| CNGCs | Calcium channel | Stripe rust | [56] |
| AP (Aspartic proteases) | Proteolysis enzymes | Powdery mildew | [57] |
| ZF_HD (Zinc finger homeodomain) | Transcription factors | Fusarium head blight | [58] |
| Xylanase inhibitor | Plant pathogen interaction | Fusarium head blight | [59] |
| Caffeoyl-coenzyme A O-methyltransferase | Lignin biosynthesis | Fusarium head blight | [13] |
| Abiotic stress | |||
| DEAD box RNA helicases | RNA metabolism | Drought, cold and salt | [60] |
| WRKY | Transcription factors | Drought and salt | [61] |
| Domain of unknown function | Uncharacterized | Salt | [62] |
| Trihelix | Plant specific transcription factors | Salt and cold | [63] |
| HSPs (Heat shock proteins) | Protein folding | Heat | [64] |
| NAC (NAM-ATAF1-2-CUC2) | Plant specific transcription factors | Heat and drought | [65] |
| bZIP (basic leucine zipper) | Transcription factors | Heat, salinity, drought and oxidative stress | [66] |
| ASR (ABA-stress-ripening) genes | Transcription factors | Salt and low temperature | [67] |
| SRO (similar to radical-induced cell death 1 proteins) | Small protein family | Various stresses | [68] |
| PLC (Phospholipase C) | Cytoplasmic membranes | Salt, low temperature and drought | [54] |
| Expansin | Cell wall component | Salt | [69] |
| BAM (B-amylase) | Sugar | Heat and drought | [70] |
| LIM (Lin-11, IsI-1 and Mec-3) | Transcription factors | Heat, drought, salt, abscisic acid | [51] |
| Growth regulating factors | Transcription factors | Osmotic stress | [71] |
| Amino acid transporter | Transporter proteins | Heat and drought | [72] |
| Trait for Transcriptome Analysis | Summary | Reference |
|---|---|---|
| Heat and drought stress | HSFs and DREBs are involved in alleviating stress effect. Further, 1328 TFs found to be responsive to stress treatment. | [111] |
| Salt stress | TF’s including WD40-like, C2H2, MYB-HB-like, genes coding for V-ATPase, glutathione S-transferases, cytochrome c oxidase and Cbl-interacting protein kinasewere over expressed in Triticum aestivum cv. Kharchia Local. | [112] |
| Drought and/or heat stress | Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 were found to confer drought and/or heat resistance in Arabidopsis. | [113] |
| High temperature stress | Identified six heat-induced MYB genes in wheat. | [46] |
| Metal stress | The expressions of ABC transporters in dwarf polish wheat played important roles in metal transport (Cd, Cu, Mg, Zn, Fe, and Ni) and detoxification. | [114] |
| Drought stress | Drought stress significantly upregulated auxin receptor (AFB2) and ABA responsive transcription factors (MYB78, WRKY18 and GBF3), ACC oxidase and 2OG-Fe(II) oxygenase in roots. Genes related to gibberellic acid, jasmonic acid and phenylpropanoid pathways were down regulated in roots. | [115] |
| Water stress | Root transcriptome profiles identified DEGs involved in carbon metabolism, flavonoid biosynthesis and phytohormone signal transduction. | [6] |
| Leaf rust | Genes involved in reactive oxygen species (ROS) homeostasis and several genes encoding TFs, most abundant being WRKY TFs, were identified along with some ncRNAs and histone variants in HD 2329 + Lr28 NIL in comparison to HD 2329. | [116] |
| Seedling salt stress | Salt tolerance was conferred by polyunsaturated fatty acid (PUFAs) by enhancing the photosynthetic system and JA-related pathways. | [45] |
| Leaf spot (Bipolaris sorokiniana) tolerance | The upregulation of hydrolase inhibitor, NAC (including NAM, ATAF1 and CUC2) transcriptional factor, and peroxidase in infected wheat tissues suggested their central roles in the defensive response of wheat to Bipolaris sorokiniana. | [117] |
| Elevated CO2 and high temperature stress | DEGs in response to stress includes protein kinases, receptor kinases, and transcription factors. | [118] |
| Stripe rust | Several regulators, including splicing and transcription factors and Hsp70 protein are responsive in Puccinia striformis induced response network. | [119] |
| Water Stress | Comparative analysis of root transcriptome revealed that transcription factors, pyroline-5-carboxylate reductase and late-embryogenesis-abundant proteins were upregulated genes in the tolerant cultivar. | [120] |
| Heat stress during grain filling | Hsp-family, ascorbate peroxidase, β-amylase, γ-gliadin-2 and LMW-glutenin were heat stress responsive and were upregulated during stress. | [121] |
| Abiotic Stresses Analysed | Research Summary | Reference |
|---|---|---|
| Drought Stress | Stress response of two genotypes SW89.5193/kAu2|SERI M 82 (Susceptible|Tolerant) was analyzed under drought. They found 40 roots; 73 leaves differentially expressed proteins (DEPs) in roots and leaves, respectively. | [151] |
| Pretreatment with 0.5 mM salicylic acid (SA) for 3 days significantly enhanced the growth and tolerance to subsequent drought stress in Yumai 34. A total of 76 proteins were found to be differentially regulated by using 2-DE, MALDI-TOF-TOF from leaf samples. | [152] | |
| Monitored the roots of two different wheat varieties, Nesser (drought-tolerant) and Opata (drought-sensitive), in the absence and presence of abscisic acid (ABA, as a proxy for drought). A total of 151 proteins were found to be differentially regulated. | [153] | |
| Monitored the stress response of two cultivars Bahar, drought-susceptible; Kavir, drought tolerant under drought stress. A total of 81 proteins were found to be differentially regulated. | [154] | |
| Monitored the stress response of two varieties, Ningchun 4 (Tolerant) and Chinese Spring (Susceptible), at grain development stage: A total of 91 proteins were found to be differentially regulated. | [155] | |
| Monitored PEPC transgenic lines for drought tolerance, expressing maize C4 phosphoenolpyruvate carboxylase (PEPC) gene. By employing the 2-DE, MALDI-TOF, a total of 75 genes were found to be differentially regulated from flag leaf samples. | [156] | |
| Monitored the stress response of two wheat cultivars, Xihan No. 2 and Longchun 23, under dehydration and rehydration. They reported 84 and 64 proteins differentially regulated in Xihan No. 2 and Longchun 23, respectively. | [157] | |
| Monitored the stress response of two varieties, Zhongmai 8601 and Zhongmai 8601-Thinopyrum intermedium 7XL/7DS translocation line YW642, under drought stress at grain development stage. They found the differential regulation of 146 proteins in response to drought. | [158] | |
| Monitored the NaHS treated seedlings under drought stress in Yumai 34. They found the differential regulation of 120 proteins. | [159] | |
| Identified drought-tolerant proteins via virus-induced gene silencing in drought-tolerant XN979 and drought-sensitive LA379 varieties. They found the differential regulation of 335 proteins in response to stress. | [160] | |
| Heat Stress | Monitored the stress response of Gaocheng 8901, winter wheat. They found the upregulation of 207 proteins. | [161] |
| Monitored the stress response of two cultivars, HD2985 (thermotolerant) and HD2329 (thermosusceptible), at pollination and grain filling stages. They identified 4271 stress-associated proteins. | [162] | |
| Monitored the BWL4444 (HD2967+ Yr10) plants during the grain filling stage to understand the effect of heat stress in heat-tolerant varieties. Differential expression of 153 proteins was found in developing grain samples. | [163] | |
| Monitored the stability of the filling rate under heat stress in two wheat varieties, Chinese Spring and Liao-10. They found 309 proteins associated with heat stress. | [164] | |
| Salinity Stress | Monitored priming-induced salt tolerance on vigor in T durum var. Waha. The ratio of seed weight to the volume of solution employed for priming was 1:5; 12 h soaking. Priming treatments: distilled water (c) and ascorbate (t). Salt stress: 10 mL of saline solution (NaCl 250 mmol L−1) or distilled water (control) and drying under shade with forced air at 27 ± 3 °C. They found 72 proteins hydroprimed and 83 proteins ascorbate primed. | [165] |
| Monitored the roots of two wheat varieties, Jing-411 (salt-tolerant) and Chinese Spring (salt-sensitive), under salt stress. They found 52 proteins in Jing-411 and 47 proteins in Chinese Spring to be differentially regulated. | [166] | |
| Monitored the roots of two wheat varieties, Kharchia-65 (highly salt-tolerant) and PBW-373 (salt-sensitive), under salt stress. They found 2520 proteins in Kharchia-65 and 1633 proteins in PBW-373 to be differentially regulated. | [167] | |
| Cold Stress | Monitored the cold stress response of seeds of one wheat cultivar, T urartu L. They found 34 proteins differentially regulated in response to cold stress in leaf samples. | [168] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sehgal, D.; Dhakate, P.; Ambreen, H.; Shaik, K.H.B.; Rathan, N.D.; Anusha, N.M.; Deshmukh, R.; Vikram, P. Wheat Omics: Advancements and Opportunities. Plants 2023, 12, 426. https://doi.org/10.3390/plants12030426
Sehgal D, Dhakate P, Ambreen H, Shaik KHB, Rathan ND, Anusha NM, Deshmukh R, Vikram P. Wheat Omics: Advancements and Opportunities. Plants. 2023; 12(3):426. https://doi.org/10.3390/plants12030426
Chicago/Turabian StyleSehgal, Deepmala, Priyanka Dhakate, Heena Ambreen, Khasim Hussain Baji Shaik, Nagenahalli Dharmegowda Rathan, Nayanahalli Munireddy Anusha, Rupesh Deshmukh, and Prashant Vikram. 2023. "Wheat Omics: Advancements and Opportunities" Plants 12, no. 3: 426. https://doi.org/10.3390/plants12030426