
Transcriptome Profiling of Gossypium anomalum Seedlings Reveals Key Regulators and Metabolic Pathways in Response to Drought Stress
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
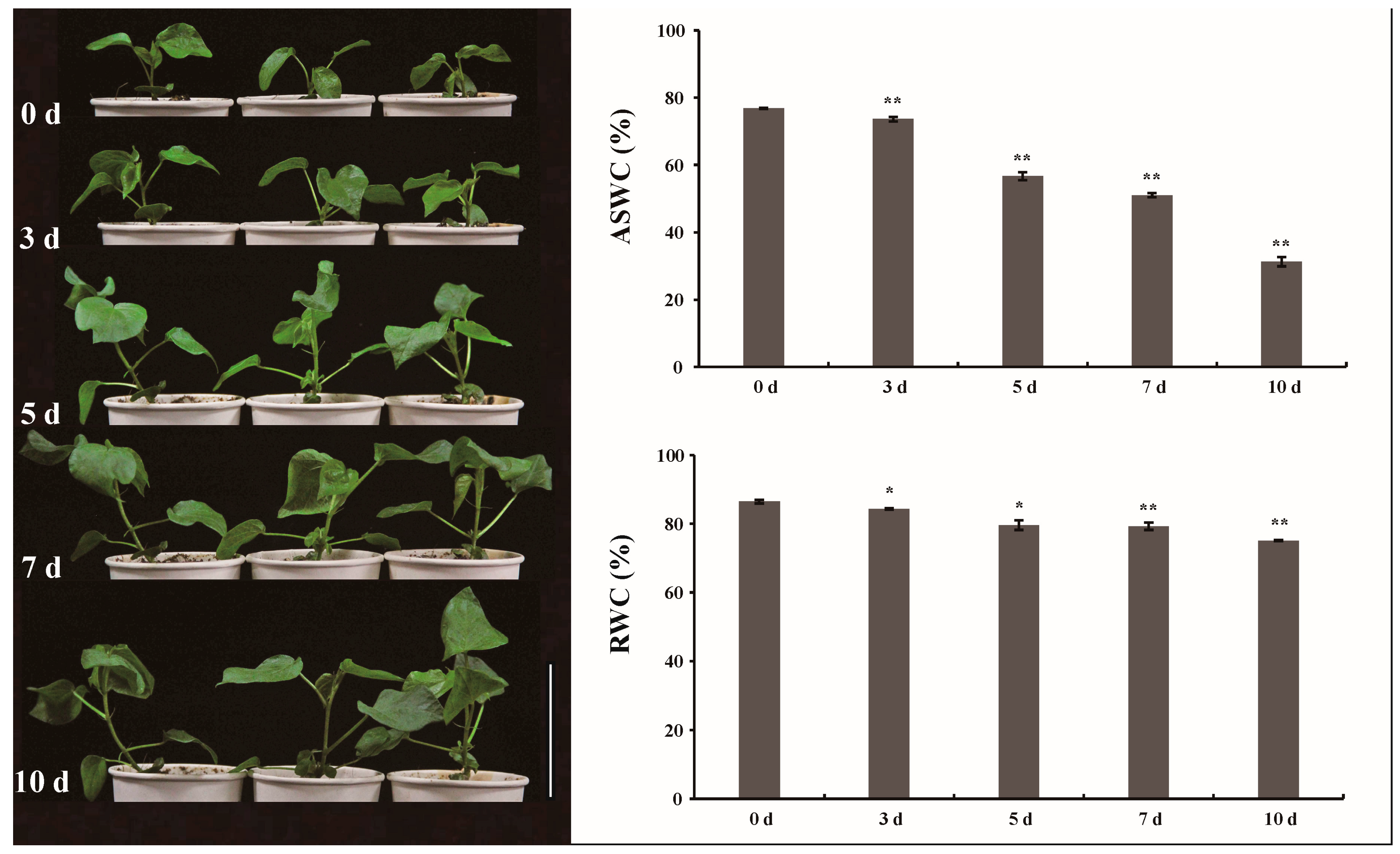
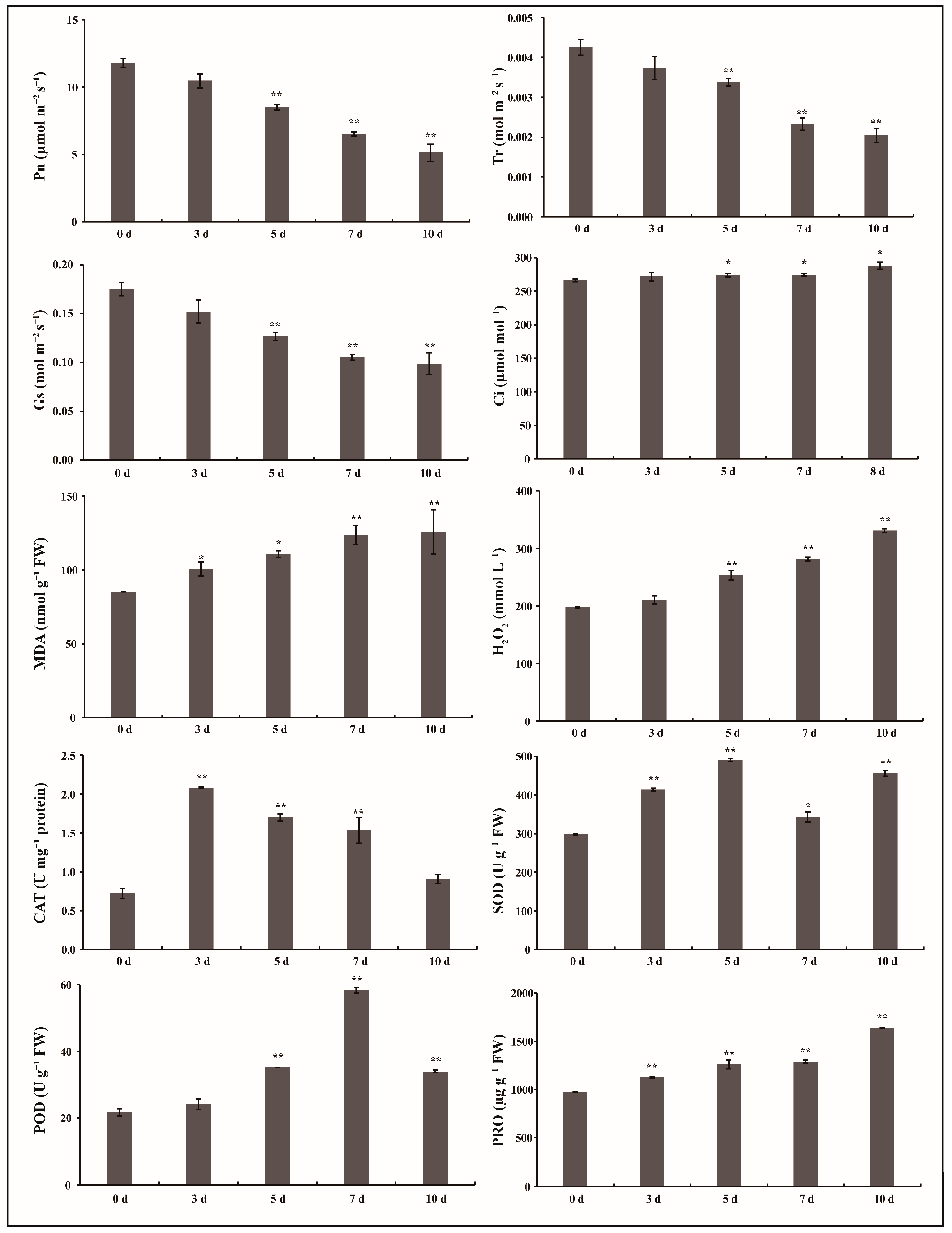
2.1. Physiological and Biochemical Evaluation of G. anomalum Seedlings in Response to Drought Treatment
2.2. Analyses of RNA-Seq Data
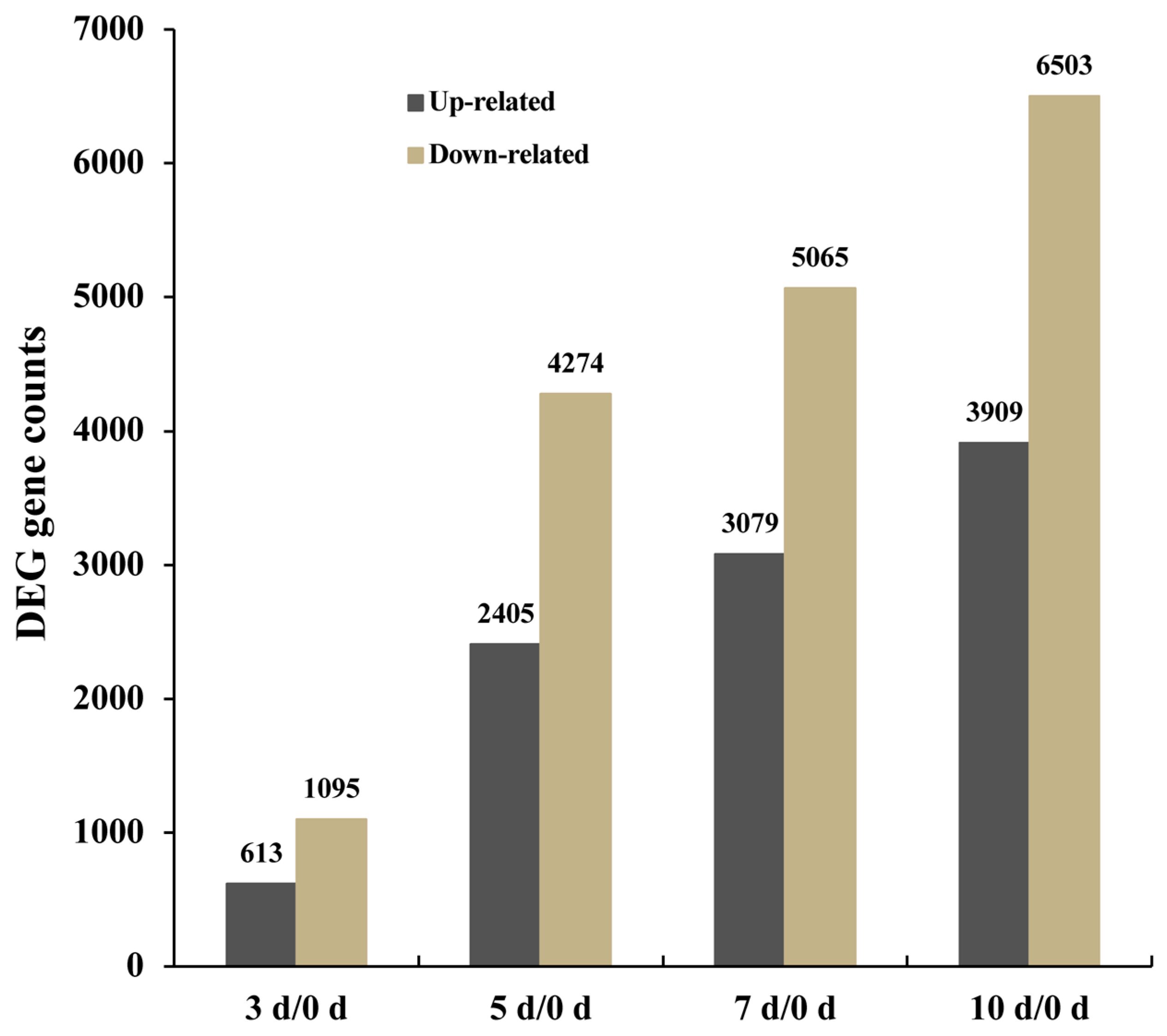
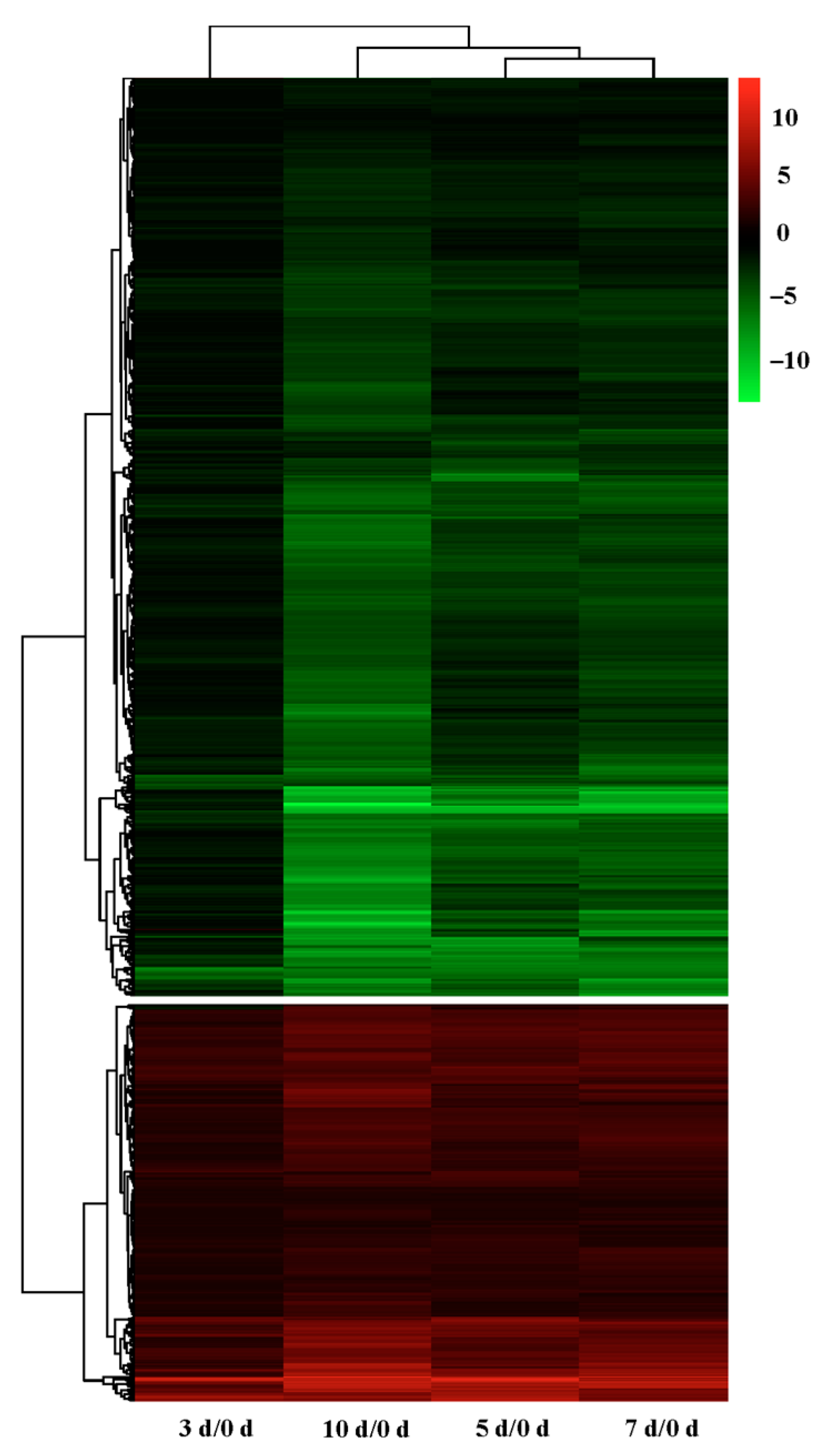
2.3. Differential Gene Expression at Different Stages of Drought Stress
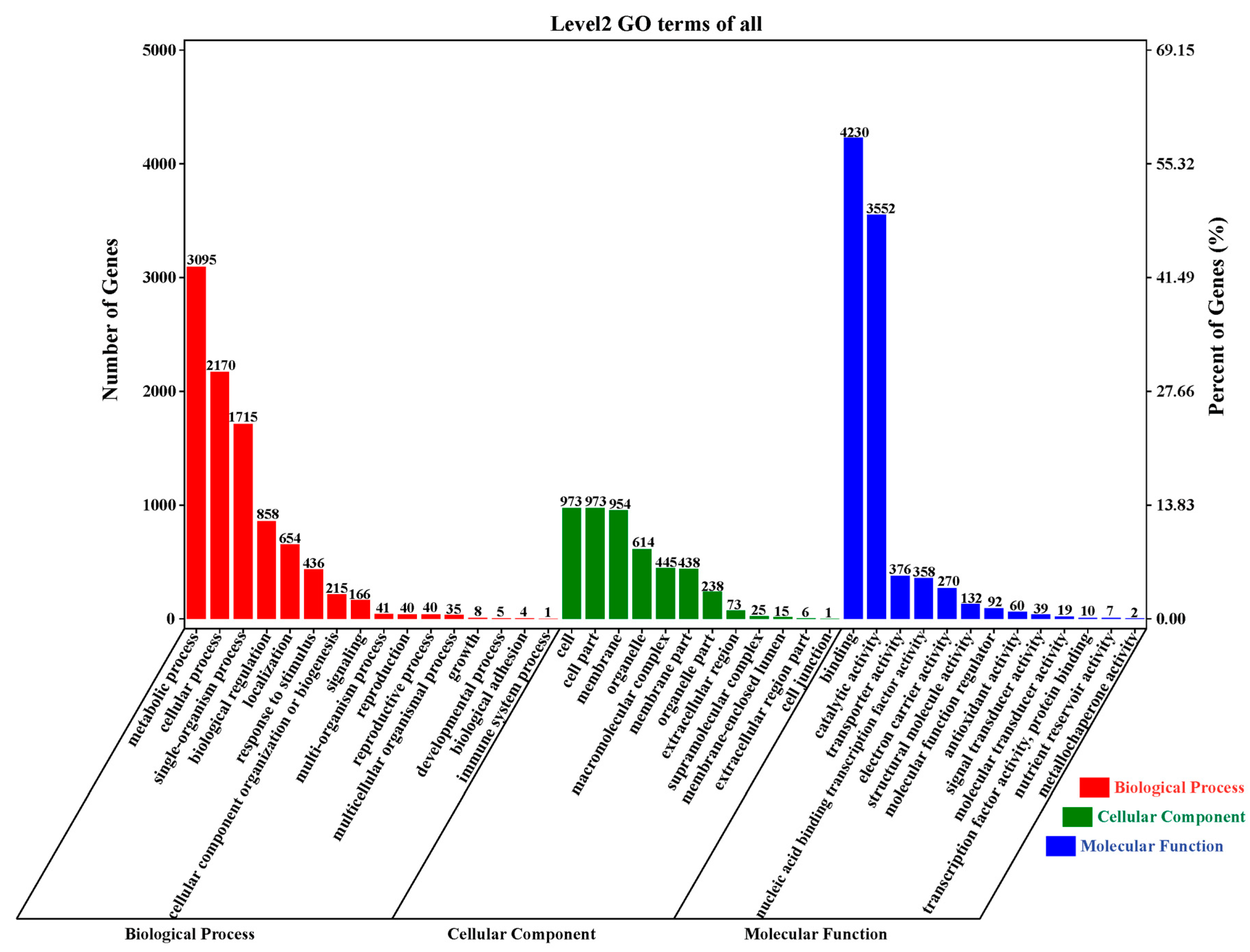
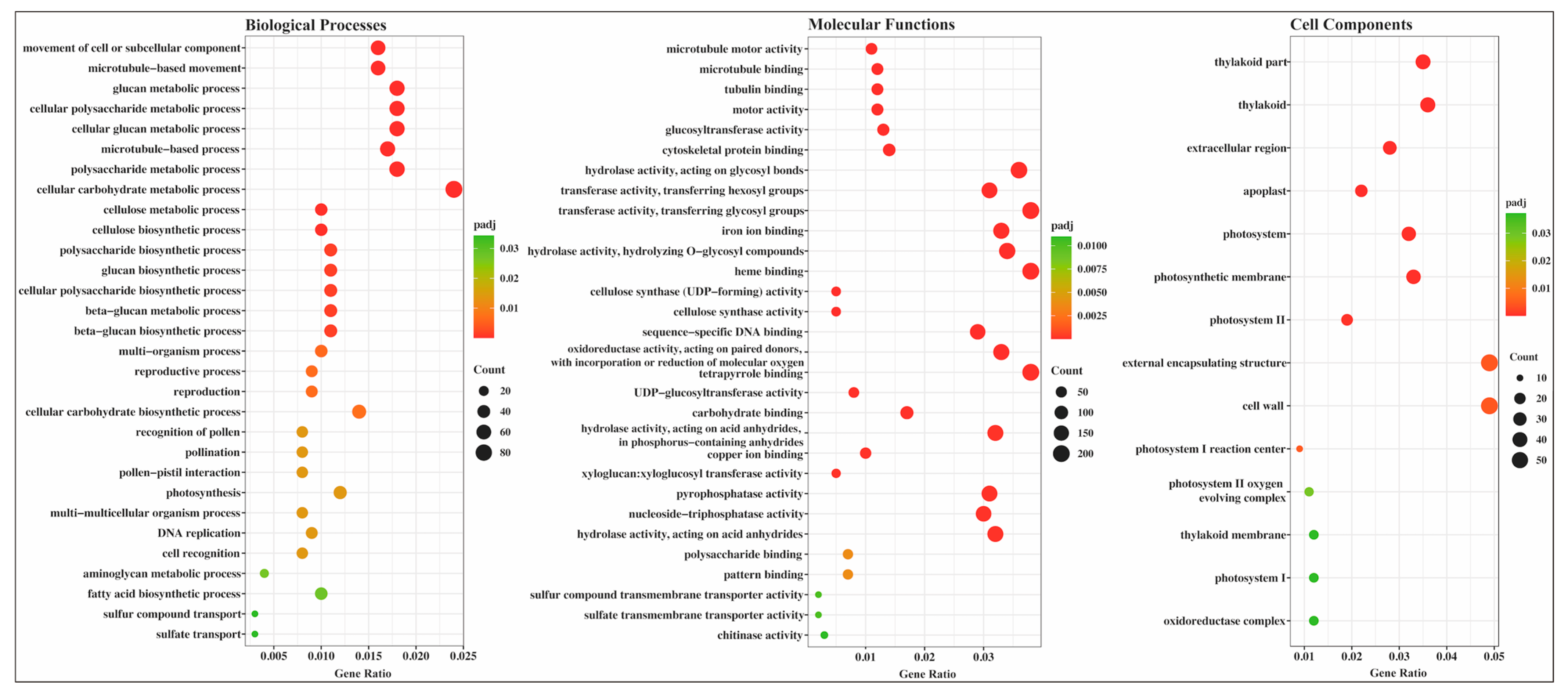
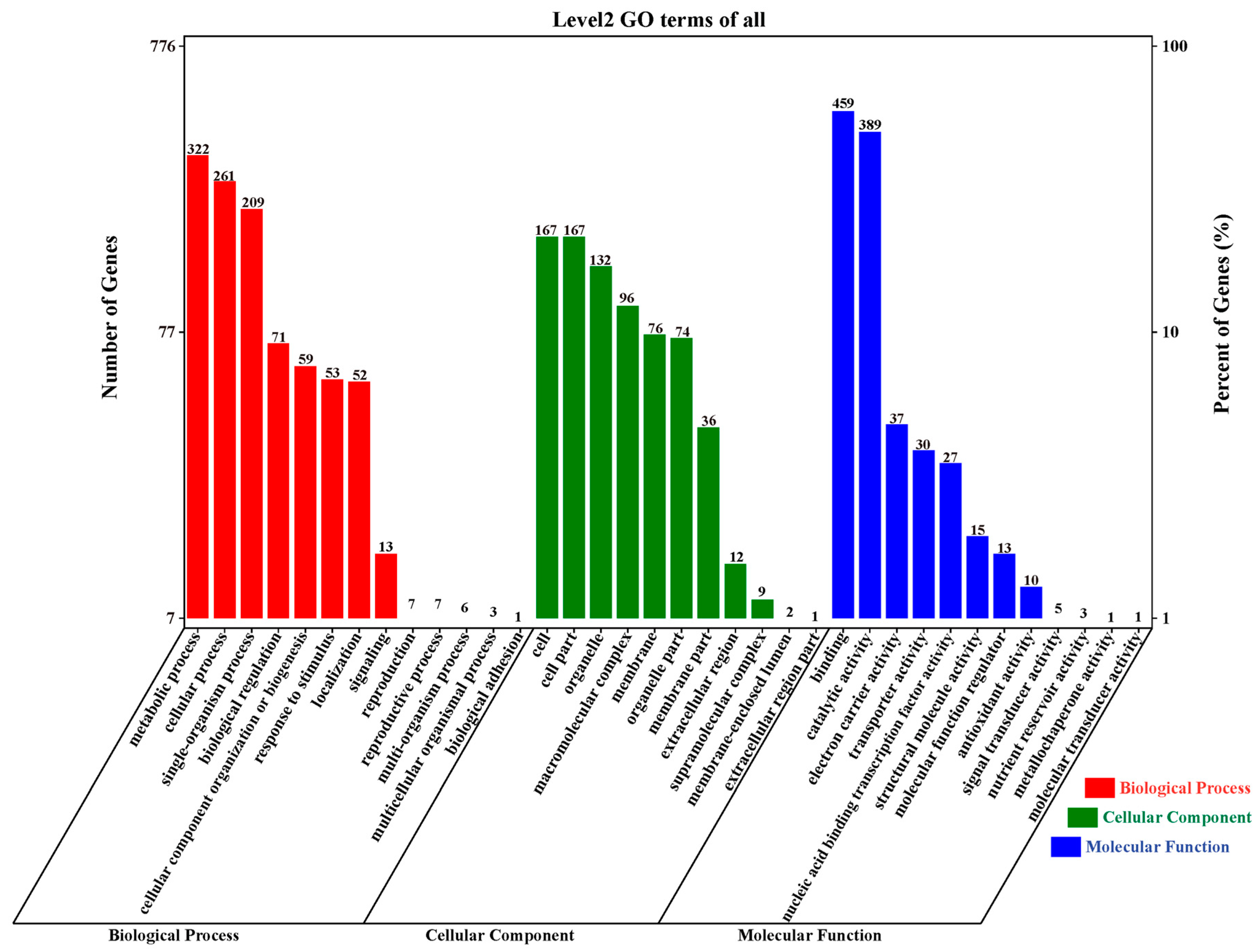
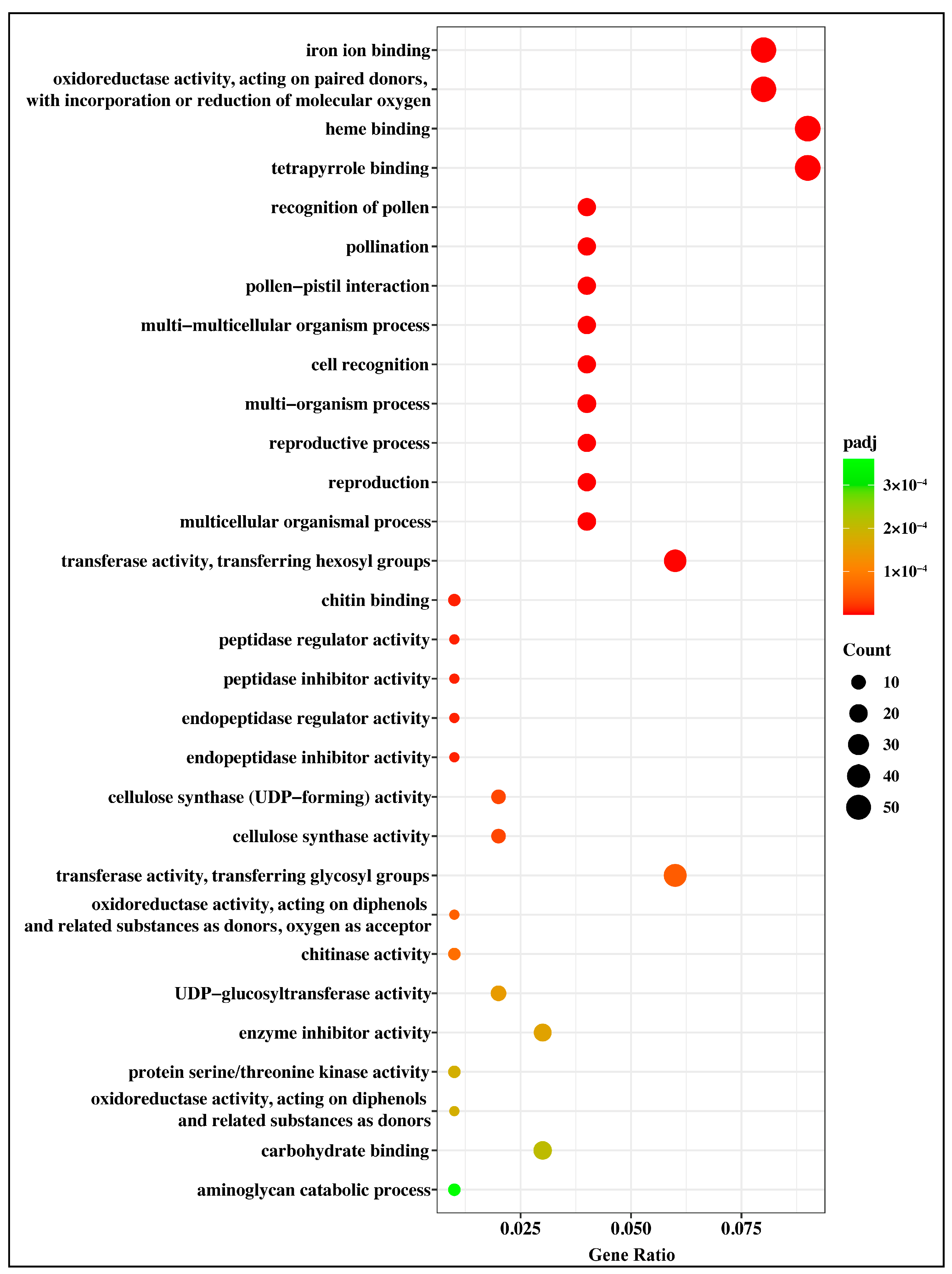
2.4. Gene Ontology (GO) Annotation and Enrichment Analysis
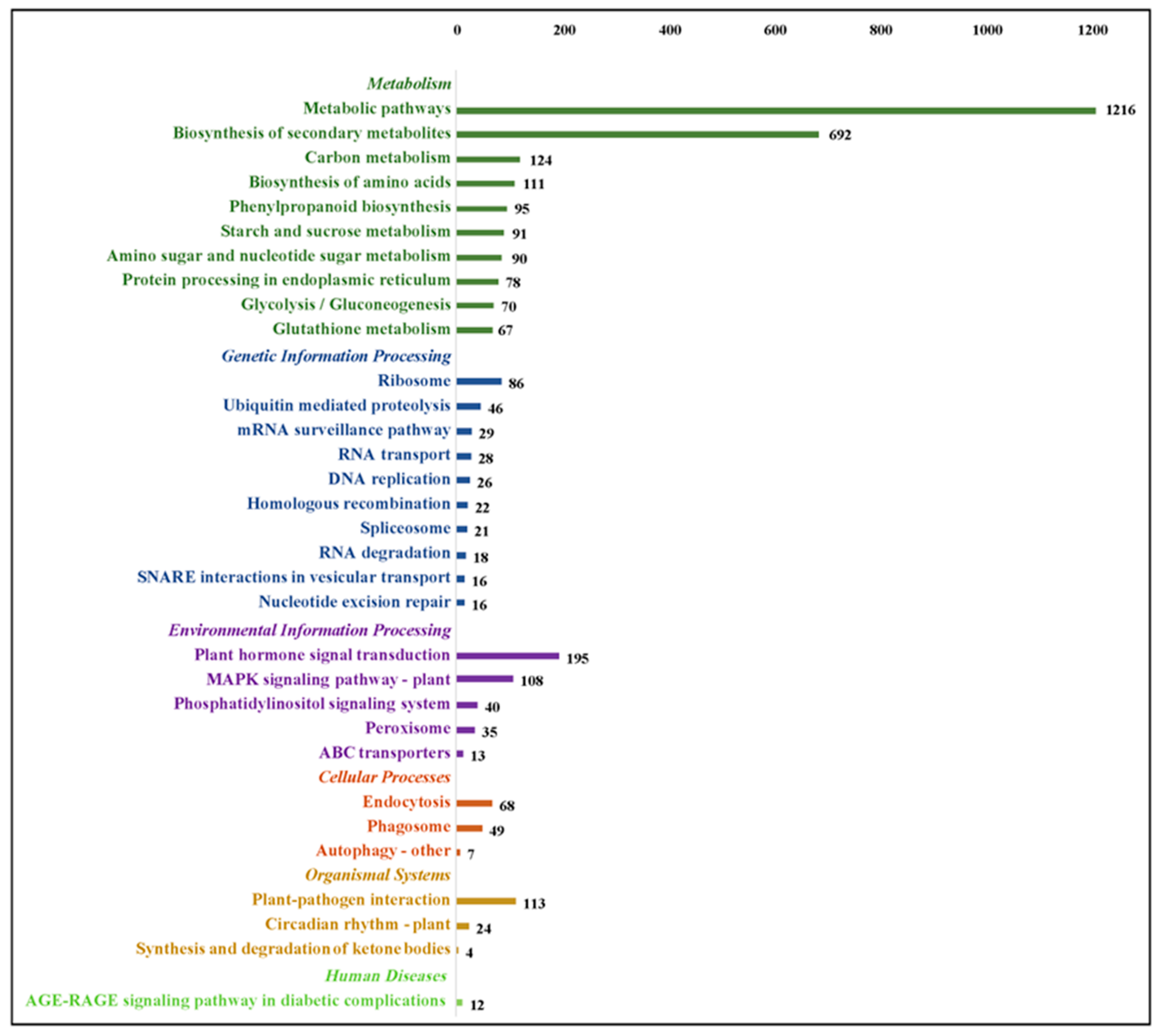
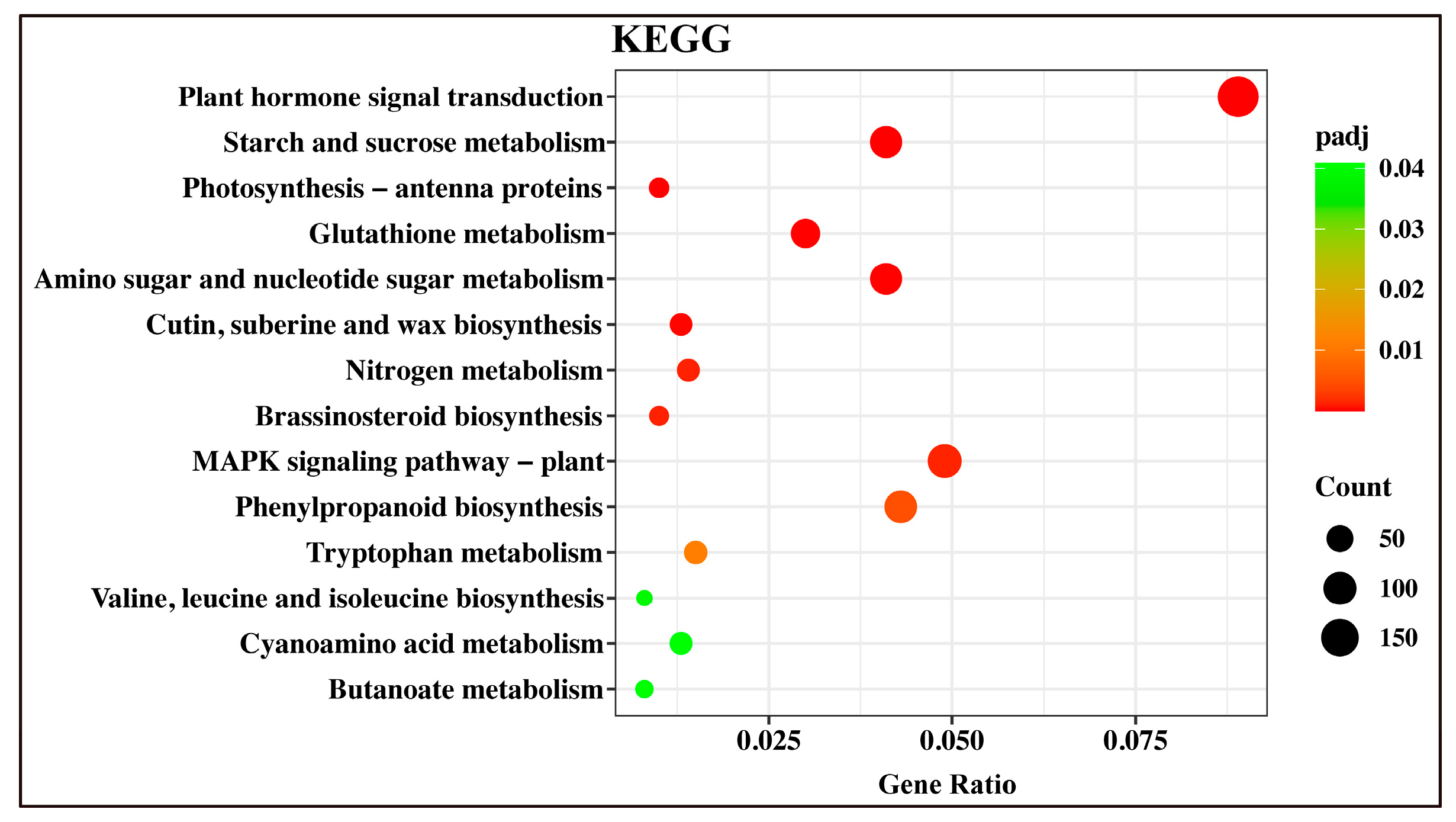
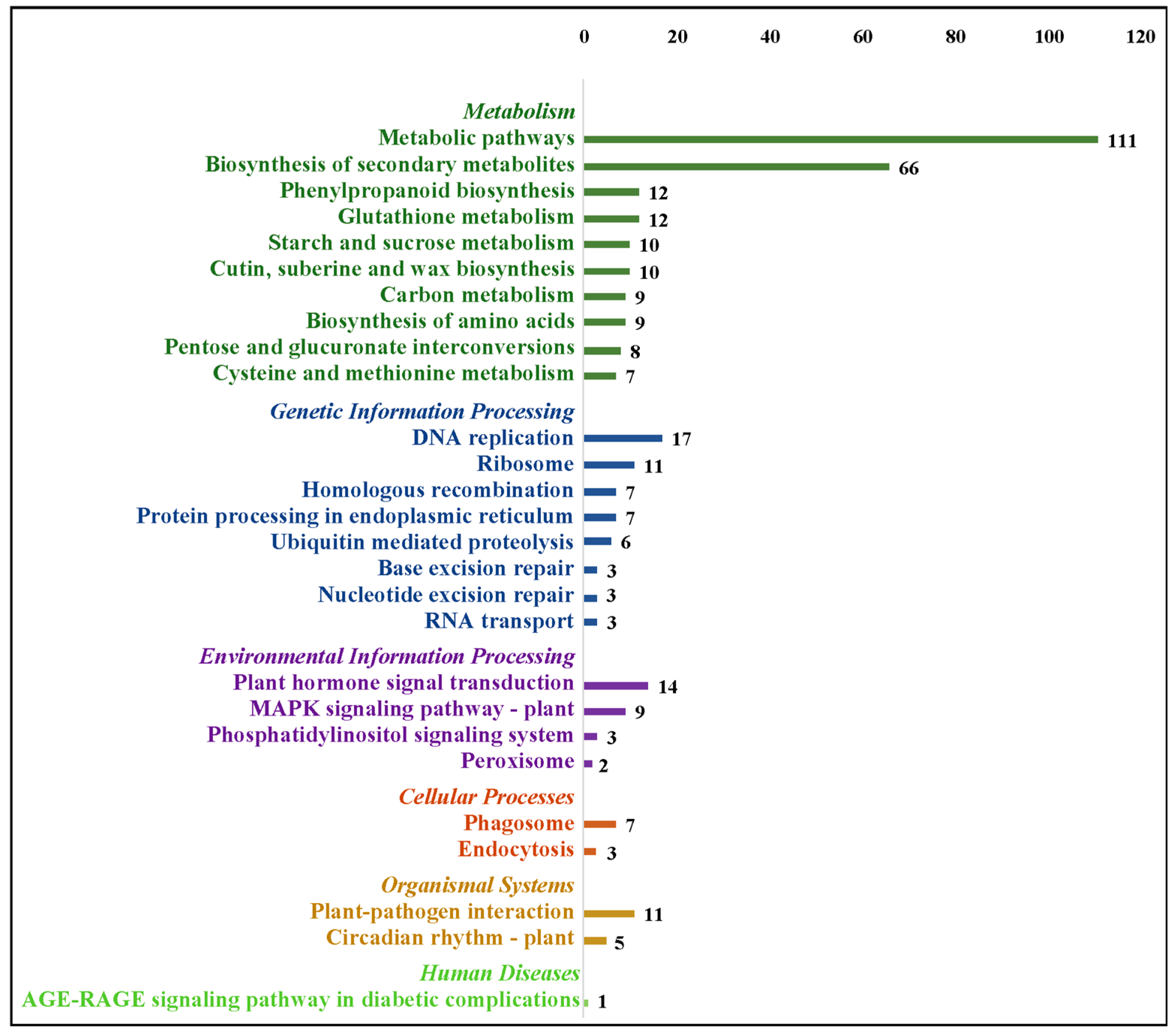
2.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Annotation and Enrichment Analysis
2.6. Functional Annotation of Common DEGs
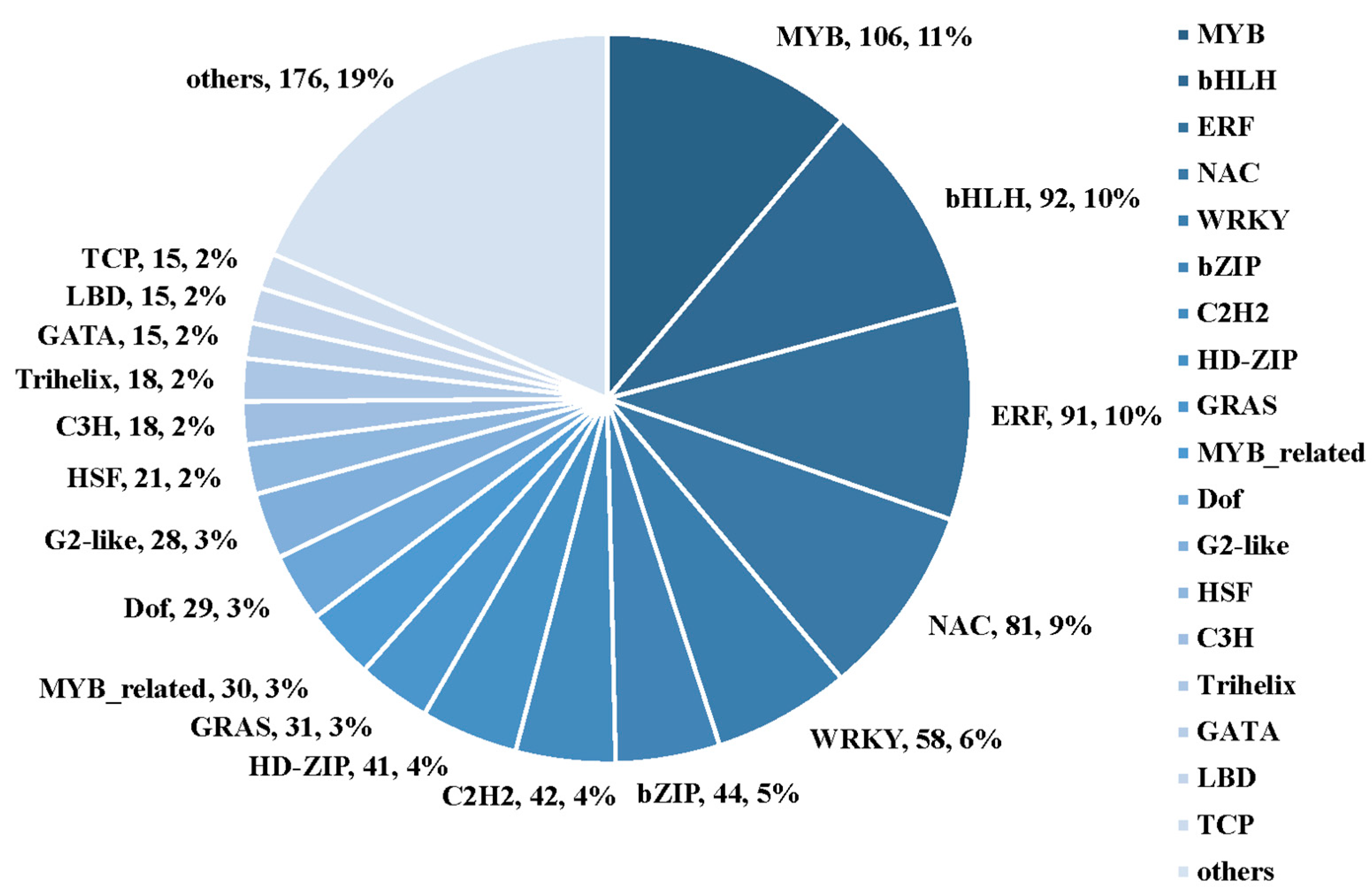
2.7. Identification of Drought-Responsive Tandemly Repeated Genes and Transcriptional Factors (TFs)
3. Discussion
4. Materials and Methods
4.1. Preparation of Plant Material
4.2. Determination of Physiological and Biochemical Indexes
4.3. Library Preparation for Transcriptome Sequencing
4.4. Differentially Expressed Genes (DEGs) Analysis
4.5. GO and KEGG Analysis
4.6. Tandemly Repeated Genes and Transcription Factors (TFs) Annotation
4.7. Data Validation by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Reynolds, M.; Tuberosa, R. Translational research impacting on crop productivity in drought-prone environments. Curr. Opin. Plant Biol. 2008, 11, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Babu, R.C.; Blum, A. Breeding for drought resistance in rice: Physiology and molecular genetics considerations. Crop Sci. 1997, 37, 1426–1434. [Google Scholar] [CrossRef]
- Chen, X.X.; Ding, Y.Y.; Yang, Y.Q.; Song, C.P.; Wang, B.S.; Yang, S.H.; Guo, Y.; Gong, Z.Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef] [PubMed]
- Garrido, Y.; Tudela, J.A.; Marin, A.; Mestre, T.; Martinez, V.; Gil, M.I. Physiological, phytochemical and structural changes of multi-leaf lettuce caused by salt stress. J. Sci. Food Agric. 2014, 94, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.M.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14 (Suppl. S1), 165–183. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Umezawa, T.; Urano, K.; Shinozaki, K. Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 2007, 10, 296–302. [Google Scholar] [CrossRef]
- Umezawa, T.; Fujita, M.; Fujita, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Engineering drought tolerance in plants: Discovering and tailoring genes to unlock the future. Curr. Opin. Biotechnol. 2006, 17, 113–122. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef]
- Hirayama, T.; Shinozak, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant J. 2010, 61, 1041–1052. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Matsui, A.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kaminuma, E.; Endo, T.A.; Okamoto, M.; Nambara, E.; Nakajima, M.; Kawashima, M.; et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008, 49, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.W.; Tai, H.H.; Li, S.S.; Gao, W.; Zhao, M.; Xie, C.X.; Li, W.X. bHLH122 is important for drought and osmotic stress resistance in Arabidopsis and in the repression of ABA catabolism. New Phytol. 2014, 201, 1192–1204. [Google Scholar] [CrossRef]
- Rasheed, S.; Bashir, K.; Matsui, A.; Tanaka, M.; Seki, M. Transcriptomic analysis of soil-grown Arabidopsis thaliana roots and shoots in response to a drought stress. Front. Plant Sci. 2016, 7, 180. [Google Scholar] [CrossRef]
- Rasheed, S.; Bashir, K.; Nakaminami, K.; Hanada, K.; Matsui, A.; Seki, M. Drought stress differentially regulates the expression of small open reading frames (sORFs) in Arabidopsis roots and shoots. Plant Signal Behav. 2016, 11, e1215792. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.E.; Gao, C.X.; Zhang, Y.H.; Yan, Q.D.; Hu, W.; Yang, L.; Wang, Z.; Li, F.G. Recent progression and future perspectives in cotton genomic breeding. J. Integr. Plant Biol. 2022. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.D.; Fang, L.; Zhang, Z.Y.; Ma, W.; Niu, Y.C.; Ju, L.Z.; Deng, J.Q.; Zhao, T.; Lian, J.M.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Parida, A.K.; Dagaonkar, V.S.; Phalak, M.S.; Umalkar, G.; Aurangabadkar, L.P. Alterations in photosynthetic pigments, protein and osmotic components in cotton genotypes subjected to short-term drought stress followed by recovery. Plant Biotechnol. Rep. 2007, 1, 37–48. [Google Scholar] [CrossRef]
- Lee, J.A. Cotton as a World Crop. In Cotton; Kohel, R.J., Lewis, C.F., Eds.; ASA, CSSA, SSSA: Madison, WI, USA, 1984; Volume 24, pp. 1–25. [Google Scholar]
- Fryxell, P.A. A revised taxonomic interpretation of Gossypium L. (Malvacea). Rheedea 1992, 2, 108–165. [Google Scholar]
- Mehetre, S.S. Wild Gossypium anomalum: A unique source of fibre fineness and strength-overview and achievements of prebreeding efforts. Curr. Sci. 2010, 99, 58–71. [Google Scholar]
- Newaskar, G.S.; Chimote, V.P.; Mehetre, S.S.; Jadhav, A.S. Interspecific hybridization in Gossypium L.: Characterization of progenies with different ploidy-confirmed multigenomic backgrounds. Plant Breed. 2013, 132, 211–216. [Google Scholar] [CrossRef]
- Silow, R.A. The comparative genetics of Gossypium anomalum and the cultivated Asiatic cottons. J. Genet. 1941, 42, 259–358. [Google Scholar] [CrossRef]
- Vieiradasilva, J.B. A contribution to the study of drought resistance in the genus Gossypium. II.-The variation of some enzyme activities. Physiol. Veg. Paris 1970, 8, 413–447. [Google Scholar]
- Narayanan, S.S.; Singh, J.; Varma, P.K. Introgressive gene transfer in Gossypium: Goals, problems, strategies and achievements. Cotton Fibres Trop. 1984, 39, 123–135. [Google Scholar]
- Sathiyaraj, G.; Lee, O.R.; Parvin, S.; Khorolragchaa, A.; Kim, Y.J.; Yang, D.C. Transcript profiling of antioxidant genes during biotic and abiotic stresses in Panax ginseng C. A. Meyer. Mol. Biol. Rep. 2011, 38, 2761–2769. [Google Scholar] [CrossRef]
- Juszczuk, I.M.; Szal, B.; Rychter, A.M. Oxidation-reduction and reactive oxygen species homeostasis in mutant plants with respiratory chain complex I dysfunction. Plant Cell Environ. 2012, 35, 296–307. [Google Scholar] [CrossRef]
- Xu, Z.Z.; Chen, J.D.; Meng, S.; Xu, P.; Zhai, C.J.; Huang, F.; Guo, Q.; Zhao, L.; Quan, Y.G.; Shangguan, Y.X.; et al. Genome sequence of Gossypium anomalum facilitates interspecific introgression breeding. Plant Commun. 2022, 3, 100350. [Google Scholar] [CrossRef]
- Li, X.Y.; Feng, B.E.; Zhang, F.J.; Tang, Y.M.; Zhang, L.P.; Ma, L.J.; Zhao, C.P.; Gao, S.Q. Bioinformatic analyses of subgroup-A members of the wheat bZIP transcription factor family and functional identification of TabZIP174 involved in drought stress response. Front. Plant Sci. 2016, 7, 1643. [Google Scholar] [CrossRef] [Green Version]
- Mosfeq-Ul Hasan, M.; Ma, F.L.; Islam, F.; Sajid, M.; Prodhan, Z.H.; Li, F.; Shen, H.; Chen, Y.D.; Wang, X.D. Comparative transcriptomic analysis of biological process and key pathway in three cotton (Gossypium spp.) species under drought stress. Int. J. Mol. Sci. 2019, 20, 2076. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.L.; Xu, Z.; Cong, K.; Zhu, X.Y.; Zhang, L.; Wang, J.F.; Wei, J.K.; Ji, P.Z. Transcriptomic responses to drought stress in Polygonatum kingianum tuber. BMC Plant Biol. 2021, 21, 537. [Google Scholar] [CrossRef]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2012, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Fracasso, A.; Trindade, L.M.; Amaducci, S. Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 2016, 16, 115. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Bai, L.J.; Wei, Z.X.; Yuan, H.J.; Wang, Y.L.; Xu, Q.J.; Tang, Y.W.; Nyima, T. Transcriptome analysis revealed the drought-responsive genes in Tibetan hulless barley. BMC Genom. 2016, 17, 386. [Google Scholar] [CrossRef]
- Min, H.W.; Chen, C.X.; Wei, S.W.; Shang, X.L.; Sun, M.Y.; Xia, R.; Liu, X.G.; Hao, D.Y.; Chen, H.B.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, T.; Khalid, S.; Abdullah, M.; Ahmed, Z.; Shah, M.K.N.; Ghafoor, A.; Du, X.M. Insights into drought stress signaling in plants and the molecular genetic basis of cotton drought tolerance. Cells 2020, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.; Park, Y.; Hwang, I. Abscisic acid: Biosynthesis, inactivation, homoeostasis and signalling. Essays Biochem. 2015, 58, 29–48. [Google Scholar]
- Ma, L.F.; Li, Y.; Chen, Y.; Li, X.B. Plant cell, tissue, and organ culture. Plant Cell Tissue Organ Cult. 2016, 124, 583–598. [Google Scholar] [CrossRef]
- Liang, C.Z.; Liu, Y.; Li, Y.Y.; Meng, Z.G.; Yan, R.; Zhu, T.; Wang, Y.; Kang, S.J.; Abid, M.A.; Malik, W.; et al. Activation of ABA receptors gene GhPYL9-11A is positively correlated with cotton drought tolerance in transgenic Arabidopsis. Front. Plant Sci. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Mao, X.G.; Wang, C.S.; Jing, R.L. Overexpression of a common wheat gene Tasnrk2.8 enhances tolerance to drought, salt and low temperature in Arabidopsis. PLoS ONE 2010, 5, e16041. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Wang, J.; Zhu, M.; Jia, H.H.; Liu, D.D.; Hao, L.L.; Guo, X.Q. A cotton Raf-like MAP3K gene, GhMAP3K40, mediates reduced tolerance to biotic and abiotic stress in Nicotiana benthamiana by negatively regulating growth and development. Plant Sci. 2015, 240, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.N.; Zhao, L.L.; Lu, R.; Li, Y.; Li, X.B. Cotton mitogen-activated protein kinase4 (GhMPK4) confers the transgenic Arabidopsis hypersensitivity to salt and osmotic stresses. Plant Cell Tissue Organ Cult. 2015, 123, 619–632. [Google Scholar] [CrossRef]
- Long, L.; Gao, W.; Xu, L.; Liu, M.; Luo, X.Y.; He, X.; Yang, X.Y.; Zhang, X.L.; Zhu, L.F. GbMPK3, a mitogen-activated protein kinase from cotton, enhances drought and oxidative stress tolerance in tobacco. Plant Cell Tissue Organ Cult. 2014, 116, 153–162. [Google Scholar] [CrossRef]
- Li, Y.Z.; Zhang, L.; Wang, X.L.; Zhang, W.; Hao, L.L.; Chu, X.Q.; Guo, X.Q. Cotton GhMPK6a negatively regulates osmotic tolerance and bacterial infection in transgenic Nicotiana benthamiana, and plays a pivotal role in development. FEBS J. 2013, 280, 5128–5144. [Google Scholar] [CrossRef]
- Lu, W.J.; Chu, X.Q.; Li, Y.Z.; Wang, C.; Guo, X.Q. Cotton GhMKK1 induces the tolerance of salt and drought stress, and mediates defence responses to pathogen infection in transgenic Nicotiana benthamiana. PLoS ONE 2013, 8, e68503. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.Z.; Lu, W.J.; Meng, F.; Wu, C.A.; Guo, X.Q. Cotton GhMKK5 affects disease resistance, induces HR-like cell death, and reduces the tolerance to salt and drought stress in transgenic Nicotiana benthamiana. J. Exp. Bot. 2012, 63, 3935–3951. [Google Scholar] [CrossRef]
- Zhang, L.; Xi, D.M.; Li, S.W.; Gao, Z.; Zhao, S.L.; Shi, J.; Wu, C.G.; Guo, X.Q. A cotton group C MAP kinase gene, GhMPK2, positively regulates salt and drought tolerance in tobacco. Plant Mol. Biol. 2011, 77, 17–31. [Google Scholar] [CrossRef]
- Chen, E.Y.; Zhang, X.Y.; Yang, Z.E.; Wang, X.Q.; Yang, Z.R.; Zhang, C.J.; Wu, Z.X.; Kong, D.P.; Liu, Z.; Zhao, G.; et al. Genome-wide analysis of the HD-ZIP IV transcription factor family in Gossypium arboreum and GaHDG11 involved in osmotic tolerance in transgenic Arabidopsis. Mol. Genet. Genom. 2017, 292, 593–609. [Google Scholar] [CrossRef]
- Sadau, S.B.; Ahmad, A.; Tajo, S.M.; Ibrahim, S.; Kazeem, B.B.; Wei, H.L.; Yu, S.X. Overexpression of GhMPK3 from cotton enhances cold, drought, and salt stress in Arabidopsis. Agronomy 2021, 11, 1049. [Google Scholar] [CrossRef]
- Liu, D.D.; Zhu, M.; Hao, L.L.; Chen, X.B.; Gao, Y.G.; Gao, X.; Li, H. GhMAPKKK49, a novel cotton (Gossypium hirsutum L.) MAPKKK gene, is involved in diverse stress responses. Acta Physiol. Plant. 2016, 38, 1–12. [Google Scholar]
- Li, F.J.; Li, M.Y.; Wang, P.; Cox, K.L.; Duan, L.S.; Dever, J.K.; Shan, L.B.; Li, Z.H.; He, P. Regulation of cotton (Gossypium hirsutum) drought responses by mitogen-activated protein (MAP) kinase cascade-mediated phosphorylation of GhWRKY59. New Phytol. 2017, 215, 1462–1475. [Google Scholar] [CrossRef] [Green Version]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E.; et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef]
- Wang, M.; Wang, S.B.; Xia, G.M. From genome to gene: A new epoch for wheat research? Trends Plant Sci. 2015, 20, 380–387. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, J.R.; Qin, L.M.; Shi, W.M.; Xia, G.M.; Liu, S.W. TaCYP81D5, one member in a wheat cytochrome P450 gene cluster, confers salinity tolerance via reactive oxygen species scavenging. Plant Biotechnol. J. 2019, 18, 791–804. [Google Scholar] [CrossRef] [Green Version]
- DaCosta, M.; Huang, B.R. Changes in antioxidant enzyme activities and lipid peroxidation for Bentgrass species in response to drought stress. J. Am. Soc. Hortic. Sci. 2007, 132, 319–326. [Google Scholar] [CrossRef]
- Turner, N.C. Imposing and maintaining soil water deficits in drought studies in pots. Plant Soil 2018, 439, 45–55. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Parallel mapping of transcriptomes to detect InDels, gene fusions, and more. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.Y.; You, Q.; Yi, X.; Du, Z.; Xu, W.Y.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, W.; Yu, H.; Shangguan, Y.; Cao, J.; Chen, X.; Zhao, L.; Guo, Q.; Xu, P.; Shen, X.; Xu, Z. Transcriptome Profiling of Gossypium anomalum Seedlings Reveals Key Regulators and Metabolic Pathways in Response to Drought Stress. Plants 2023, 12, 312. https://doi.org/10.3390/plants12020312
Ji W, Yu H, Shangguan Y, Cao J, Chen X, Zhao L, Guo Q, Xu P, Shen X, Xu Z. Transcriptome Profiling of Gossypium anomalum Seedlings Reveals Key Regulators and Metabolic Pathways in Response to Drought Stress. Plants. 2023; 12(2):312. https://doi.org/10.3390/plants12020312
Chicago/Turabian StyleJi, Wei, Huan Yu, Yixin Shangguan, Jing Cao, Xianglong Chen, Liang Zhao, Qi Guo, Peng Xu, Xinlian Shen, and Zhenzhen Xu. 2023. "Transcriptome Profiling of Gossypium anomalum Seedlings Reveals Key Regulators and Metabolic Pathways in Response to Drought Stress" Plants 12, no. 2: 312. https://doi.org/10.3390/plants12020312