
Comparative Analysis and Identification of Terpene Synthase Genes in Convallaria keiskei Leaf, Flower and Root Using RNA-Sequencing Profiling


Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Isolation, cDNA Library Construction, and Illumina Sequencing
2.2. De Novo Assembly and Functional Annotation
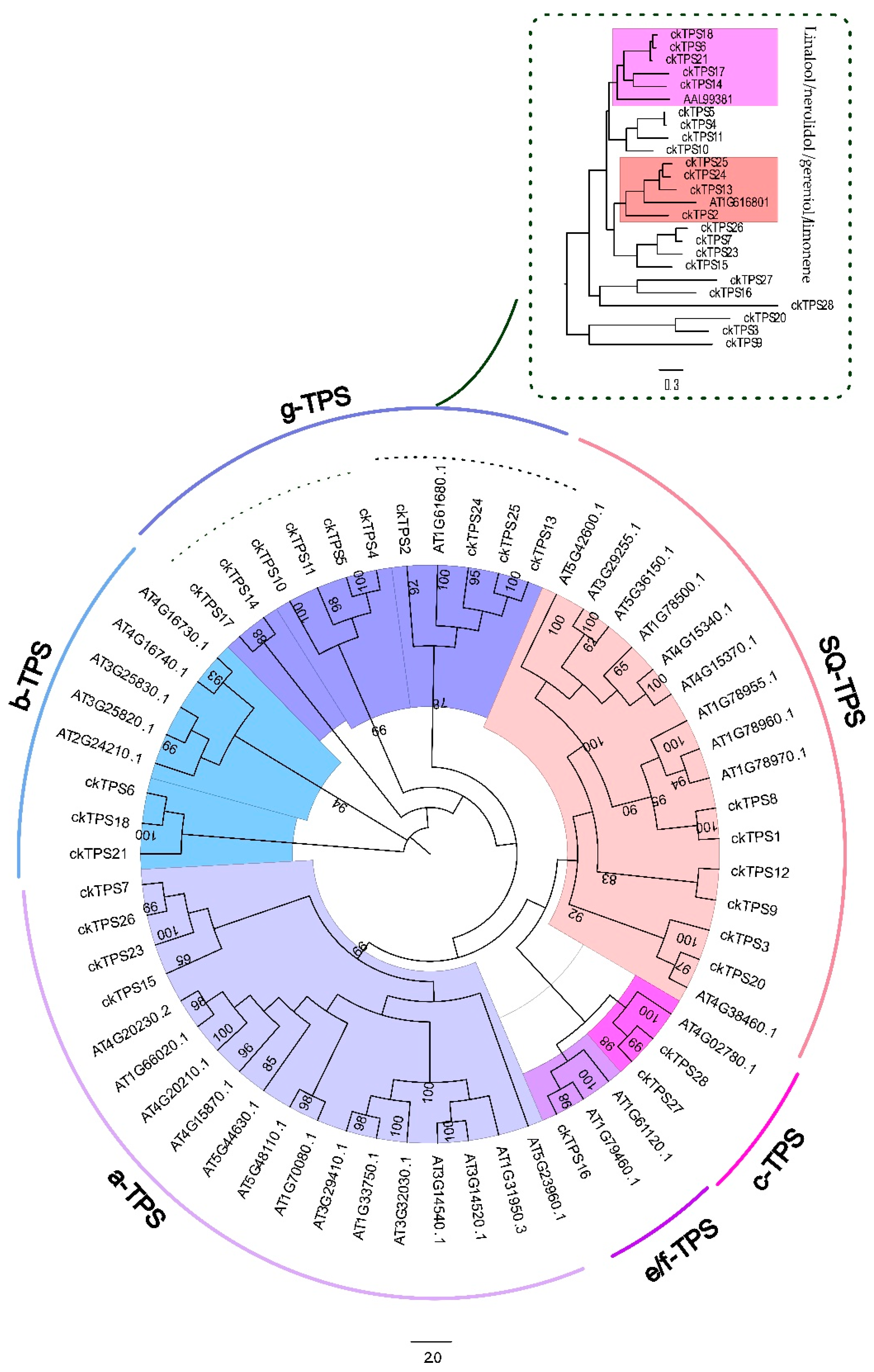
2.3. Identification and Phylogenetic Tree Analysis of Terpene Synthase Unigenes
2.4. Differentially Expressed Gene (DEG) Analysis
2.5. Quantative PCR Analysis in C. keiskei
3. Results
3.1. De Novo Assembly and Annotation Stats for C. keiskei
3.2. Gene Ontology and KEGG Annotation
3.3. Transcriptome-Wide Identification of TPS Genes Using Phylogenetic Tree Analysis
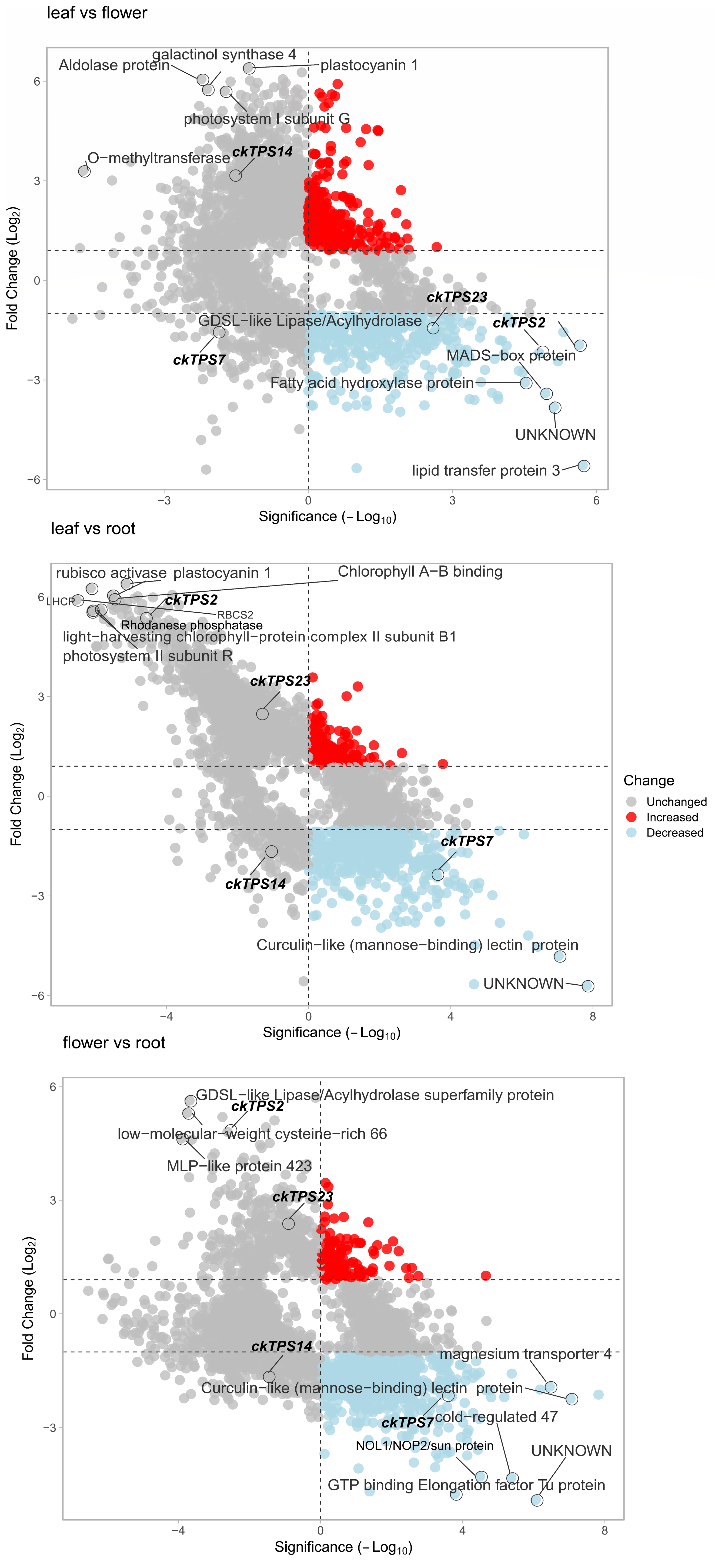
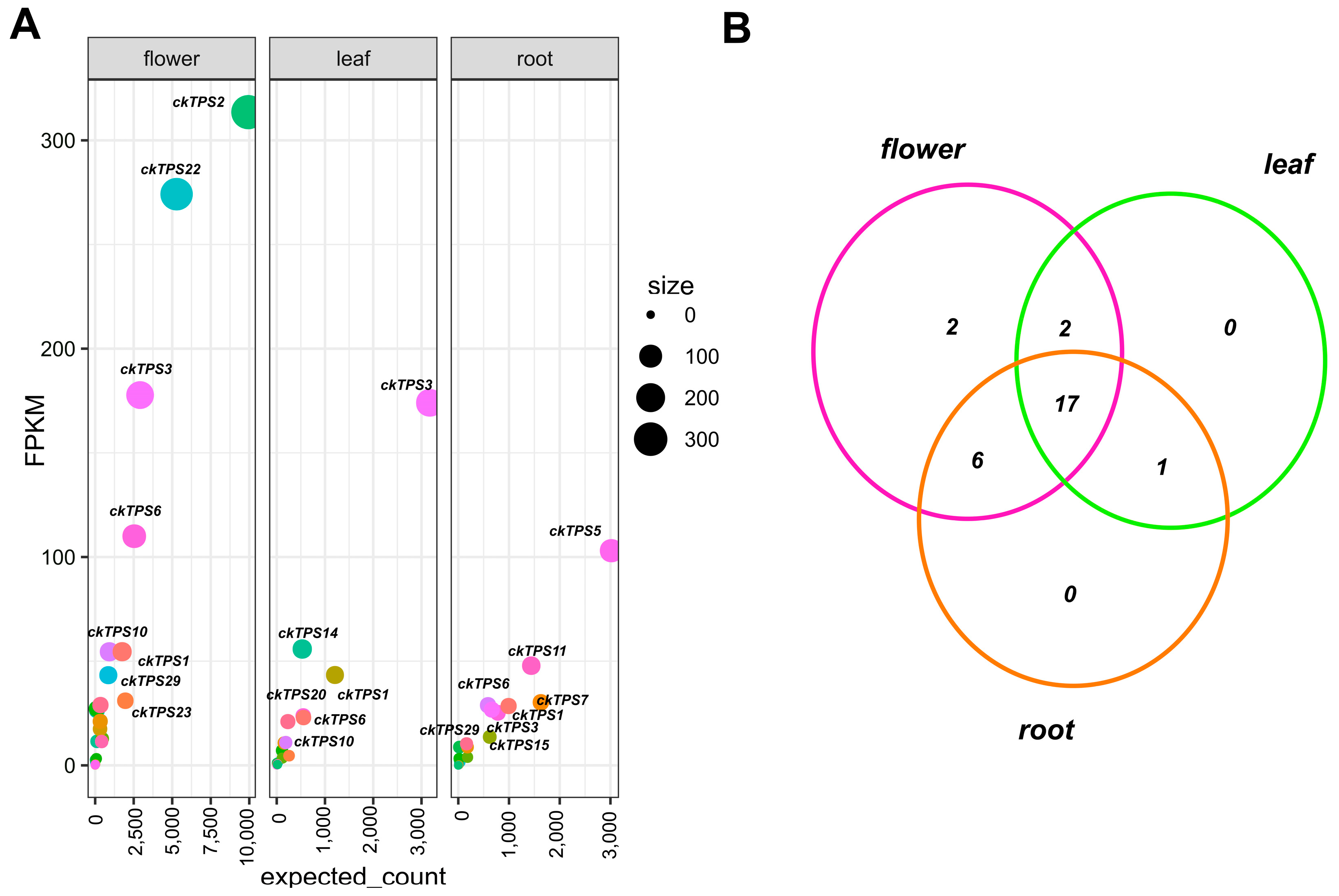
3.4. Identification of Floral Scent-Related ckTPS
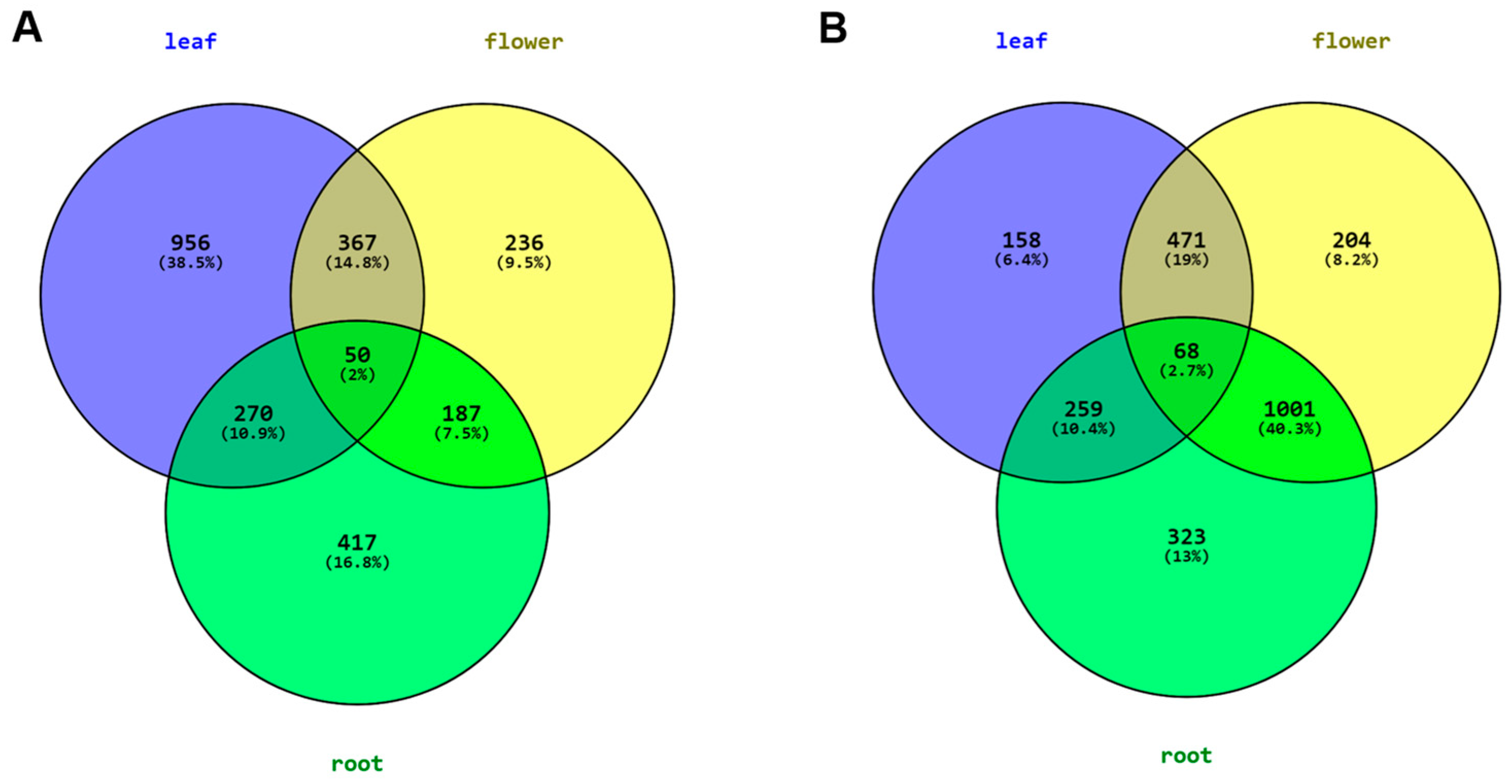
3.5. Differential Expression Analysis of the Leaf, Flower and Root Samples of C. keiskei
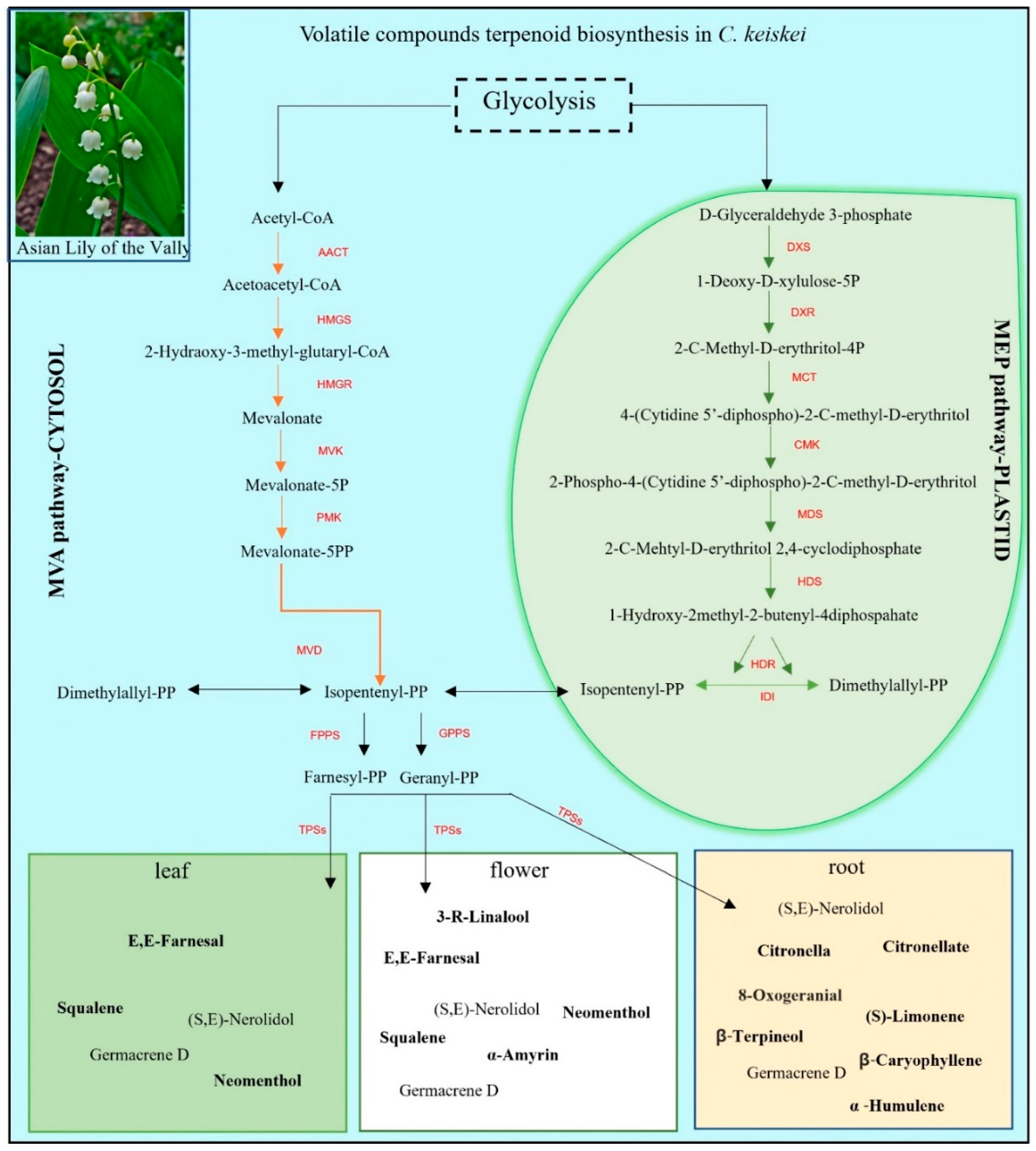
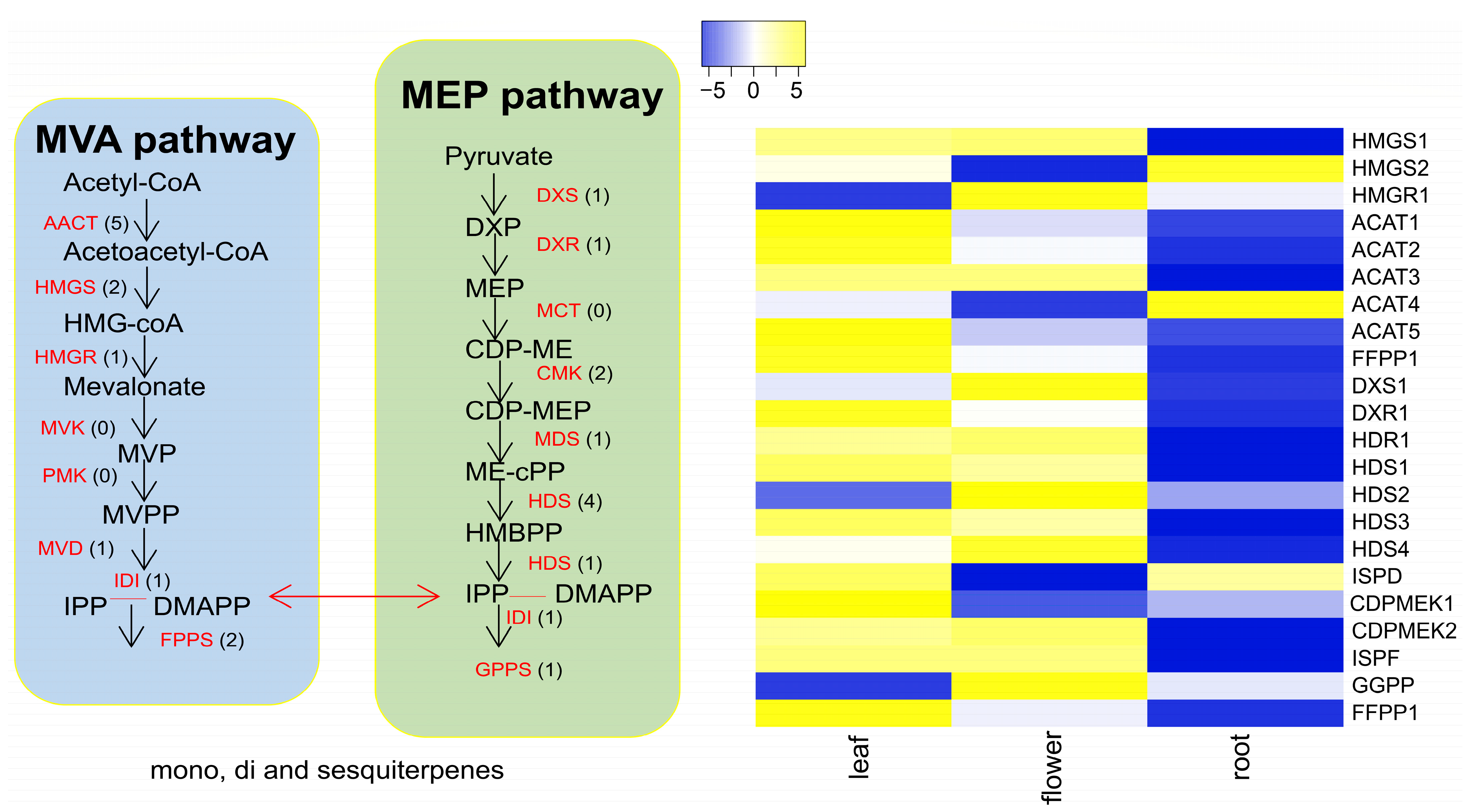
3.6. Terpene Backbone Pathway and Terpene Synthase Expression Patterns in C. keiskei
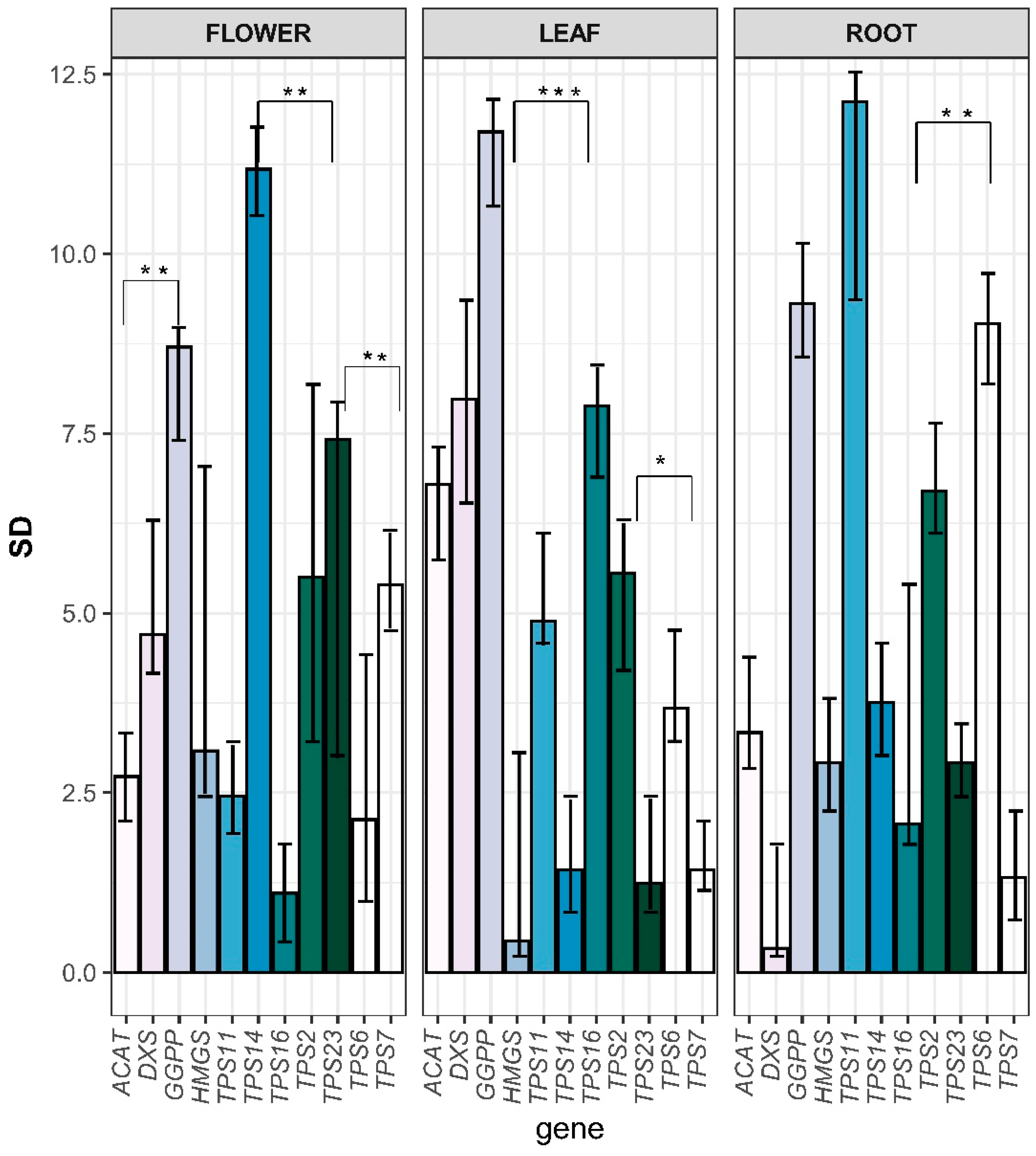
3.7. Quantitative Assay of Terpene Backbone Pathway Genes in C. keiskei
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yeshi, K.; Crayn, D.; Ritmejeryte, E.; Wangchuk, P. Plant secondary metabolites produced in response to abiotic stresses has potential application in pharmaceutical product development. Molecules 2022, 27, 313. [Google Scholar] [CrossRef] [PubMed]
- Biala, W.; Jasinski, M. The phenylpropanoid case—It is transport that matters. Front. Plant Sci. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecerova, K.; Klem, K.; Vesela, B.; Holub, P.; Grace, J.; Urban, O. Combined effect of altitude, season and light on the accumulation of extractable terpenes in Norway spruce needles. Forests 2021, 12, 1737. [Google Scholar] [CrossRef]
- Pichersky, E.; Gershenzon, J. The formation and function of plant volatiles: Perfumes for pollinator attraction and defense. Curr. Opin. Plant Biol. 2002, 5, 237–243. [Google Scholar] [CrossRef]
- Abbas, F.; Rothenberg, D.O.; Zhou, Y.W.; Ke, Y.G.; Wang, H.C. Volatile organic compounds as mediators of plant communication and adaptation to climate change. Physiol. Plant. 2022, 174, e13840. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.S.; de Jesus, B.; da Silva Luiz, S.R.; Viana, C.B.; Adão Malafaia, C.R.; Figueiredo, F.S.; Carvalho, T.; Silva, M.L.; Londero, V.S.; da Costa-Silva, T.A.; et al. Antiinflammatory activity of natural triterpenes—An overview from 2006 to 2021. Phytother. Res. 2022, 36, 1459–1506. [Google Scholar] [CrossRef]
- Tang, H.V.; Berryman, D.L.; Mendoza, J.; Yactayo-Chang, J.P.; Li, Q.B.; Christensen, S.A.; Hunter, C.T.; Best, N.; Soubeyrand, E.; Akhtar, T.A.; et al. Dedicated farnesyl diphosphate synthases circumvent isoprenoid-derived growth-defense tradeoffs in Zea mays. Plant J. 2022, 112, 207–220. [Google Scholar] [CrossRef]
- Ramya, M.; Jang, S.; An, H.R.; Lee, S.Y.; Park, P.M.; Park, P.H. Volatile organic compounds from Orchids: From synthesis and function to gene regulation. Int. J. Mol. Sci. 2020, 21, 1160. [Google Scholar] [CrossRef] [Green Version]
- Pathak, G.; Dudhagi, S.S.; Raizada, S.; Singh, R.K.; Sane, A.P.; Sane, V.A. Phosphomevalonate kinase regulates the MVA/MEP pathway in mango during ripening. Plant Physiol. Biochem. 2023, 196, 174–185. [Google Scholar] [CrossRef]
- Kempinski, C.; Jiang, Z.; Zinck, G.; Sato, S.J.; Ge, Z.; Clemente, T.E.; Chappell, J. Engineering linear, branched-chain triterpene metabolism in monocots. Plant Biotechnol. J. 2019, 17, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Jongedijk, E.; Bouwmeester, H.; Van Der Krol, A. Monoterpene biosynthesis potential of plant subcellular compartments. New Phytol. 2016, 209, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Dornelas, M.C.; Mazzafera, P. A genomic approach to characterization of the Citrus terpene synthase gene family. Genet. Mol. Biol. 2007, 30, 832–840. [Google Scholar] [CrossRef]
- Yahyaa, M.; Tholl, D.; Cormier, G.; Jensen, R.; Simon, P.W.; Ibdah, M. Identification and characterization of terpene synthases potentially involved in the formation of volatile terpenes in Carrot (Daucus carota L.) roots. J. Agric. Food Chem. 2015, 63, 4870–4878. [Google Scholar] [CrossRef]
- Zhou, H.C.; Shamala, L.F.; Yi, X.K.; Yan, Z.; Wei, S. Analysis of terpene synthase family genes in Camellia sinensis with an emphasis on abiotic stress conditions. Sci. Rep. 2020, 10, 933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, T.T.; Shadrack, K.; Yang, S.; Xue, X.X.; Li, S.Y.; Wang, N.; Wang, Q.Y.; Wang, L.; Gao, X.; Cronk, Q. Functional characterization of terpene synthases accounting for the volatilized-terpene heterogeneity in Lathyrus odoratus cultivar flowers. Plant Cell Physiol. 2020, 61, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Jin, J.J.; Sarojam, R.; Ramachandran, S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 2019, 11, 2078–2098. [Google Scholar] [CrossRef] [Green Version]
- Aubourg, S.; Lecharny, A.; Bohlmann, J. Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol. Genet. Genom. 2002, 267, 730–745. [Google Scholar] [CrossRef]
- Keilwagen, J.; Lehnert, H.; Berner, T.; Budahn, H.; Nothnagel, T.; Ulrich, D.; Dunemann, F. The terpene synthase gene family of Carrot (Daucus carota L.): Identification of QTLs and candidate genes associated with terpenoid volatile compounds. Front. Plant Sci. 2017, 8, 1930. [Google Scholar] [CrossRef] [Green Version]
- Kulheim, C.; Padovan, A.; Hefer, C.; Krause, S.T.; Kollner, T.G.; Myburg, A.A.; Degenhardt, J.; Foley, W.J. The Eucalyptus terpene synthase gene family. BMC Genom. 2015, 16, 450. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuizen, N.J.; Green, S.A.; Chen, X.Y.; Bailleul, E.J.D.; Matich, A.J.; Wang, M.Y.; Atkinson, R.G. Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol. 2013, 161, 787–804. [Google Scholar] [CrossRef] [Green Version]
- Falara, V.; Akhtar, T.A.; Nguyen, T.T.H.; Spyropoulou, E.A.; Bleeker, P.M.; Schauvinhold, I.; Matsuba, Y.; Bonini, M.E.; Schilmiller, A.L.; Last, R.L.; et al. The tomato terpene synthase gene family. Plant Physiol. 2011, 157, 770–789. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Kollner, T.G.; Li, G.L.; Wei, G.; Chen, X.L.; Zeng, D.L.; Qian, Q.; Chen, F. Combinatorial evolution of a terpene synthase gene cluster explains terpene variations in Oryza. Plant Physiol. 2020, 182, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Bremer, B.; Bremer, K.; Chase, M.W.; Fay, M.F.; Reveal, J.L.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F.; Anderberg, A.A.; Moore, M.J.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Lee, E.M.; Park, S. Intracellular DNA transfer events restricted to the genus Convallaria within the Asparagaceae family: Possible mechanisms and potential as genetic markers for biographical studies. Genomics 2021, 113, 2906–2918. [Google Scholar] [CrossRef]
- Raman, G.; Park, S.; Lee, E.M.; Park, S. Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the Asparagales. Sci. Rep. 2019, 9, 5028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, M.; Araki, K.; Yamada, E.; Kawano, S. Life-history monographs of Japanese plants. 6: Convallaria keiskei Miq. (Convallariaceae). Plant Spec. Biol. 2006, 21, 119–126. [Google Scholar] [CrossRef]
- Yang, S.Y.; Kim, N.H.; Cho, Y.S.; Lee, H.; Kwon, H.J. Convallatoxin, a dual inducer of autophagy and apoptosis, inhibits angiogenesis in vitro and in vivo. PLoS ONE 2014, 9, e91094. [Google Scholar] [CrossRef]
- Choi, D.H.; Kang, D.G.; Cul, X.; Cho, K.W.; Sohn, E.J.; Kim, J.S.; Lee, H.S. The positive inotropic effect of the aqueous extract of Convallaria keiskei in beating rabbit atria. Life Sci. 2006, 79, 1178–1185. [Google Scholar] [CrossRef]
- Dorrich, S.; Mahler, C.; Tacke, R.; Kraft, P. Synthesis and olfactory characterization of silicon-containing derivatives of the acyclic Lily-of-the-Valley odorant 5,7,7-trimethyl-4-methylideneoctanal. Chem. Biodivers. 2014, 11, 1675–1687. [Google Scholar] [CrossRef]
- Men, W.X.; Song, Y.Y.; Xing, Y.P.; Bian, C.; Xue, H.F.; Xu, L.; Xie, M.; Kang, T.G. The complete chloroplast genome sequence of Convallaria majalis L. Mitochondrial DNA Part B 2022, 7, 692–693. [Google Scholar] [CrossRef]
- Ramachanderan, R.; Schaefer, B. Lily-of-the-valley fragrances. Chemtexts 2019, 5, 11. [Google Scholar] [CrossRef]
- Hwang, S.H.; Jang, Y.S.; Wang, Z.Q.; Kim, Y.D.; Lim, S.S. Quantification of the volatile constituents found in Convallaria keiskei. Chem. Nat. Compd. 2017, 53, 377–378. [Google Scholar] [CrossRef]
- Guo, X.L.; Yu, C.; Luo, L.; Wan, H.H.; Li, Y.S.; Wang, J.; Cheng, T.R.; Pan, H.T.; Zhang, Q.X. Comparative transcriptome analysis of the floral transition in Rosa chinensis ‘Old Blush’ and R. odorata var. gigantea. Sci. Rep. 2017, 7, 6068. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.X.; Zhang, J.; Zhang, B.H.; Jin, X.F.; Zhang, H.Y.; Jin, Z.N. Transcriptional analysis of metabolic pathways and regulatory mechanisms of essential oil biosynthesis in the leaves of Cinnamomum camphora (L.) Presl. Front. Genet. 2020, 11, 598714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yan, Y.; Zhou, W.H.; Feng, R.Z.; Shuai, Y.K.; Yang, L.; Liu, M.J.; He, X.Y.; Wei, Q. Transcriptome and metabolome reveal the accumulation of secondary metabolites in different varieties of Cinnamomum longepaniculatum. BMC Plant Biol. 2022, 22, 243. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.L.; Kang, K.C.; Wang, P.; Li, M.; Huang, X.Z. Transcriptome profiling of spike provides expression features of genes related to terpene biosynthesis in lavender. Sci. Rep. 2020, 10, 6933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Arya, S.K.; Singh, B.; Singh, S.; Dhar, Y.V.; Verma, P.C.; Ganjewala, D. Transcriptome analysis of the palmarosa Cymbopogon martinii inflorescence with emphasis on genes involved in essential oil biosynthesis. Ind. Crop. Prod. 2019, 140, 111602. [Google Scholar] [CrossRef]
- Breitler, J.C.; Campa, C.; Georget, F.; Bertrand, B.; Etienne, H. A single-step method for RNA isolation from tropical crops in the field. Sci. Rep. 2016, 6, 38368. [Google Scholar] [CrossRef] [Green Version]
- Roser, L.G.; Aguero, F.; Sanchez, D.O. FastqCleaner: An interactive Bioconductor application for quality-control, filtering and trimming of FASTQ files. BMC Bioinform. 2019, 20, 361. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Iijima, Y.; Gang, D.R.; Fridman, E.; Lewinsohn, E.; Pichersky, E. Characterization of geraniol synthase from the peltate glands of sweet basil. Plant Physiol. 2004, 134, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Colby, S.M.; Alonso, W.R.; Katahira, E.J.; McGarvey, D.J.; Croteau, R. 4S-limonene synthase from the oil glands of spearmint (Mentha spicata). cDNA isolation, characterization, and bacterial expression of the catalytically active monoterpene cyclase. J. Biol. Chem. 1993, 268, 23016–23024. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate Maximum-Likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.Y.; Fernie, A.R.; Luo, J. Exploring the diversity of plant metabolism. Trends Plant Sci. 2019, 24, 83–98. [Google Scholar] [CrossRef]
- Nagegowda, D.A. Plant volatile terpenoid metabolism: Biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett. 2010, 584, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Schiestl, F.P. Ecology and evolution of floral volatile-mediated information transfer in plants. New Phytol. 2015, 206, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.R.; Sosenski, P.; Raguso, R.A. Phenotypic plasticity of floral volatiles in response to increasing drought stress. Ann. Bot. 2019, 123, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negre-Zakharov, F.; Long, M.C.; Dudareva, N. Floral scents and fruit aromas inspired by nature. In Plant-Derived Natural Products: Synthesis, Function, and Application; Osbourn, A.E., Lanzotti, V., Eds.; Springer: New York, NY, USA, 2009; pp. 405–431. [Google Scholar]
- Koley, S.; Grafahrend-Belau, E.; Raorane, M.L.; Junker, B.H. The mevalonate pathway contributes to monoterpene production in peppermint. bioRxiv 2020. bioRxiv:2029.124016. [Google Scholar] [CrossRef]
- Opitz, S.; Nes, W.D.; Gershenzon, J. Both methylerythritol phosphate and mevalonate pathways contribute to biosynthesis of each of the major isoprenoid classes in young cotton seedlings. Phytochemistry 2014, 98, 110–119. [Google Scholar] [CrossRef]
- Wei, C.; Liu, H.; Cao, X.; Zhang, M.; Li, X.; Chen, K.; Zhang, B. Synthesis of flavour-related linalool is regulated by PpbHLH1 and associated with changes in DNA methylation during peach fruit ripening. Plant Biotechnol. J. 2021, 19, 2082–2096. [Google Scholar] [CrossRef]
- May, B.; Lange, B.M.; Wüst, M. Biosynthesis of sesquiterpenes in grape berry exocarp of Vitis vinifera L.: Evidence for a transport of farnesyl diphosphate precursors from plastids to the cytosol. Phytochemistry 2013, 95, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.R.; Rai, A.; Bomzan, D.P.; Kumar, K.; Hemmerlin, A.; Dwivedi, V.; Godbole, R.C.; Barvkar, V.; Shanker, K.; Shilpashree, H.B.; et al. A plastid-localized bona fide geranylgeranyl diphosphate synthase plays a necessary role in monoterpene indole alkaloid biosynthesis in Catharanthus roseus. Plant J. 2020, 103, 248–265. [Google Scholar] [CrossRef]
- Liu, G.H.; Fu, J.Y. Squalene synthase cloning and functional identification in wintersweet plant (Chimonanthus zhejiangensis). Bot. Stud. 2018, 59, 30. [Google Scholar] [CrossRef] [Green Version]
- Landmann, C.; Fink, B.; Festner, M.; Dregus, M.; Engel, K.H.; Schwab, W. Cloning and functional characterization of three terpene synthases from lavender (Lavandula angustifolia). Arch. Biochem. Biophys. 2007, 465, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.C.; Maiti, P.; Chanotiya, C.S.; Shanker, K.; Ghosh, S. Methyl jasmonate-elicited transcriptional responses and pentacyclic triterpene biosynthesis in sweet basil. Plant Physiol. 2014, 164, 1028–1044. [Google Scholar] [CrossRef] [Green Version]
- Stashenko, E.E.; Martinez, J.R. Sampling flower scent for chromatographic analysis. J. Sep. Sci. 2008, 31, 2022–2031. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Chen, J.M.; Xiao, L.J.; Zhang, Y.; Wang, F.; Li, X. Efficient biosynthesis of R-(–)-linalool through adjusting the expression strategy and increasing GPP supply in Escherichia coli. J. Agric. Food Chem. 2020, 68, 8381–8390. [Google Scholar] [CrossRef]
- Lucker, J.; Bowen, P.; Bohlmann, J. Vitis vinifera terpenoid cyclases: Functional identification of two sesquiterpene synthase cDNAs encoding (+)-valencene synthase and (−)-germacrene D synthase and expression of mono- and sesquiterpene synthases in grapevine flowers and berries. Phytochemistry 2004, 65, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Kim, M.J.; Dhandapani, S.; Tjhang, J.G.; Yin, J.L.; Wong, L.; Sarojam, R.; Chua, N.H.; Jang, I.C. The floral transcriptome of ylang ylang (Cananga odorata var. fruticosa) uncovers biosynthetic pathways for volatile organic compounds and a multifunctional and novel sesquiterpene synthase. J. Exp. Bot. 2015, 66, 3959–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guterman, I.; Shalit, M.; Menda, N.; Piestun, D.; Dafny-Yelin, M.; Shalev, G.; Bar, E.; Davydov, O.; Ovadis, M.; Emanuel, M.; et al. Rose scent: Genomics approach to discovering novel floral fragrance-related genes. Plant Cell 2002, 14, 2325–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, F.; Ke, Y.G.; Yu, R.C.; Fan, Y.P. Functional characterization and expression analysis of two terpene synthases involved in floral scent formation in Lilium ‘Siberia’. Planta 2019, 249, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Farré-Armengol, G.; Filella, I.; Llusià, J.; Peñuelas, J. β-Ocimene, a key floral and foliar volatile involved in multiple interactions between plants and other organisms. Molecules 2017, 22, 1148. [Google Scholar] [CrossRef] [Green Version]
- Ashaari, N.S.; Ab Rahim, M.H.; Sabri, S.; Lai, K.S.; Song, A.A.L.; Rahim, R.A.; Abdullah, J.O. Kinetic studies and homology modeling of a dual-substrate linalool/nerolidol synthase from Plectranthus amboinicus. Sci. Rep. 2021, 11, 17094. [Google Scholar] [CrossRef]
- Azzi, J.; Auezova, L.; Danjou, P.E.; Fourmentin, S.; Greige-Gerges, H. First evaluation of drug-in-cyclodextrin-in-liposomes as an encapsulating system for nerolidol. Food Chem. 2018, 255, 399–404. [Google Scholar] [CrossRef]
- Chan, W.K.; Tan, L.T.H.; Chan, K.G.; Lee, L.H.; Goh, B.H. Nerolidol: A Sesquiterpene Alcohol with Multi-Faceted Pharmacological and Biological Activities. Molecules 2016, 21, 529. [Google Scholar] [CrossRef] [Green Version]
- Pacifico, S.; D’Abrosca, B.; Golino, A.; Mastellone, C.; Piccolella, S.; Fiorentino, A.; Monaco, P. Antioxidant evaluation of polyhydroxylated nerolidols from redroot pigweed (Amaranthus retroflexus) leaves. LWT-Food Sci. Technol. 2008, 41, 1665–1671. [Google Scholar] [CrossRef]
- Meeran, M.F.N.; Azimullah, S.; Mamoudh, H.H.; Sharma, C.; Kumar, S.; Goyal, S.N.; Ojha, S. Nerolidol, a Sesquiterpene from the Essential Oils of Aromatic Plants, Attenuates Doxorubicin-Induced Chronic Cardiotoxicity in Rats. J. Agric. Food Chem. 2021, 69, 7334–7343. [Google Scholar] [CrossRef]
- Benelli, G.; Pavela, R.; Drenaggi, E.; Desneux, N.; Maggi, F. Phytol, (E)-nerolidol and spathulenol from Stevia rebaudiana leaf essential oil as effective and eco-friendly botanical insecticides against Metopolophium dirhodum. Ind. Crop. Prod. 2020, 155, 112844. [Google Scholar] [CrossRef]
- Di Campli, E.; Di Bartolomeo, S.; Pizzi, P.D.; Di Giulio, M.; Grande, R.; Nostro, A.; Cellini, L. Activity of tea tree oil and nerolidol alone or in combination against Pediculus capitis (head lice) and its eggs. Parasitol. Res. 2012, 111, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Chappell, J. Biochemical and genomic characterization of terpene synthases in Magnolia grandiflora. Plant Physiol. 2008, 147, 1017–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.A.; Wildung, M.R.; Williams, D.C.; Hyatt, D.C.; Croteau, R. cDNA isolation, functional expression, and characterization of (+)-alpha-pinene synthase and (–)-alpha-pinene synthase from loblolly pine (Pinus taeda): Stereocontrol in pinene biosynthesis. Arch. Biochem. Biophys. 2003, 411, 267–276. [Google Scholar] [CrossRef]
- MacMillan, J. Occurrence of gibberellins in vascular plants, fungi, and bacteria. J. Plant Growth Regul. 2001, 20, 387–442. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Saito, T.; Abe, H.; Yamane, H.; Murofushi, N.; Kamiya, Y. Molecular cloning and characterization of a cDNA encoding the gibberellin biosynthetic enzyme ent-kaurene synthase B from pumpkin (Cucurbita maxima L.). Plant J. 1996, 10, 203–213. [Google Scholar] [CrossRef]
- Tudzynski, B.; Hedden, P.; Carrera, E.; Gaskin, P. The P450-4 gene of Gibberella fujikuroi encodes ent-kaurene oxidase in the gibberellin biosynthesis pathway. Appl. Environ. Microbiol. 2001, 67, 3514–3522. [Google Scholar] [CrossRef] [Green Version]
- Li, W.G.; Xu, R.R.; Yan, X.G.; Liang, D.M.; Zhang, L.; Qin, X.Y.; Caiyin, Q.G.L.; Zhao, G.R.; Xiao, W.H.; Hu, Z.N.; et al. De novo leaf and root transcriptome analysis to explore biosynthetic pathway of Celangulin V in Celastrus angulatus maxim. BMC Genom. 2019, 20, 7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. no | C. keiskei Tissue | Unigene | N50 | TAIR | Pfam | KEGG |
|---|---|---|---|---|---|---|
| 1 | leaf | 72,044 | 1058.54 | 14,413 | 46,578 | 42,221 |
| 2 | flower | 116,434 | 972.03 | 15,679 | 55,244 | 50,597 |
| 3 | root | 146,550 | 802.03 | 14,880 | 58,909 | 45,363 |
| all | 237,129 | 811.21 | 33,021 | 59,121 | 68,282 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claude, S.-J.; Raman, G.; Park, S.-J. Comparative Analysis and Identification of Terpene Synthase Genes in Convallaria keiskei Leaf, Flower and Root Using RNA-Sequencing Profiling. Plants 2023, 12, 2797. https://doi.org/10.3390/plants12152797
Claude S-J, Raman G, Park S-J. Comparative Analysis and Identification of Terpene Synthase Genes in Convallaria keiskei Leaf, Flower and Root Using RNA-Sequencing Profiling. Plants. 2023; 12(15):2797. https://doi.org/10.3390/plants12152797
Chicago/Turabian StyleClaude, Sivagami-Jean, Gurusamy Raman, and Seon-Joo Park. 2023. "Comparative Analysis and Identification of Terpene Synthase Genes in Convallaria keiskei Leaf, Flower and Root Using RNA-Sequencing Profiling" Plants 12, no. 15: 2797. https://doi.org/10.3390/plants12152797