

Identification of Crucial Genes and Regulatory Pathways in Alfalfa against Fusarium Root Rot
 , , and
, , and 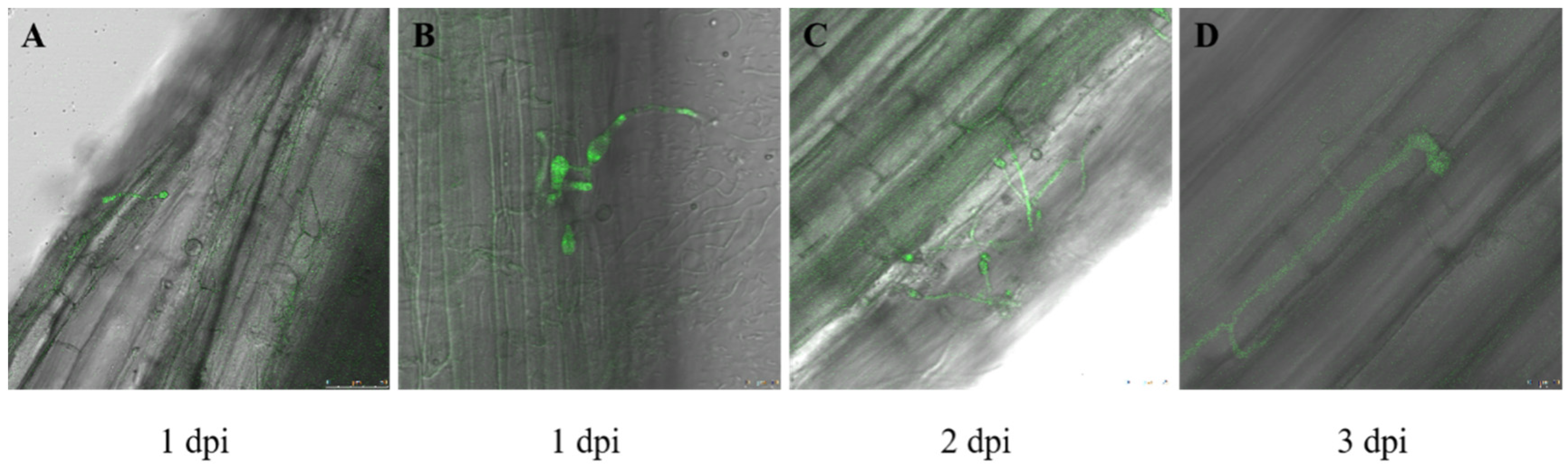{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Inoculation of Alfalfa Roots with Fusarium acuminatum
2.2. Determination of Physiological and Biochemical Indicators
2.3. Transcriptome Sequencing
2.4. WGCNA Analysis
2.5. Quantitative Real-Time PCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Fusarium acuminatum Infection on Medicago sativa
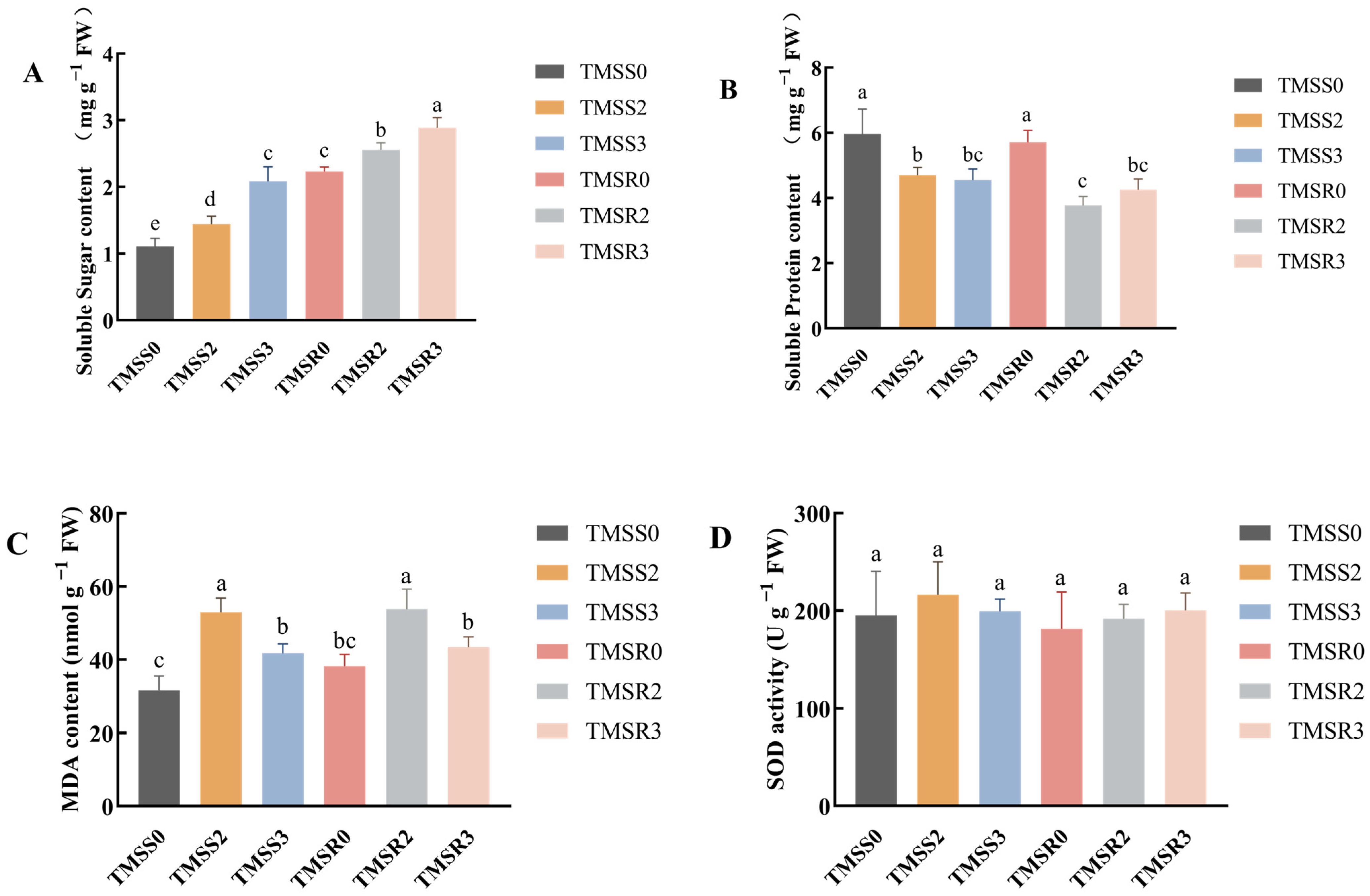
3.2. Physiological and Biochemical Activities after Plant Infection
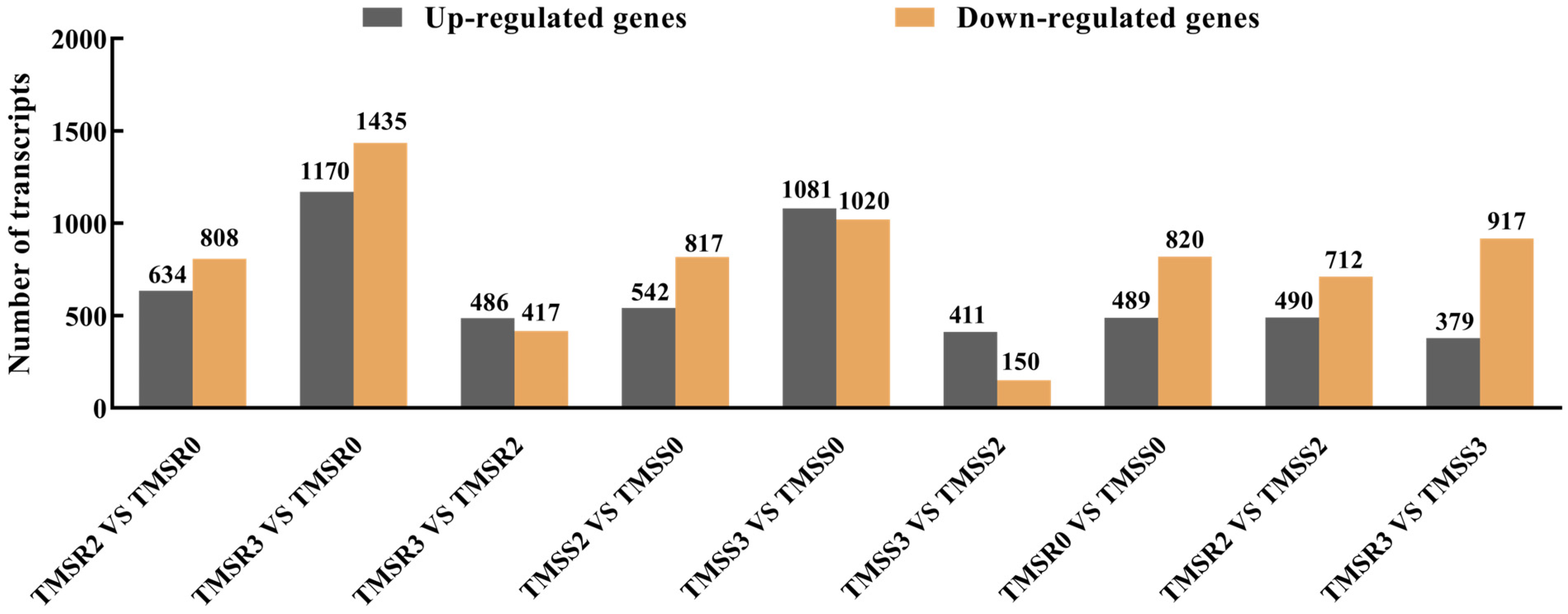
3.3. Transcriptome Analysis of TMSR and TMSS in Response to Fusarium acuminatum Infection
3.4. Identification of Genes with Changes in Expression following Fusarium acuminatum Infection in Resistant (TMSR) and Susceptible (TMSS) Alfalfa Varieties
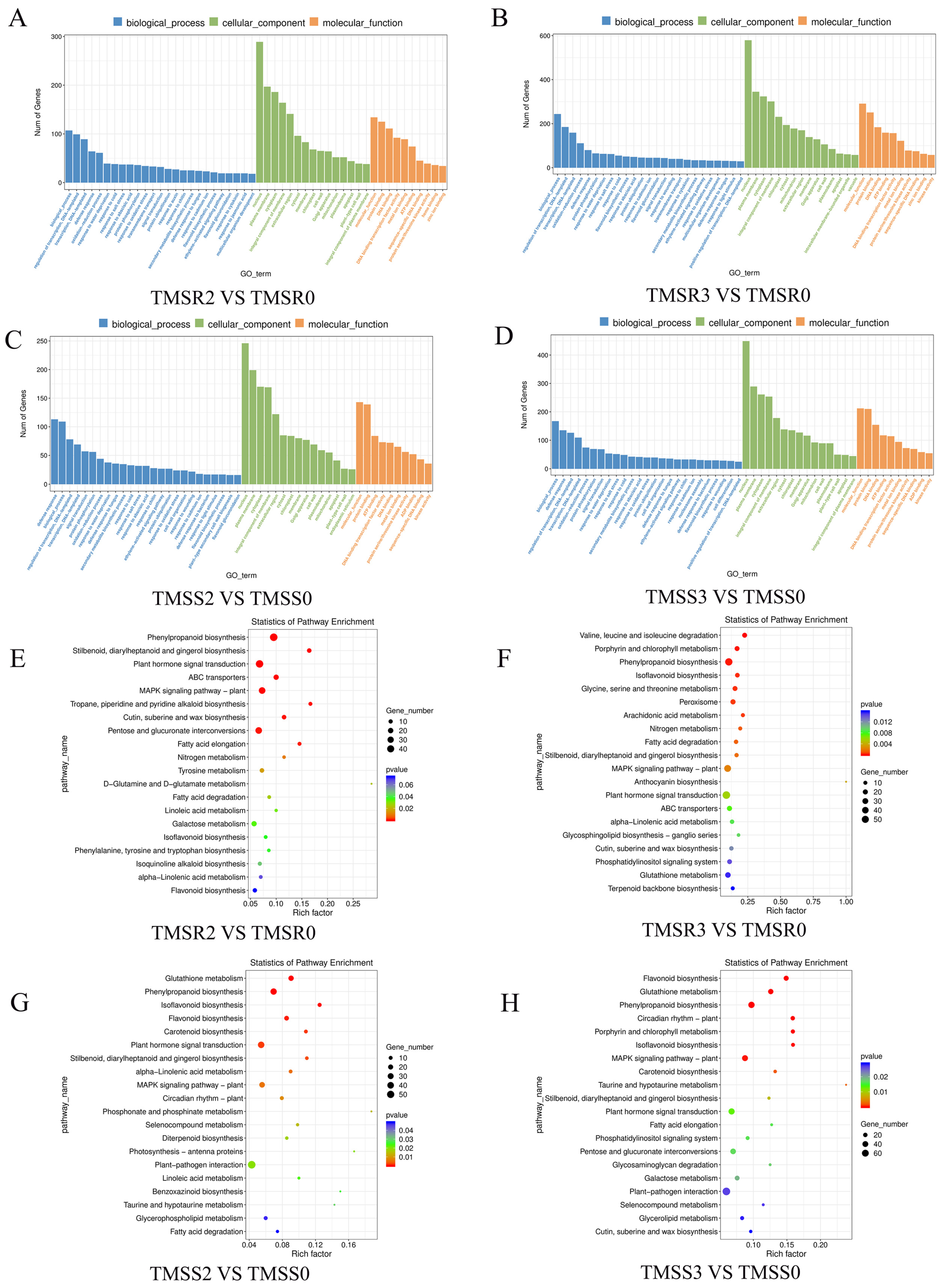
3.5. Identification and Functional Annotation of DEGs in TMSR and TMSS in Response to Fusarium acuminatum Infection
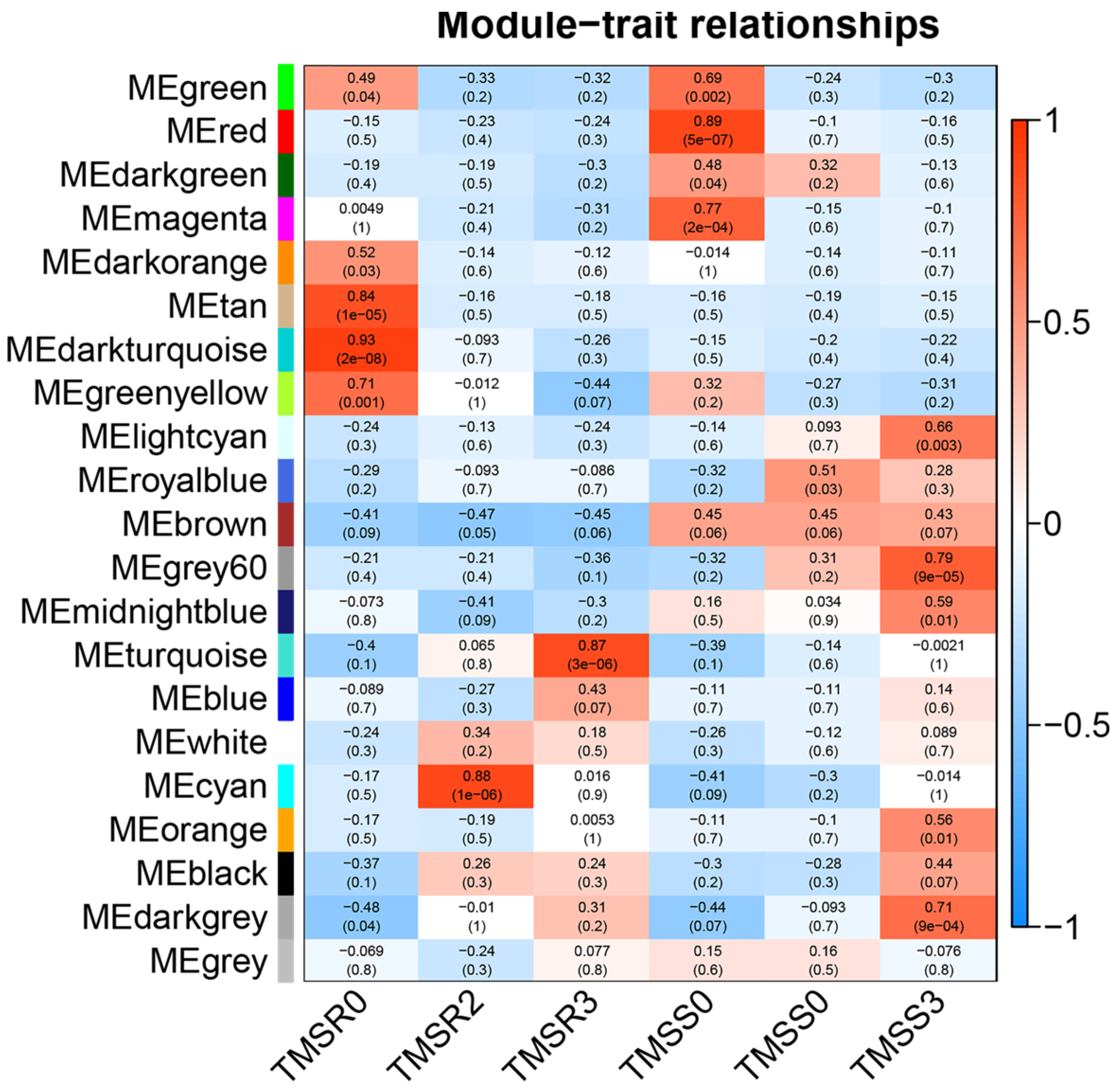
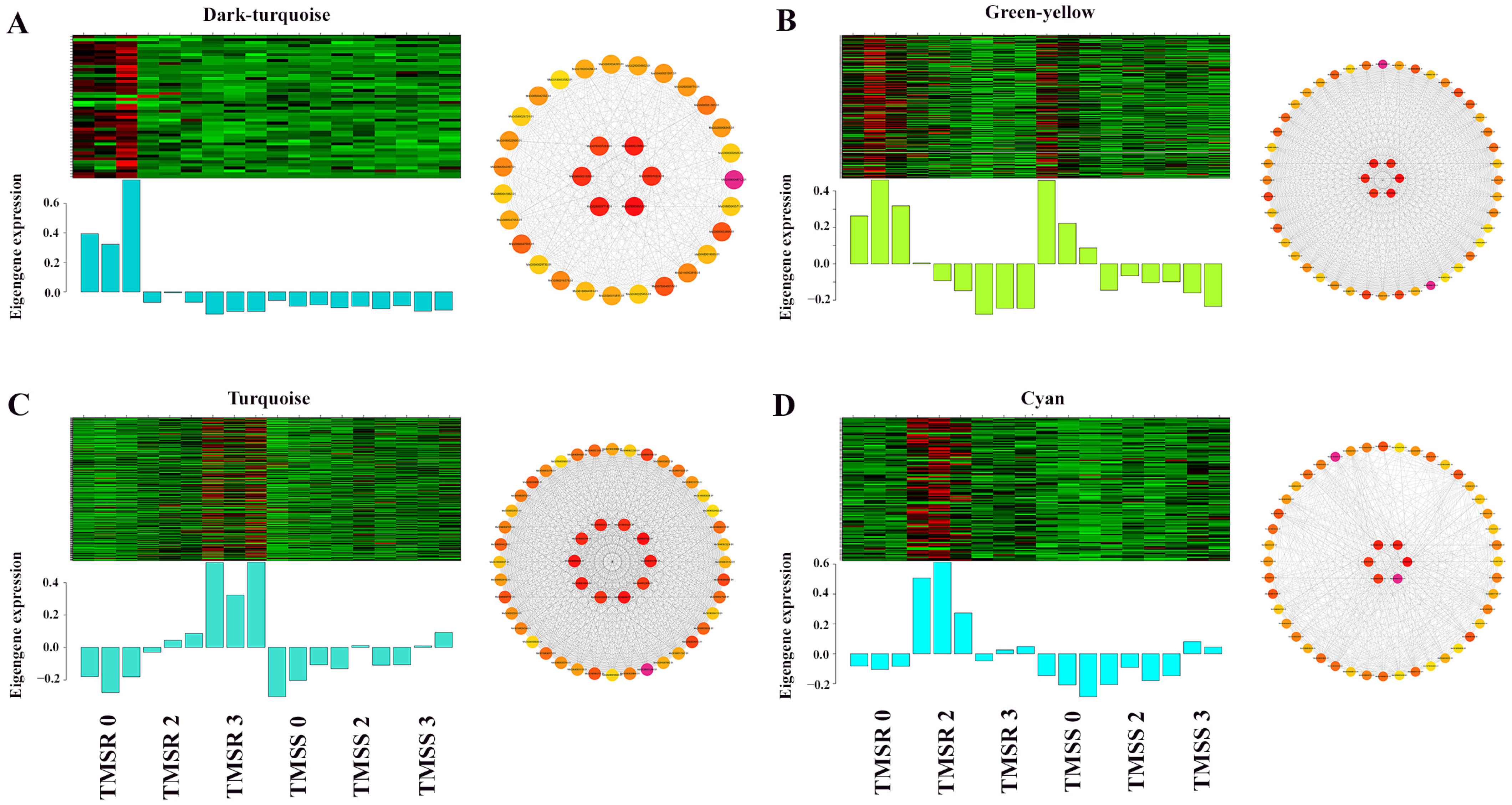
3.6. Identification and Functional Annotation of Core Resistance Genes in TMSR and TMSS
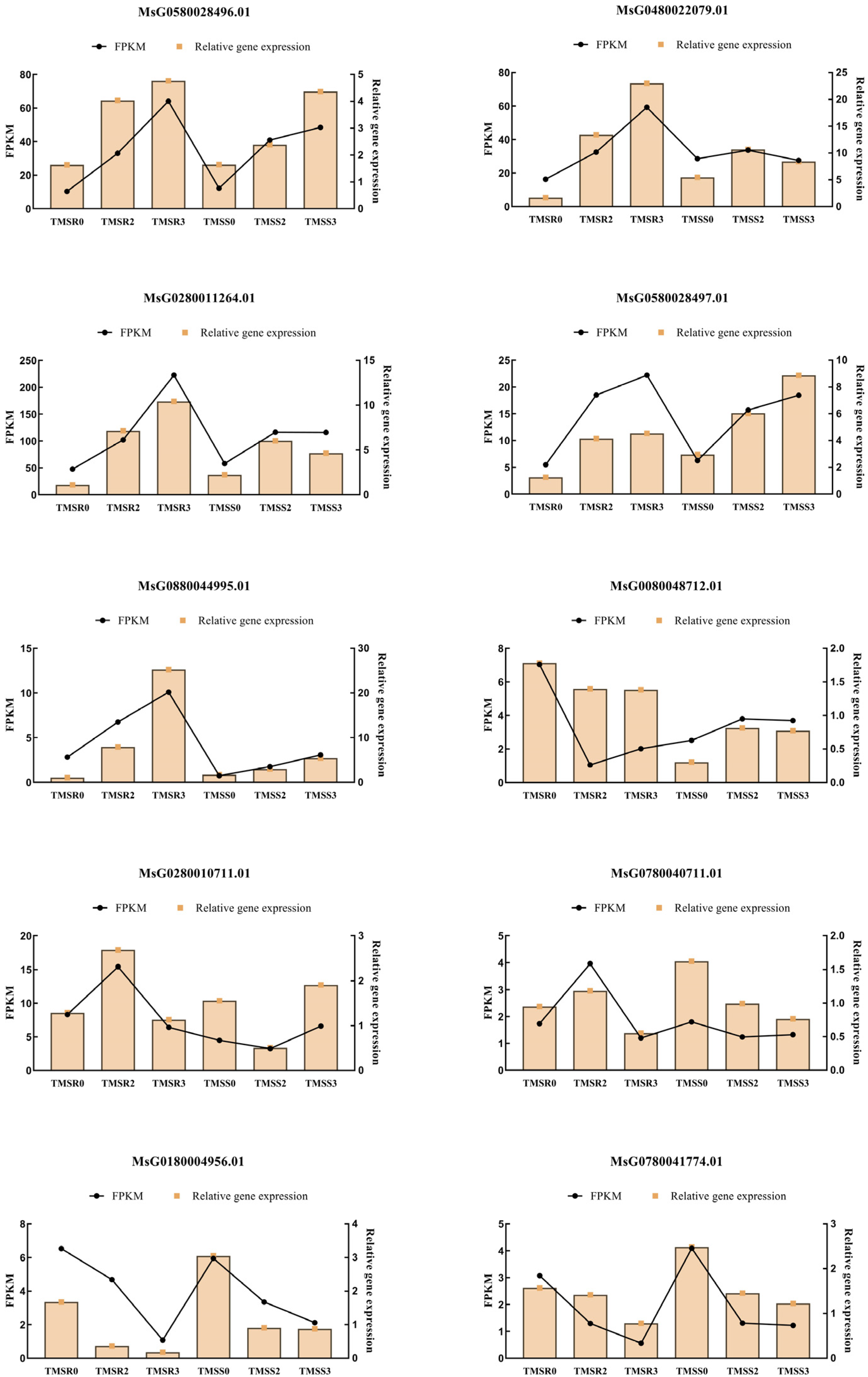
3.7. Verification of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, Y.; Guo, Z.; Zhang, J.; Wang, M.; Qu, G.; Yan, X.; Zhang, M. Study on the pathogenicity of F. acuminatum and disease resistance of alfalfa varieties. Acta Phytopathol. Sin. 2018, 48, 213–222. [Google Scholar]
- Fang, X.; Zhang, C.; Nan, Z. Research advances in Fusarium root rot of alfalfa (Medicago sativa). Acta Prataculturae Sin. 2019, 28, 169–183. [Google Scholar]
- Tao, S.; Wang, Y.T.; Zhang, Q. Situation of domestic alfalfa market and the impact from Sino-US trade frictions and its countermeasures. Anim. Husb. Feed Sci. 2019, 40, 46–50. [Google Scholar] [CrossRef]
- Yi, M. National alfalfa industry development plan. China Agric. Inf. 2017, 4, 9–13. [Google Scholar]
- Shi, Z.Z.; Wang, M.L.; Liu, Y.Z. A study on international competitiveness of China’s forage industry. Pratacultural Sci. 2018, 35, 2530–2539. [Google Scholar] [CrossRef]
- Nyvall, R.F. Field Crop Diseases Handbook; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Pan, L.Q.; Zhang, L.; Yang, C.D.; Yuan, Q.H.; Wang, Y.; Miao, L.H. Identification and biological characteristics of Fusarium sporotrichioide isolated from Medicago sativa root. Acta Prataculturae Sin. 2015, 24, 88. [Google Scholar]
- Cong, L.L.; Kang, J.M.; Zhang, T.J.; Long, R.C.; Yang, Q.C. Identification and pathogenicity test of pathogenic Fusarium of alfalfa root rot. Acta Agrestia Sin. 2017, 25, 857. [Google Scholar]
- Huang, N.; Lu, X. Research and resistance evaluation progress on alfalfa leaf and root diseases. Chin. Agric. Sci. Bull. 2012, 28, 1–7. [Google Scholar]
- Wang, Y.; Yuan, Q.; Miao, L.; Zhang, L.; Pan, L. The major types and epidemic trends of alfalfa diseases in Northeast and North China. Acta Prataculturae Sin. 2016, 25, 52–59. [Google Scholar]
- Yuan, Q.H. Advances in alfalfa diseases in China. Plant Prot. Beijing 2007, 33, 6. [Google Scholar]
- Cao, L.; Zhao, C.; Bai, Q.; Shao, Z. Identification of the pathogens causing root rot of alfalfa in Inner Mongolia. Acta Agric. Boreali-Sin. 2008, 23, 105–107. [Google Scholar]
- Cong, L.L.; Sun, Y.; Wang, Z.; Kang, J.M.; Zhang, T.J.; Biligetu, B.; Yang, Q.C. A rapid screening method for evaluating resistance of alfalfa (Medicago sativa L.) to Fusarium root rot. Can. J. Plant Pathol. 2018, 40, 61–69. [Google Scholar] [CrossRef]
- Tu, X.B.; Du, G.L.; Li, C.J.; Wei, Y.H.; Zhang, W.G.; Yuan, X.J.; Hong, J.; LI, Y.Z.; Wang, B.H.; Zhao, L.; et al. Research progress in biological control of rangeland pests in China. Chin. J. Biol. Control 2015, 31, 780. [Google Scholar]
- Bani, M.; Rispail, N.; Evidente, A.; Rubiales, D.; Cimmino, A. Identification of the main toxins isolated from Fusarium oxysporum f. sp. pisi race 2 and their relation with isolates’ pathogenicity. J. Agric. Food Chem. 2014, 62, 2574–2580. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Wiesner-Hanks, T.; Wisser, R.; Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018, 19, 21–33. [Google Scholar] [CrossRef]
- Arnab, P.; Sanatan, G.; Shreeparna, G.; Singh, N.M.; Bhushan, T.S.; Kundu, C.R.; Dipankar, C. Comparative transcriptomic profiling of susceptible and resistant cultivars of pigeonpea demonstrates early molecular responses during Fusarium udum infection. Sci. Rep. 2021, 11, 22319. [Google Scholar]
- Krnjaja, V.; Tomić, Z.; Lević, J.; Nešić, Z. Resistance of certain alfalfa cultivars (Medicago sativa L.) to root rot in field conditions. Biotechnol. Anim. Husb. 2005, 21, 305–308. [Google Scholar] [CrossRef]
- Berrocal-Lobo, M.; Molina, A. Arabidopsis defense response against Fusarium oxysporum. Trends Plant Sci. 2008, 13, 145–150. [Google Scholar] [CrossRef]
- Wu, H.S.; Zhou, X.D.; Shi, X.; Liu, Y.D.; Wang, M.Y.; Shang, X.X.; Gu, D.L.; Wang, W.Z.; Wu, C.W. In vitro responses of Fusarium oxysporum f. sp. niveum to phenolic acids in decaying watermelon tissues. Phytochem. Lett. 2014, 8, 171–178. [Google Scholar] [CrossRef]
- Pan, L.Q. Studies on the Root Rot of Fusarium oxysporum in Alfalfa. Master’s Thesis, Gansu Agricultural University, Lanzhou, China, 2015. [Google Scholar]
- Yu, Y.Z. Studies on Small Black Spot Disease of Chinese Cabbage and Its Physiological Basis of Disease Resistance. Master’s Thesis, Shandong Agricultural University, Taian, China, 2007. [Google Scholar]
- Li, Y.L.; Long, S.S.; Guo, J.Z.; Zhang, Y.H.; Li, Q.; Wang, W. Changes of PAL and POD activity and isozymogram in Maize infected with Fusarium graminearum. Acta Bot. Boreali-Occident. Sin. 2003, 23, 1927–1931. [Google Scholar] [CrossRef]
- Cui, X.P. Study on Pathogen Identification and Biochemical Mechanism of Resistance to Ramie anthracnose. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2009. [Google Scholar]
- Gao, N.X.; Mu, Y.C.; Jiang, B.X.; Li, M.M.; Gao, Z.M. Physiological and biochemical analysis of resistance to Sclerotinia sclerotiorum in different rape varieties. J. Anhui Agric. Univ. 2012, 39, 672–676. [Google Scholar] [CrossRef]
- Li, J. Studies on the Pathogen and Physiological and Biochemical Mechanism of Root Rot of Lycium barbarum in Gansu Province. Master’s Thesis, Gansu Agricultural University, Lanzhou, China, 2015. [Google Scholar]
- Fang, T.; Hou, L.; Bai, L. Analysis of pathogenesis to Lycium barbarum root rot on transcriptome sequencing. J. Arid Land Resour. Environ. 2022, 36, 133–140. [Google Scholar]
- Yang, D.; Bao, Y.; Chen, L.M.; Cui, X.M.; Li, K.Z. Molecular cloning, bacterial expression analysis and functional characterization of pathogenesis-related PR10-2 gene in Panax notoginseng. China J. Chin. Mater. Medica 2017, 42, 3106–3111. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Wang, Z.C.; Dan, Z.C.; Zhang, L.X.; Xu, M.; Yang, G.F.; Chai, M.F.; Li, Z.Y.; Xie, H.L.; Cong, L.L. Transcriptome Analysis of Fusarium Root-Rot-Resistant and -Susceptible Alfalfa (Medicago sativa L.) Plants during Plant-Pathogen Interactions. Genes 2022, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, S. Decoding neuron transcriptome by SAGE. In Encyclopedia of Neuroscience; Elsevier Ltd.: Amsterdam, The Netherlands, 2009; pp. 357–363. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J. Determination of the content of soluble sugar in sweet corn with optimized anthrone colorimetric method. Storage Process 2013, 13, 24–27. [Google Scholar]
- Bainor, A.; Chang, L.; McQuade, T.J.; Webb, B.; Gestwicki, J.E. Bicinchoninic acid (BCA) assay in low volume. Anal. Biochem. 2011, 410, 310–312. [Google Scholar] [CrossRef]
- Draper, H.H.; Squires, E.J.; Mahmoodi, H.; Wu, J.; Agarwal, S.; Hadley, M. A comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials. Free Radic. Biol. Med. 1993, 15, 353–363. [Google Scholar] [CrossRef]
- Ukeda, H.; Kawana, D.; Maeda, S.; Sawamura, M. Spectrophotometric assay for superoxide dismutase based on the reduction of highly water-soluble tetrazolium salts by xanthine-xanthine oxidase. Biosci. Biotechnol. Biochem. 1999, 63, 485–488. [Google Scholar] [CrossRef]
- Chen, F.H.; Wang, L.; Hu, L.H. Selection of internal reference genes in real-time fluorescence quantitative RT-PCR. Chin. J. Clin. Lab. Sci. 2005, 15, 393–395. [Google Scholar] [CrossRef]
- Ge, X.X. Preliminary Study on the Mechanism of Resistance to Phytophthora Root Rot in Soybean. Ph.D. Thesis, Northeast Agricultural University, Haerbin, China, 2001. [Google Scholar]
- Perry, J.W.; Evert, R.F. The effect of colonization by Verticillium ahlia on the root tips of Russet Burbank potatoes. Can. J. Bot. 1983, 61, 3422–3429. [Google Scholar] [CrossRef]
- Garber, R. Penetration and development of Verticillium albo-atrum in the cotton plant. Phytopathology 1966, 56, 1121–1126. [Google Scholar]
- Pantelides, I.S.; Tjamos, S.E.; Striglis, I.A.; Chatzipavlidis, I.; Paplomatas, E.J. Mode of action of a non-pathogenic Fusarium oxysporum strain against Verticillium ahlia using Real Time QPCR analysis and biomarker transformation. Biol. Control 2009, 50, 30–36. [Google Scholar] [CrossRef]
- El-Fiki, A.; El-Deeb, A.; Mohamed, F.; Khalifa, M. Controlling Sesame charcoal Rot Incited by Macrophomina phaseolina under field conditions by using the resistant cultivars and some seed and soil treatments. Egypt J. Phytopathol. 2004, 32, 103–118. [Google Scholar]
- Li, Z.; Jin, X.; Zhang, Y.; Wu, C. The relationship between soluble protein, soluble sugar content and rice blast resistance of rice seedlings. N. Rice 2009, 39, 6–9. [Google Scholar]
- Durrant, W.E.; Dong, X. Systemic acquired resistance. Annu. Rev. Phytopathol. 2004, 42, 185–209. [Google Scholar] [CrossRef]
- Zhang, X.Y. Study on Identification Technique and Resistance Mechanism of Potato to Black Mole Disease. Ph.D. Thesis, Inner Mongolia Agricultural University, Huhehaote, China, 2012. [Google Scholar]
- Mu, X.Y.; Zhang, H.Y.; Zhao, Y. M Effects of rough dwarf disease on leaf physiological characteristics of different resistant maize varieties. Plant Physiol. J. 2014, 4, 801–804. [Google Scholar] [CrossRef]
- Liu, Z.D.; Zhang, S.W.; Liu, J.; Xu, B.L.; Jia, X.L.; Xue, Y.Y. Screening of powdery mildew-resistant varieties of Cucurbita pepo and its correlation with physiological traits. J. Gansu Agric. Univ. 2023, 58, 63–68. [Google Scholar] [CrossRef]
- Chen, Y.P. Pathogenicity of Root Rot Pathogen of Astragalus membranaceus and Its Effect on Physiological and Biochemical Indexes of Plant Stress Resistance. Master’s Thesis, Northwest Normal University, Lanzhou, China, 2013. [Google Scholar]
- Sun, R. Studies on the Response of Ginseng to Fusarium oxysporum Infection and the Changes of Metabolism. Master’s Thesis, Changchun University of Chinese Medicine, Changchun, China, 2022. [Google Scholar]
- Stadnik, M.J.; Buchenauer, H. Inhibition of phenylalanine ammonia-lyase suppresses the resistance induced by benzothiadiazole in wheat to Blumeria graminis f. sp. tritici. Physiol. Mol. Plant Pathol. 2000, 57, 25–34. [Google Scholar] [CrossRef]
- Aghajanzadeh, T.A.A.; Watanabe, M.; Tohge, T.; Hawkesford, M.J.J.; Fernie, A.R.R.; Hoefgen, R.; Elzenga, J.T.M.; De Kok, L.J.J. Necrotrophic fungal infection affects indolic glucosinolate metabolism in Brassica rapa. Acta Physiol. Plant. 2023, 45, 64. [Google Scholar] [CrossRef]
- Feng, J. Molecular Regulation Mechanism of Resistance to Powdery Mildew of Strawberry Mediated by Salicylic Acid. Ph.D. Thesis, Beijing Forestry University, Beijing, China, 2020. [Google Scholar]
- Girardi, C.L.; Rombaldi, C.V.; Dal Cero, J.; Nobile, P.M.; Laurens, F.; Bouzayen, M.; Quecini, V. Genome-wide analysis of the AP2/ERF superfamily in apple and transcriptional evidence of ERF involvement in scab pathogenesis. Sci. Hortic. 2013, 151, 112–121. [Google Scholar] [CrossRef]
- Pinot, F.; Benveniste, I.; Salaun, J.P.; Loreau, O.; Noel, J.P.; Schreiber, L.; Durst, F. Production in vitro by the cytochrome P450CYP94A1 of major C-18 cutin monomers and potential messengers in plant-pathogen interactions: Enantioselectivity studies. Biochem. J. 1999, 342, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Yang, Q. Study on the relationship between resistance of soybean and activity of PAL in leaves of soybean infected by Cercospora sojina Hara. Soybean Sci. 2002, 21, 195–198. [Google Scholar]
- Ma, J.; Li, M.; Zhang, Z.; Chai, Z. Study on relationship between phenylalanine ammonia-lyase (PAL) and resistance to crown and root rot in alfalfa cultivars. Acta Prataculturae Sin. 2003, 12, 35–39. [Google Scholar]
- Wang, R.; Wang, G.L.; Ning, Y. PALs: Emerging Key Players in Broad-Spectrum Disease Resistance. Trends Plant Sci. 2019, 24, 785–787. [Google Scholar] [CrossRef]
- Xu, J.; Huang, D.; Yue, E. Relationship of PAL activity and transfer of resistance to Fusarium oxysporum f. sp. niveum in grafted watermelon. J. Shanghai Jiaotong Univ. Agric. Sci. 2004, 22, 12–16. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Han, H.; Zhang, B.; Wang, L.; Wu, J.; Chen, Z.; Lin, K.; Hao, J.; Jia, R.; Zhang, Y. Identification of Crucial Genes and Regulatory Pathways in Alfalfa against Fusarium Root Rot. Plants 2023, 12, 3634. https://doi.org/10.3390/plants12203634
Wang S, Han H, Zhang B, Wang L, Wu J, Chen Z, Lin K, Hao J, Jia R, Zhang Y. Identification of Crucial Genes and Regulatory Pathways in Alfalfa against Fusarium Root Rot. Plants. 2023; 12(20):3634. https://doi.org/10.3390/plants12203634
Chicago/Turabian StyleWang, Shengze, Haibin Han, Bo Zhang, Le Wang, Jie Wu, Zhengqiang Chen, Kejian Lin, Jianjun Hao, Ruifang Jia, and Yuanyuan Zhang. 2023. "Identification of Crucial Genes and Regulatory Pathways in Alfalfa against Fusarium Root Rot" Plants 12, no. 20: 3634. https://doi.org/10.3390/plants12203634