1. Introduction
Salt stress is known to negatively impact crop growth and productivity. When soil salinity increases, it creates an osmotic imbalance that limits water uptake by plants and leads to ionic toxicity, resulting in reduced growth, decreased yield, and even death of the plant. As the salt concentration in the soil increases, the plant responds by activating various defense mechanisms at the molecular and physiological levels. For example, plants can synthesize compatible solutes, such as proline and glycine betaine, to maintain osmotic balance, and they can also activate antioxidant systems to scavenge reactive oxygen species (ROS) that accumulate under salt stress [
1]. However, prolonged exposure to high salt concentrations can lead to irreversible damage to plant tissues, affecting various physiological processes such as photosynthesis, respiration, and nutrient uptake. For example, salt stress can result in a decrease in chlorophyll content, stomatal conductance, and CO
2 assimilation, ultimately leading to reduced growth and yield [
2,
3,
4]. At the molecular level, salt stress triggers the activation of various signaling pathways, including the mitogen-activated protein kinase cascade, the calcium-dependent protein kinase pathway, and the abscisic acid signaling pathway. These pathways mediate the expression of stress-responsive genes, such as those encoding transporters, enzymes, and transcription factors, which regulate various physiological and biochemical processes involved in salt stress tolerance [
5,
6,
7].
S. bicolor is an important cereal crop that is widely cultivated around the world, with a global production of 57.6 million tons. It is valued for its drought tolerance and adaptability to a wide range of agro-climatic conditions [
8]. Primarily used as food and feed, it has sustained millions of people in developing countries. Its considerable drought tolerance sets it as a vital contributor to global food security by offering a resilient alternative to other cereal crops susceptible to climate extremes. Sorghum’s environmental value lies in its ecological benefits, such as enhancing soil fertility, reducing soil erosion, and its potential as a bioenergy source. This crop’s cultivation and use trace back to thousands of years, playing a significant role in human civilization’s growth, especially in Africa and Asia [
9]. Beyond its conventional uses,
S. bicolor serves as an advantageous model organism for genetic and genomic studies of cereal crops. The ease of studying its diploid genome and the high natural genetic variation offers a rich pool of traits for crop improvement and breeding. Genome assembly and annotation, as well as transcriptomic studies under various conditions such as abiotic stress and pathogen infection, have yielded critical insights into its stress tolerance mechanisms and potential breeding targets [
10,
11,
12]. These multifaceted perspectives on
S. bicolor, spanning genetics, genomics, and transcriptomics, equip us with a comprehensive understanding of its genetic diversity and its potential for crop improvement. Information extracted from these studies can guide the development of new varieties with improved yield, quality, and resilience to environmental stresses, further enhancing its contribution to global food security [
13,
14].
Sorghum, a salt-tolerant species, employs adaptive mechanisms at the physiological, biochemical, and molecular levels to mitigate the adverse impacts of salinity stress. These include the maintenance of photosynthetic apparatus, osmotic balance, ionic equilibrium, and redox homeostasis [
15,
16,
17,
18]. Salinity-tolerant sorghum genotypes exhibit superior germination and growth parameters, maintaining efficient photosynthetic machinery and pigments under salt stress conditions [
19,
20,
21]. These genotypes also possess the ability to regulate toxic and beneficial ion concentrations in response to salinity, thus ensuring ion homeostasis. This involves various membrane transport systems, which help retain essential ions and exclude or compartmentalize sodium ions [
22]. Furthermore, alterations in cell wall constituents and cellular membrane structures play a pivotal role in the salinity adaptation of sorghum. However, the exact workings of membrane lipid remodeling in relation to sorghum salinity tolerance are not yet fully understood, necessitating further research [
23,
24]. Osmotic homeostasis, crucial for plant salinity tolerance, enables the regulation of internal water balance under saline conditions. This involves lowering the cellular water potential through accumulation of compatible solutes and/or inorganic ions, which helps maintain water absorption under saline conditions. Consequently, this osmotic adjustment is integral to sorghum’s adaption to salinity [
25,
26]. Finally, salinity stress disrupts cellular metabolism, leading to excessive reactive oxygen species (ROS) production. These ROS can damage proteins, nucleic acids, and other cellular components, causing membrane leakage. In response, plants activate antioxidant redox systems to control ROS homeostasis, a key adaptation mechanism to high salinity [
27]. Understanding of sorghum’s salinity tolerance mechanisms is continually evolving, emphasizing the need for ongoing research in this area.
Mutation breeding has been effectively employed as an approach for enhancing crop diversity, particularly through the use of gamma-ray irradiation [
28]. Numerous studies have highlighted the efficacy of gamma rays in improving the nutritional and functional characteristics of grain mutants [
29,
30]. For instance, sorghum mutants generated through gamma-ray exposure have shown increased concentrations of essential compounds such as amino acids [
31], along with improved storage attributes due to decreased mold growth and free fatty acid content [
32]. However, the technique has its downsides. While gamma rays are potent mutagens, their application could potentially lead to unexpected genetic changes, thereby raising stability concerns. Furthermore, mutation breeding often requires meticulous screening processes to identify favorable mutants, resulting in a labor-intensive and time-consuming process. Still, gamma-ray-induced mutation breeding has brought about significant advances, as shown in a study by [
33], where 29 elite sorghum genetic resources were compared for their salt tolerance characteristics. More recently, a follow-up study developed 28 M6 mutant lines from eight original sorghum accessions/cultivars with enhanced salt tolerance [
34]. Quantitative real-time polymerase chain reaction (qRT-PCR) analyses in these mutants revealed several genes likely involved in salt tolerance. Further, among 36 sorghum materials investigated, 10 gamma-irradiated mutants showed significantly increased biomass compared to their original accessions. Overall, radiation breeding, including gamma-ray-induced mutation, has been extensively utilized for generating novel genetic diversity, bypassing the long and arduous process of traditional breeding [
35]. Such methods have been particularly focused on improving grain yields under environmental stress and enhancing biomass for the bioenergy industry.
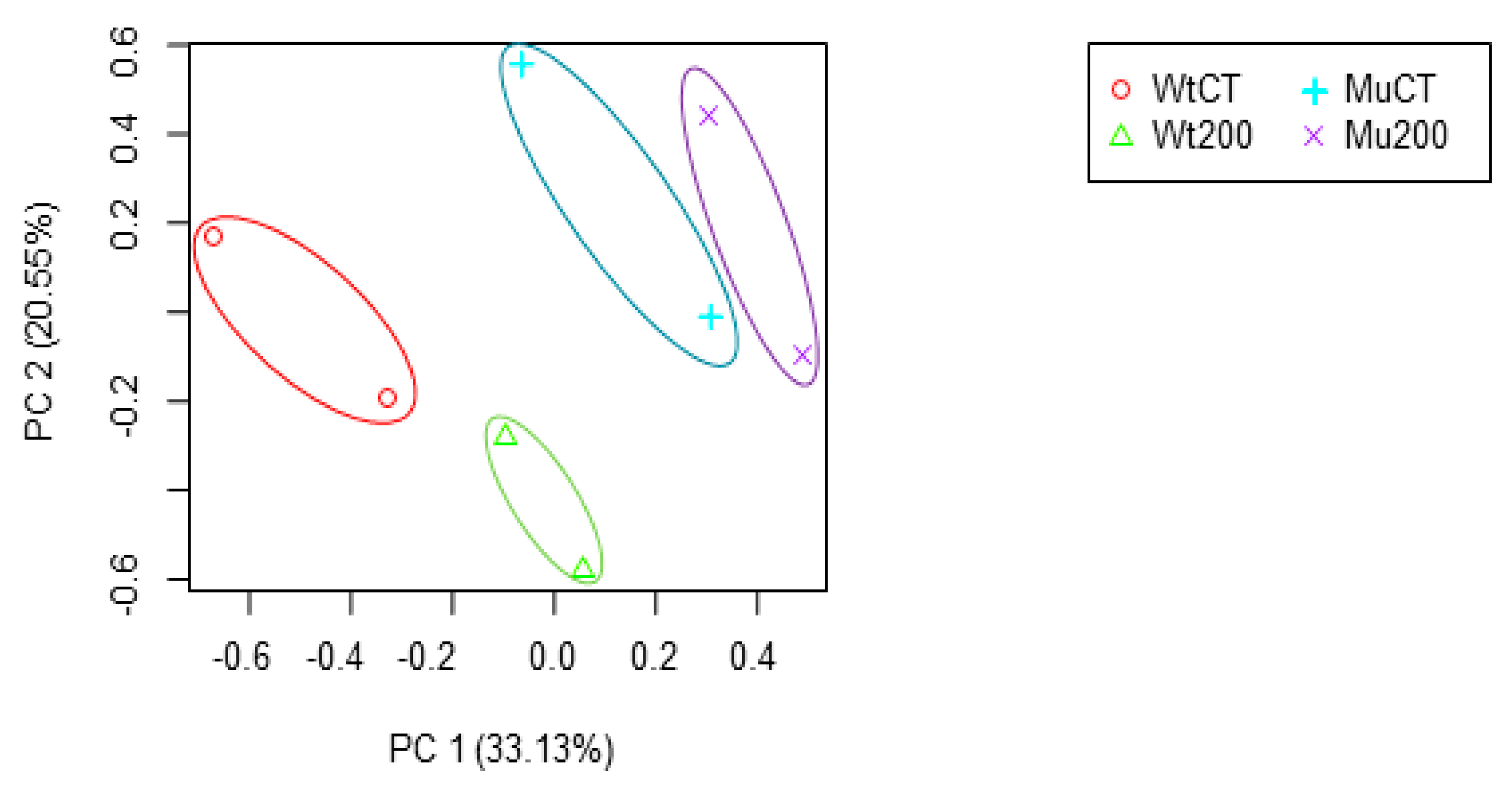
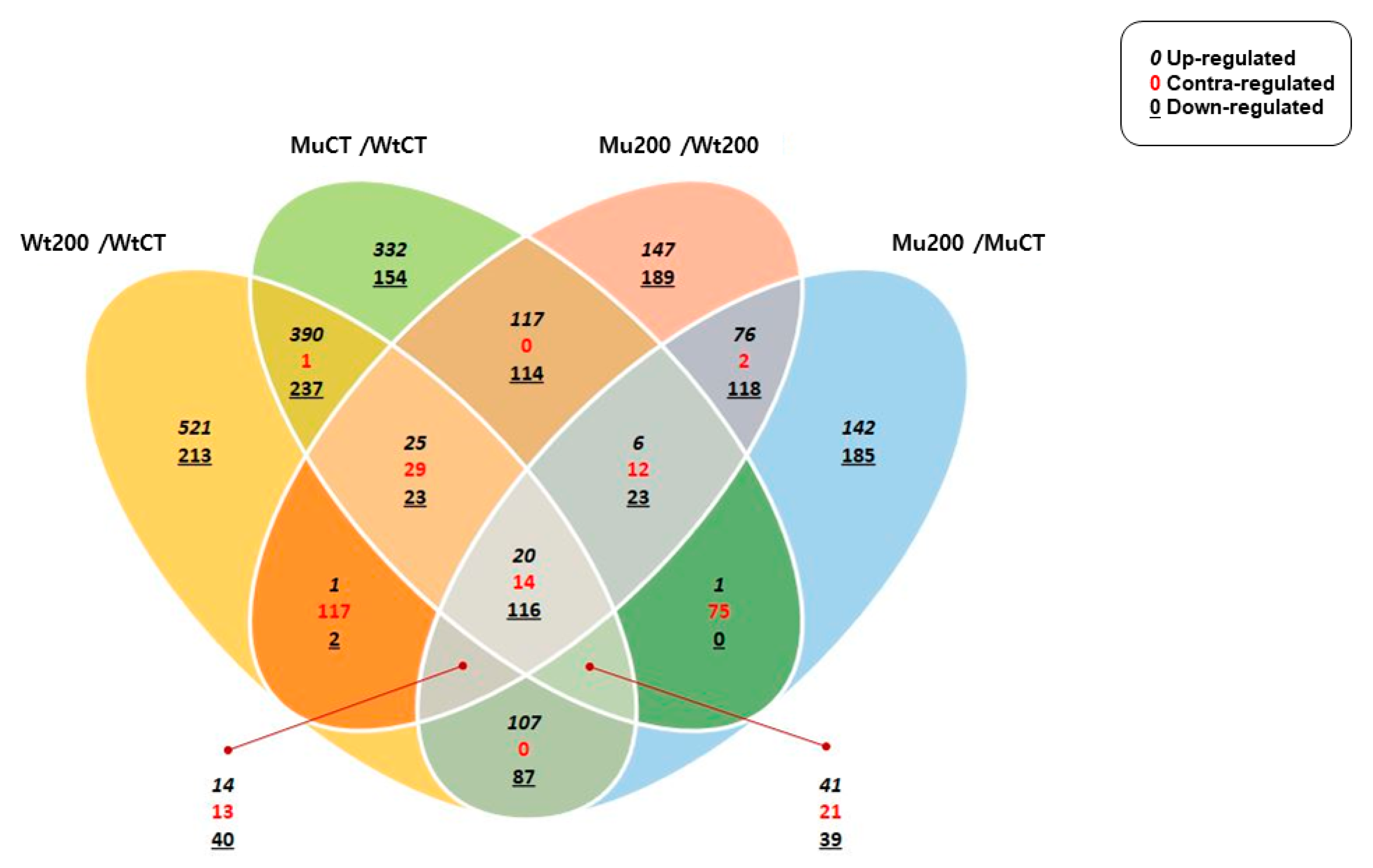
This study performed a comparative transcriptome analysis of S. bicolor wild type and mutant lines with salinity tolerance to identify differentially expressed genes (DEGs) and nsSNPs that are crucial for salt tolerance, and also provides a better understanding of the genetic mechanisms underlying salinity tolerance in sorghum and identifies genes and SNPs that are associated with salt tolerance. Moreover, this study will be useful for improving sorghum breeding strategies aimed at increasing salinity tolerance and ultimately enhancing crop productivity.
3. Discussion
Salinity stress is one of the major abiotic stress factors that limit crop productivity worldwide. High salt concentrations in the soil can lead to ionic and osmotic imbalances in plants, resulting in reduced water uptake, nutrient deficiency, and cell damage. These physiological effects ultimately impair plant growth and development, resulting in decreased crop yields and quality. To address these challenges, developing salt-tolerant crops has become increasingly important. By understanding the mechanisms of the salt stress response in plants, researchers can identify genes and molecular pathways that contribute to salinity tolerance [
1]. This knowledge can be used to develop breeding strategies that produce crops with improved salinity tolerance, thus increasing agricultural productivity in saline soils and helping to ensure global food security. Sorghum is an important crop in Korea because it can grow well in marginal lands, such as reclaimed tidal lands. However, these lands are often saline, which can limit crop growth and productivity. Therefore, developing sorghum cultivars with salt tolerance is essential for sustainable agriculture in these areas. By breeding sorghum with an enhanced salt tolerance, we can ensure stable crop yields and food security, even in challenging environments such as marginal lands. Moreover, sorghum’s small genome size of approximately 730 Mb makes it an appealing model for functional genomics of Saccharinae and other C4 grasses [
10]. Despite its tolerance to salt stress, only a limited number of salt-responsive genes have been identified in sorghum.
In this study, we used the transcriptome of sorghum to examine differential gene expression. The mutation used in this study was generated by gamma-ray irradiation of seeds and selected as a salt-tolerant individual through field tests (the selection data are not provided in this paper). We hypothesized that the DEGs between the mutant and wild type under salt stress conditions may correspond to salt tolerance-related genes. To gain a more comprehensive understanding of the transcriptome expression, we also examined the impact of nsSNPs. The nsSNPs are genetic variations that result in a change in the amino acid sequence of a protein encoded by a gene. These variations can affect the structure and function of the protein, and therefore may have downstream effects on biological processes such as gene expression. In the context of DEGs, nsSNPs may be associated with differential gene expression between different samples or conditions. For example, if nsSNPs result in a protein variant that interacts differently with other proteins or regulatory factors, it could lead to changes in the gene expression levels or patterns. This, in turn, could impact cellular processes and phenotypic traits. Furthermore, nsSNPs can be used as markers for studying genetic variations and diversity in populations. By analyzing the distribution of nsSNPs in DEGs across populations, researchers can gain insights into the evolutionary history of different populations and how genetic variations may contribute to adaptation and disease susceptibility [
36].
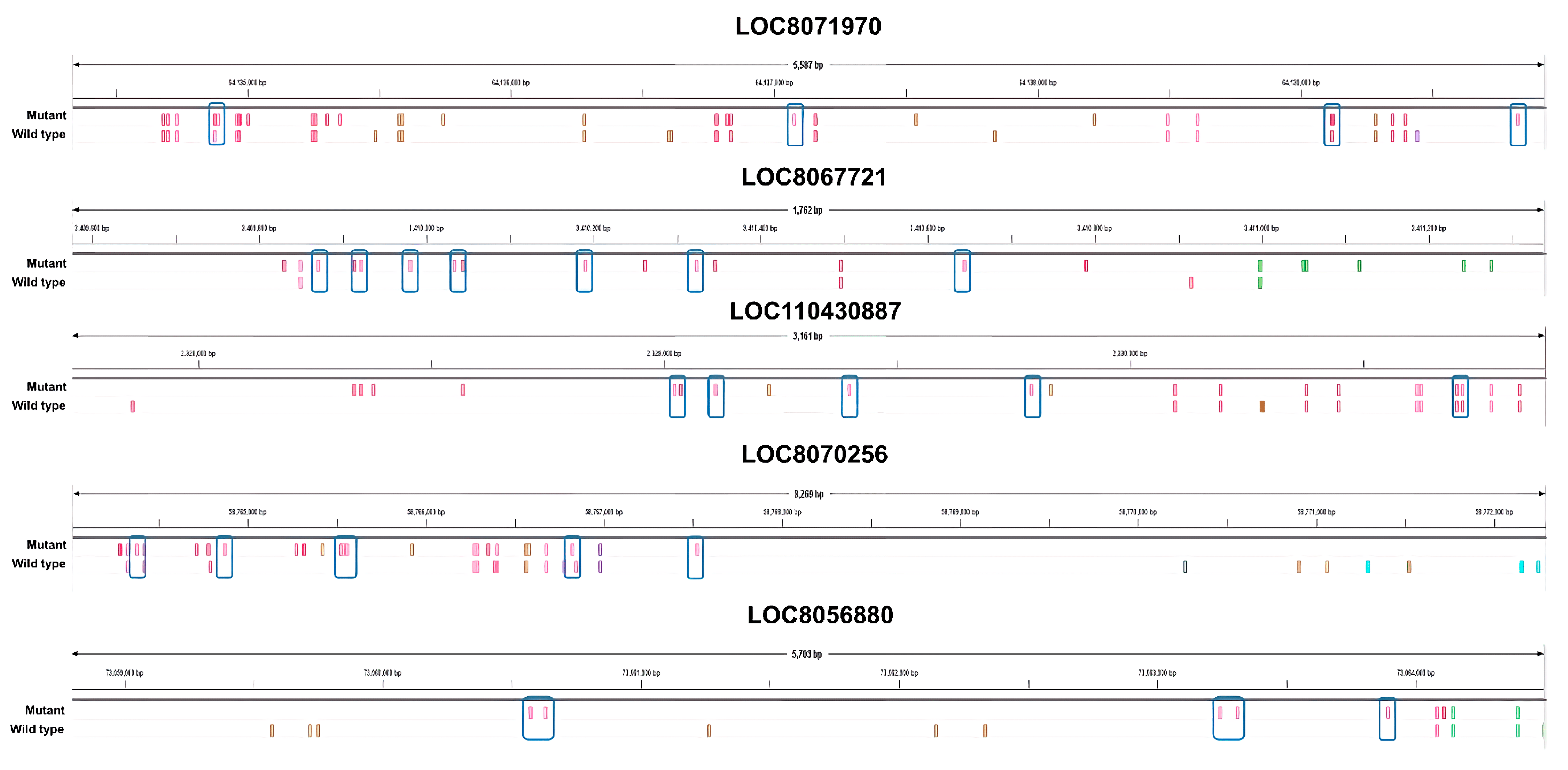
Specifically, we identified genes with a large difference in the number of nsSNPs between the mutant and wild type plants. We selected several candidate genes that showed a large difference in the nsSNPs (
Figure 4). Based on our analysis, the
LOC8071970 gene, which encodes for the subtilisin-like protease SBT3.9 and is up-regulated in response to salt treatment, has the highest number of nsSNPs among all the up-regulated genes. Specifically, the mutant plant was found to have nine nsSNPs in
LOC8071970, while the wild type plant had five nsSNPs. These differences in nsSNPs may be responsible for the up-regulation of the gene in the mutant in response to salt treatment. Subtilisin-like proteases, also known as subtilases, are a diverse family of serine peptidases found in many organisms, primarily in plants. They have a wide range of biological roles, including protein turnover, plant development, and interactions with the environment. Subtilases are known for their involvement in plant responses to both abiotic and biotic environmental stimuli [
37,
38,
39]. Previous studies reported that subtilases are involved in drought and salt tolerance mechanisms following abiotic stimuli. For example, the
Arabidopsis thaliana subtilase AtSBT6.1 was well-characterized for its association with the unfolded protein response during salt stress through the cleavage of an ER-resident type II membrane protein (bZIP28) [
40].
LOC8067721 functions as an acyl transferase 15. In the mutations, eight nsSNPs were expressed, which is eight times higher than the wild type. Numerous proteins, potentially acylated, have been pinpointed as contributors to processes such as vesicle trafficking, signal conveyance, primary and secondary metabolic functions, as well as responses to stress [
41,
42]. Overexpression of the Glycerol-3-phosphate acyltransferase (GPAT) gene in Arabidopsis plants resulted in increased salt tolerance at the germination stage with higher germination rates and longer root lengths than those of the wild type plants under salt stress conditions. Additionally, in the seedling stage, overexpressed plants showed a decrease in the chlorophyll content, and the unsaturated fatty acid content of phosphatidylglycerol (PG) decreased less than in the wild type plants under salt stress. These results suggest that GPAT plays a role in the production of unsaturated fatty acids in PG, which are important for maintaining membrane fluidity and function under salt stress. Therefore, the overexpression of GPAT could enhance plant salt tolerance by increasing the production of unsaturated fatty acids and improving membrane function under high salinity conditions [
43].
LOC110430887 is a down-regulated DEG that functions as cysteine-rich receptor-like (CRK) protein kinase 10. In the mutants, eight nsSNPs were detected, which is five more than the three nsSNPs in the wild type. CRKs undergo transcriptional induction under abiotic stress conditions, including exposure to salicylic acid, ozone, UV light, as well as during drought and salt treatments [
44,
45,
46]. Another study reported that a specific type of receptor-like kinase, called CRK5, is involved in abscisic acid (ABA) signaling in
Arabidopsis thaliana. Overexpression of CRK5 increased ABA sensitivity and enhanced plant drought tolerance without affecting plant growth or productivity. The loss-of-function mutation of the CRK5 gene did not affect the ABA response, while overexpression of two homologs of CRK5 conferred ABA responses, suggesting redundancy [
47].
LOC8070256 is a protein that functions as a linoleate 9S-lipoxygenase. In the mutant, 12 nsSNPs were detected, which is five more than the seven found in the wild type. Lipoxygenases (LOXs) are enzymes that occur naturally and are found widely in plants and animals. LOX enzymes, which are essential for plant growth and development, are the products of a multigene family. There is a noted correlation between enhanced LOX activity and improved salt tolerance [
48,
49]. These enzymes can be non-sulfur iron, non-heme iron, or manganese-containing dioxygenase redox enzymes. LOXs catalyze the oxidation of polyunsaturated fatty acids into fatty acid hydroperoxides. This oxidation process converts linolenic acid, a precursor in the biosynthesis of jasmonic acid (JA), into 12-oxo-phytodienoic acid, which is carried out through oxygenation with LOX, allene oxide synthase, and allene oxide cyclase. JA is involved in various physiological and biochemical processes such as seed germination, fruit ripening, senescence, and others. Additionally, LOXs play a vital role in defense responses against biotic stress, including pests, insects, and pathogenic attacks and abiotic stress such as wounding, extreme temperatures, UV-rays, oxidative stress, and drought [
50].
LOC8056880 is a functional sugar transport protein 13. Six nsSNPs were detected in the mutant, which is six times higher than in the wild type. SWEETs (Sugars Will Eventually be Exported Transporters) are a family of sugar transporters that facilitate the diffusion of sugar molecules across cell membranes. In plants, SWEETs play important roles in various physiological processes including phloem loading, senescence, pollen nutrition, grain filling, nectar secretion, and abiotic and biotic stress regulation. SWEET transporters are involved in plant development and abiotic stress as well as in their gene expression dynamics under different stresses in different plant species. SWEET could lead to improvements in crop productivity and stress tolerance [
51].
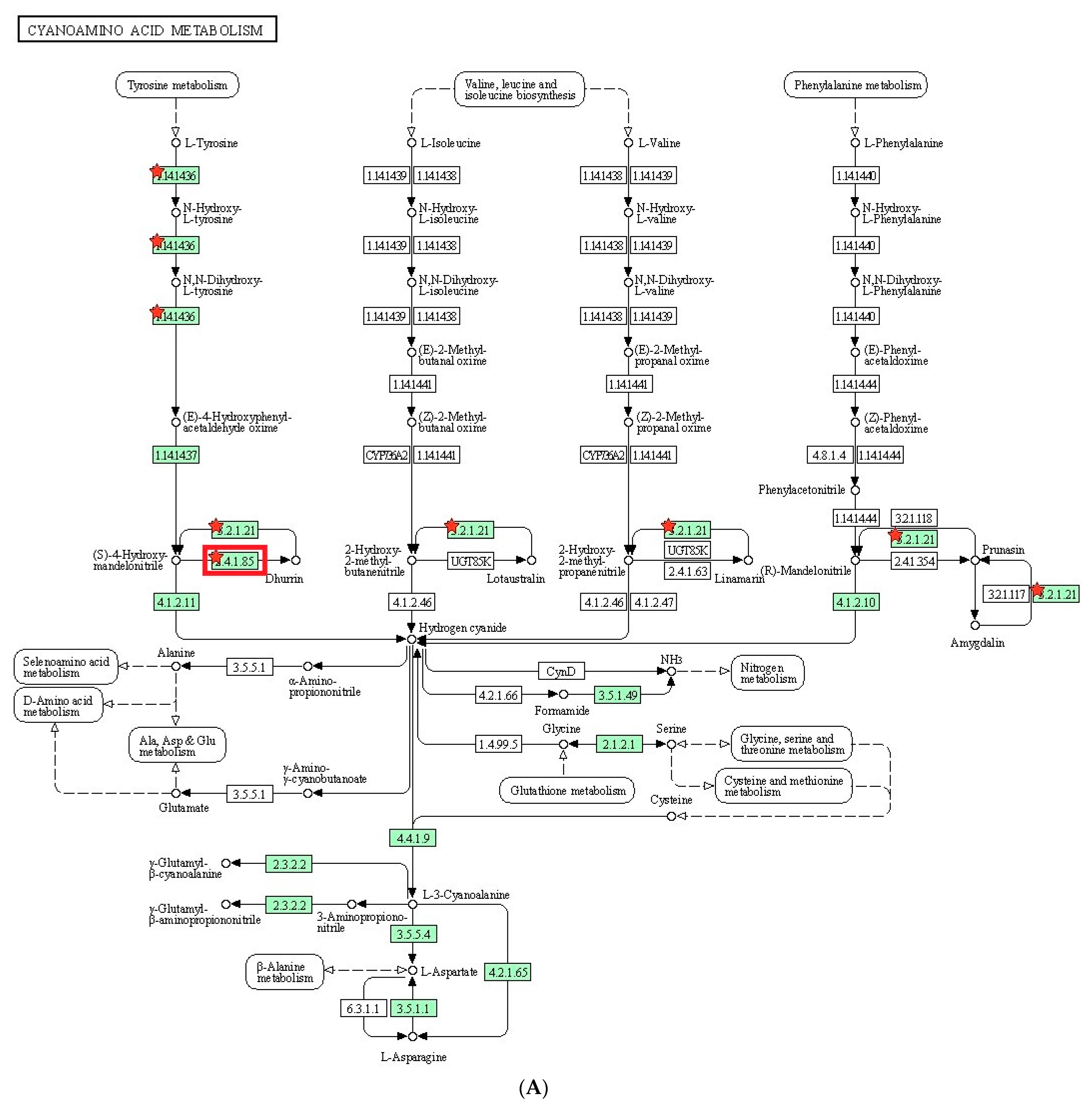
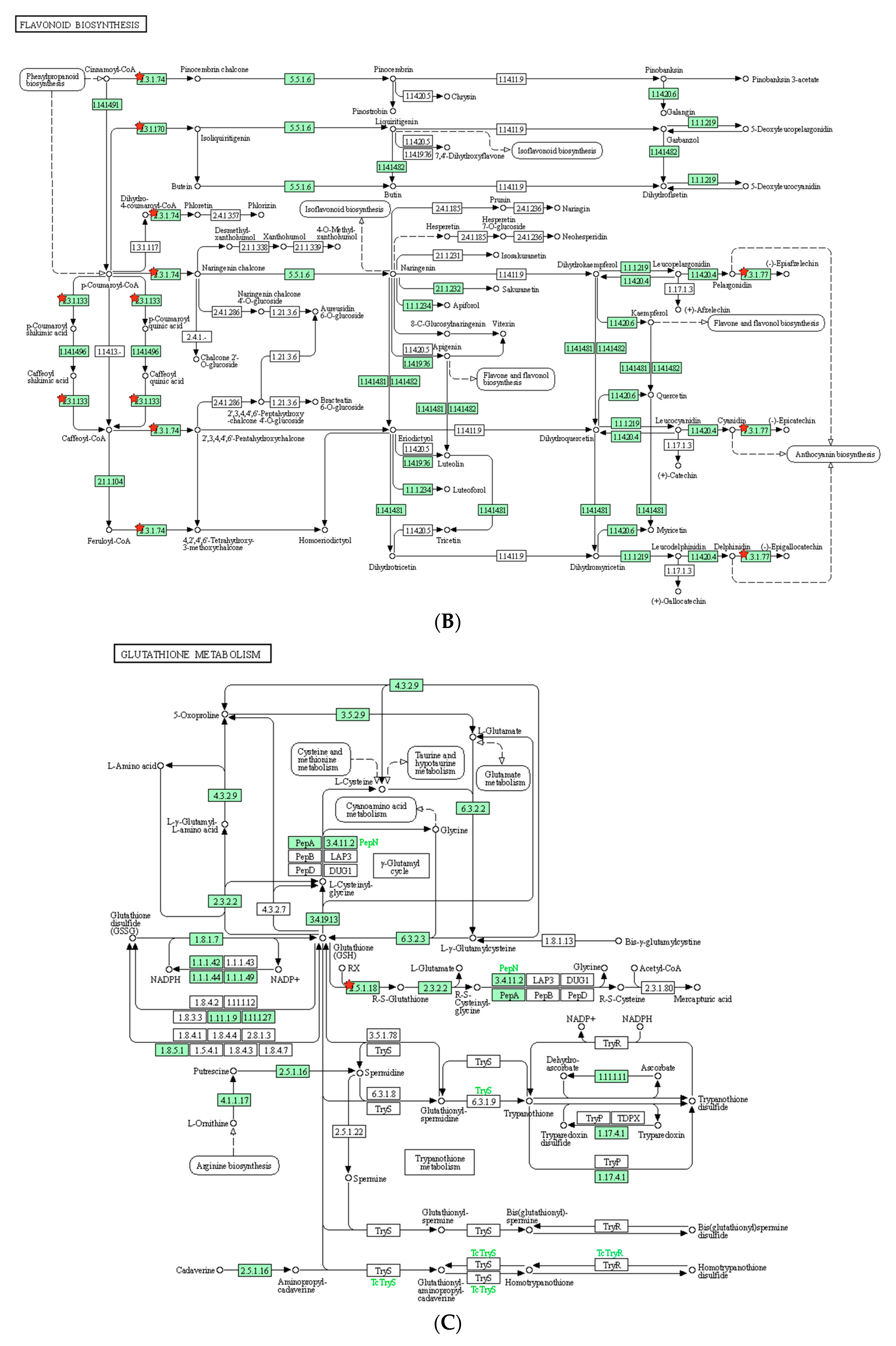
To understand the metabolic processes in plants, we conducted KEGG pathway analysis on the DEGs. In the identified pathway, cyanoamino acid metabolism, flavonoid biosynthesis, and glutathione metabolism were reported to play a role in salt tolerance in plants. Cyanoamino acid metabolism is involved in the biosynthesis of cyanogenic glycosides, which are known to act as a defense mechanism against herbivores and pathogens. In addition, cyanogenic glycosides have been shown to help plants cope with salt stress by regulating ion homeostasis [
52]. Flavonoid biosynthesis is a metabolic pathway that produces flavonoids, which are known to have antioxidant properties. Flavonoids have been found to be involved in salt stress tolerance by protecting plants against oxidative damage caused by high salt concentrations [
53]. Glutathione metabolism is a crucial pathway for maintaining cellular redox homeostasis. Glutathione plays an important role in protecting cells from oxidative damage caused by salt stress. In addition, glutathione metabolism has been found to regulate ion transport and osmotic balance in response to salt stress [
54]. Overall, these metabolic pathways are involved in regulating ion homeostasis, protecting against oxidative damage, and maintaining cellular redox balance, which are all important factors in salt stress tolerance in plants.
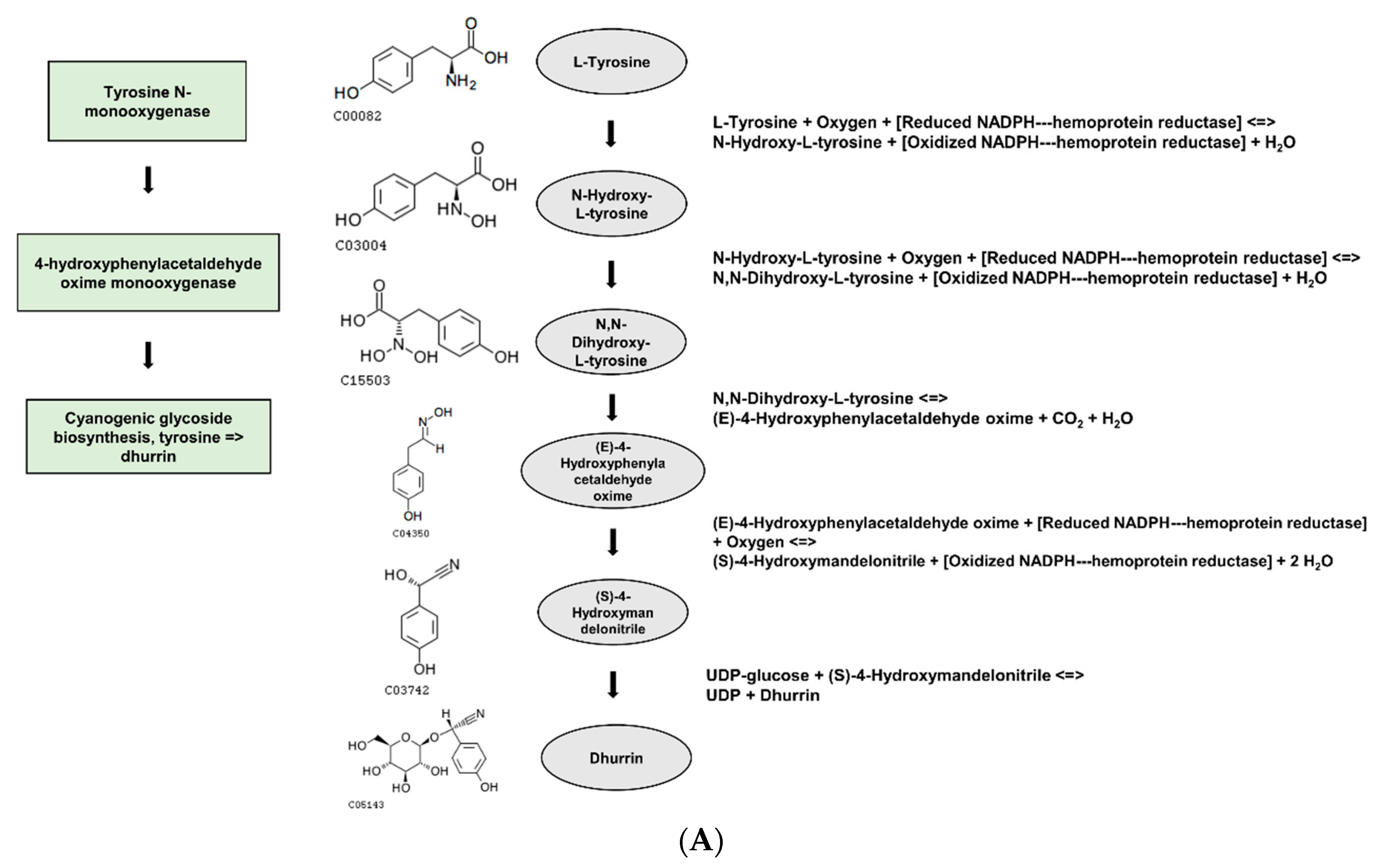
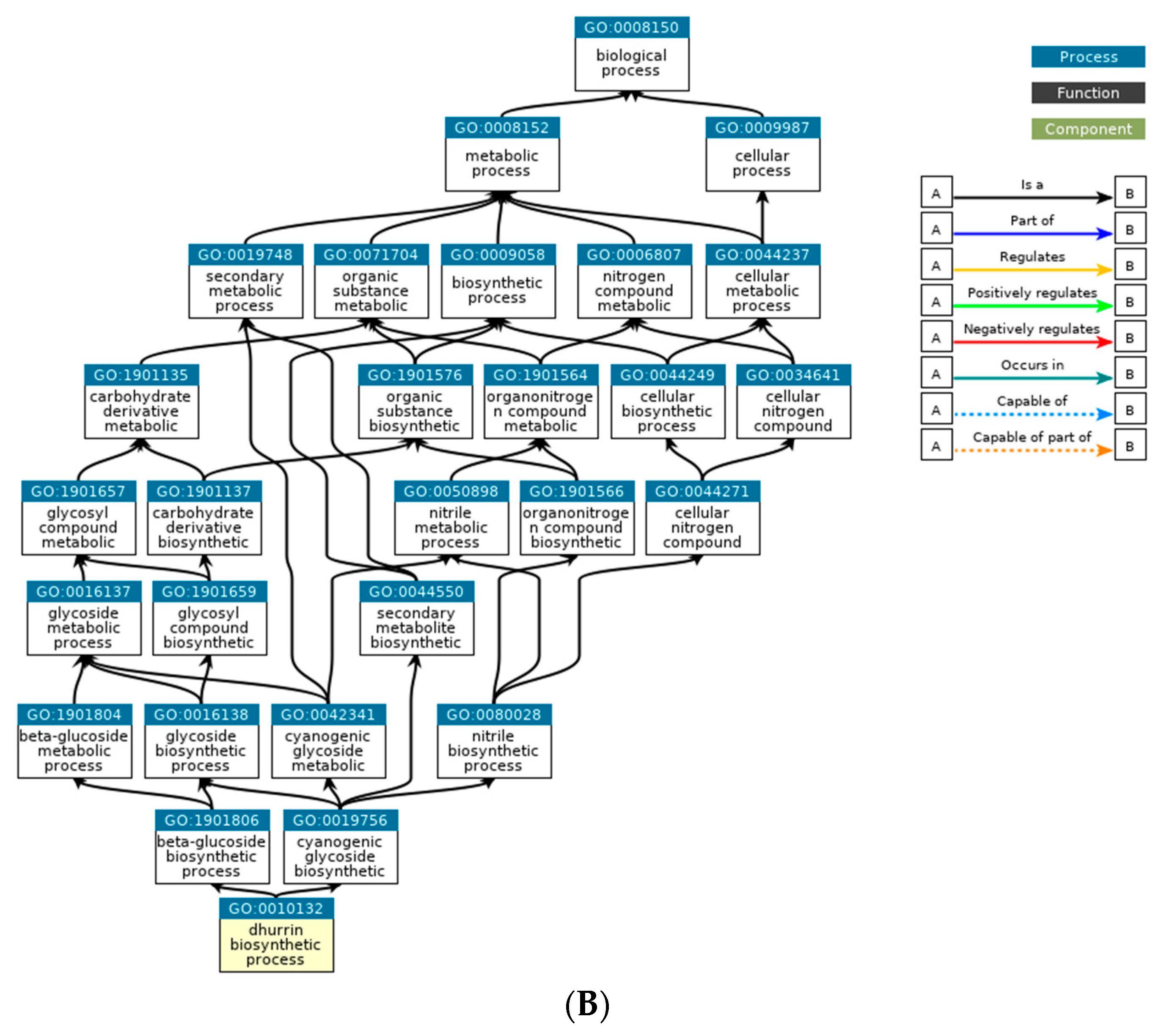
Based on the DEGs with nsSNPs, we conducted GO analysis to identify significant GO terms. Out of the nine significantly enriched GO terms for up-regulated genes, it was identified that seven GO terms were related to glycosides. Interestingly, three of these terms were specifically connected to cyanogenic glycosides, including the dhurrin biosynthetic process (GO:0010132), cyanogenic glycoside biosynthetic process (GO:0019756), and cyanogenic glycoside metabolic process (GO:0042341). Cyanogenic glycosides are a class of secondary metabolites found in various plant species. They contain a cyanide group bonded to a sugar molecule and can release toxic hydrogen cyanide when plant tissue is damaged. Dhurrin is one of the most well-studied cyanogenic glycosides and is predominantly found in
S. bicolor and other related plant species [
55]. Osmotic stress is a form of environmental stress that plants experience when there is an imbalance in water potential between the plant cells and their surroundings. This can be due to high salt concentrations, drought conditions, or even low temperatures. Osmotic stress can lead to cellular dehydration and negatively impact plant growth and development [
56]. The relationship between cyanogenic glycosides such as dhurrin and osmotic stress is complex and multifaceted. During osmotic stress, plants are more vulnerable to damage; therefore, accumulating cyanogenic glycosides may serve as an additional layer of protection. Some research suggests that cyanogenic glycosides might also function as osmoprotectants, helping plants to tolerate osmotic stress. They might do this by stabilizing proteins and cellular structures, reducing cellular damage caused by stress, and maintaining cellular water balance. Plant exposure to osmotic stress may lead to changes in the accumulation patterns of cyanogenic glycosides including dhurrin. These changes may result from stress-induced alterations in the expression of genes involved in the biosynthesis and metabolism of cyanogenic glycosides. This can lead to a differential distribution of these compounds within the plant, potentially as a response to stress conditions [
57,
58,
59]. Although the relationship between cyanogenic glycosides such as dhurrin and osmotic stress is not yet fully understood, it appears that they might play a role in plant defense mechanisms, possibly act as osmoprotectants, and have altered accumulation patterns under stress conditions.
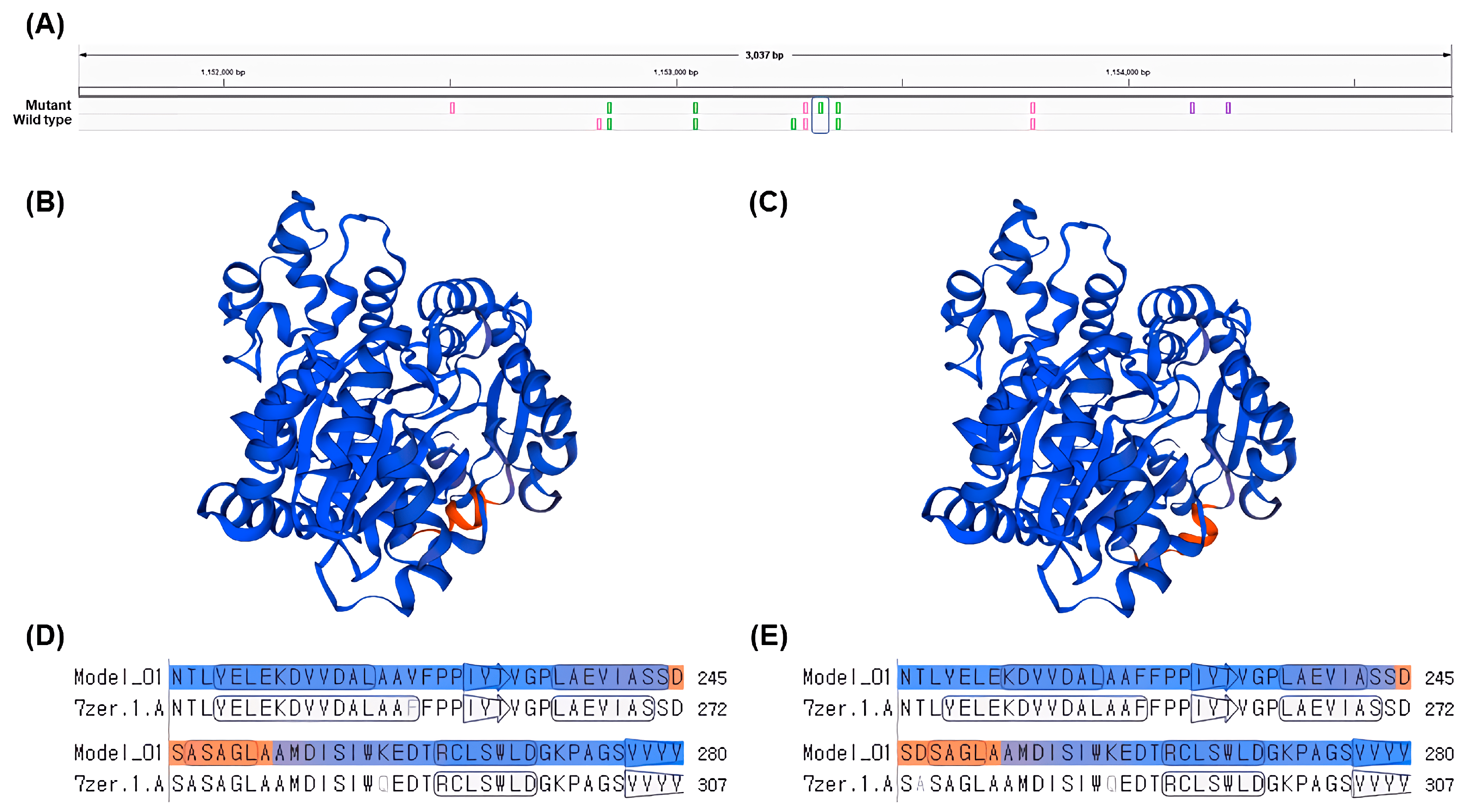
The gene
LOC8060874 (cyanohydrin beta-glucosyltransferase), which falls under the GO term: dhurrin biosynthetic process (GO:0010132), had a single nsSNP and higher gene expression levels in the gamma-ray-induced salt-tolerant mutant compared to the wild type under salt stress conditions (
Table 5 and
Figure 5). The nsSNP in
LOC8060874 leads to an amino acid substitution from alanine to aspartic acid, altering the protein’s chemical properties due to the differences between the non-polar, hydrophobic alanine and the polar, negatively charged aspartic acid due to its carboxyl side chain (
Figure 5). This difference in chemical properties could potentially affect the protein’s folding, stability, or interaction with other molecules, which might be one of the contributing factors for the observed salt tolerance in the mutant plant, considering the role of dhurrin under osmotic stress conditions [
10,
60].
Several mechanisms were proposed to help plants lower the concentrations of these detrimental free radicals. One such mechanism involves increasing the production of secondary metabolites such as phenolics and cyanogenic glucosides [
61]. This increase could be a result of diverting excess NADPH
+, which accumulates due to stomatal closure and reduced CO
2 flux. There may also be a more direct association. For instance, previous research reported that the production of amides derived from cyanogenic glucosides rises with increasing H
2O
2 levels [
62]. The proposed dhurrin turnover pathway in sorghum also implies amide formation, suggesting a possible role for dhurrin in mitigating ROS-mediated stress. Therefore, amide formation from cyanogenic glucosides might serve as a non-enzymatic scavenger mechanism in cyanogenic plants, helping them cope with oxidative stress and improve their drought tolerance [
63].
Moreover, recent research suggests that dhurrin may play a role in plant growth and development in addition to defense. Sorghum generates the cyanogenic glucoside known as dhurrin. In the early stages of growth, young sorghum plants contain high concentrations of dhurrin, which gradually decrease as the plant matures [
64]. Three-day-old seedlings of sorghum can have dhurrin concentrations as high as 6% with the highest concentration localized in the growing shoot tips where it may account for up to 30% of the total shoot dry weight [
65]. Moreover, a previous study reported that plants lacking dhurrin tend to be smaller compared to their wild-type counterparts. The absence of dhurrin appears to hinder growth during early developmental stages. Furthermore, plants deficient in dhurrin take longer to mature, potentially due to delayed flowering. Dhurrin generation may benefit plant growth, particularly during critical growth stages such as germination, by providing a readily available source of reduced nitrogen [
66]. This is consistent with our research findings, which show that the germination rate of the mutant plants was significantly higher than that of the wild-type plants. The increase in dhurrin expression and subsequent elevation in dhurrin levels might be closely related to the initial germination and growth stages (
Table 1).
However, it is important to note that this study does not provide definitive evidence that the gene
LOC8060874 (cyanohydrin beta-glucosyltransferase) directly confers salt tolerance. Instead, it suggests that the gene could be linked to the salt tolerance phenotype through its involvement in the dhurrin biosynthesis process. While there have been several studies exploring the relationship between dhurrin and osmotic stress in plants, it has been difficult to establish a clear correlation. This research proposes a possible connection between dhurrin and osmotic stress, particularly salt tolerance, in plants from the perspective of amino acid changes (
Figure 6). This study highlights the potential significance of amino acid substitutions in determining the function of proteins involved in dhurrin biosynthesis and suggests that leveraging such genetic variations could improve crop performance under stress conditions, such as high salinity. Further investigation into the molecular mechanisms underlying this salt tolerance and the specific structural changes in the protein could provide valuable insights into plant stress response pathways and help in the development of more resilient crop varieties. Additionally, generating transgenic plants expressing the mutated gene and assessing their salt tolerance would provide more conclusive evidence for the role of this amino acid change in contributing to salt tolerance.
Our results suggest that the differences in nsSNPs between the mutant and wild type plants may have a significant impact on the expression of DEGs and may contribute to salt tolerance in S. bicolor. The differential expression of genes in response to the difference in nsSNPs between the mutant and wild type plants suggests that these nsSNPs may have an impact on the salt tolerance of S. bicolor. Specifically, we observed that several genes showed a significant change in expression level with some genes being up-regulated and others being down-regulated. These findings suggest that nsSNPs may play a crucial role in the regulation of gene expression and the plant’s ability to withstand salt stress. In light of our findings, we suggest that further in-depth study of the candidate genes is critical. Subsequent studies, especially those employing reverse genetics experiments, could provide definitive insights into these genes’ roles in the salinity tolerance of S. bicolor. The impacts of nsSNPs on gene expression and salt tolerance could be more comprehensively understood through such rigorous validation. This research could guide the breeding of resilient S. bicolor cultivars for saline environments. Consequently, this study could potentially provide a foundation for the development of new varieties to prepare for future food crises.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}