
Differential RNA-Seq Analysis Predicts Genes Related to Terpene Tailoring in Caryopteris × clandonensis

Abstract
:1. Introduction
2. Results and Discussion
2.1. RNA Sequencing and Mapping Quality
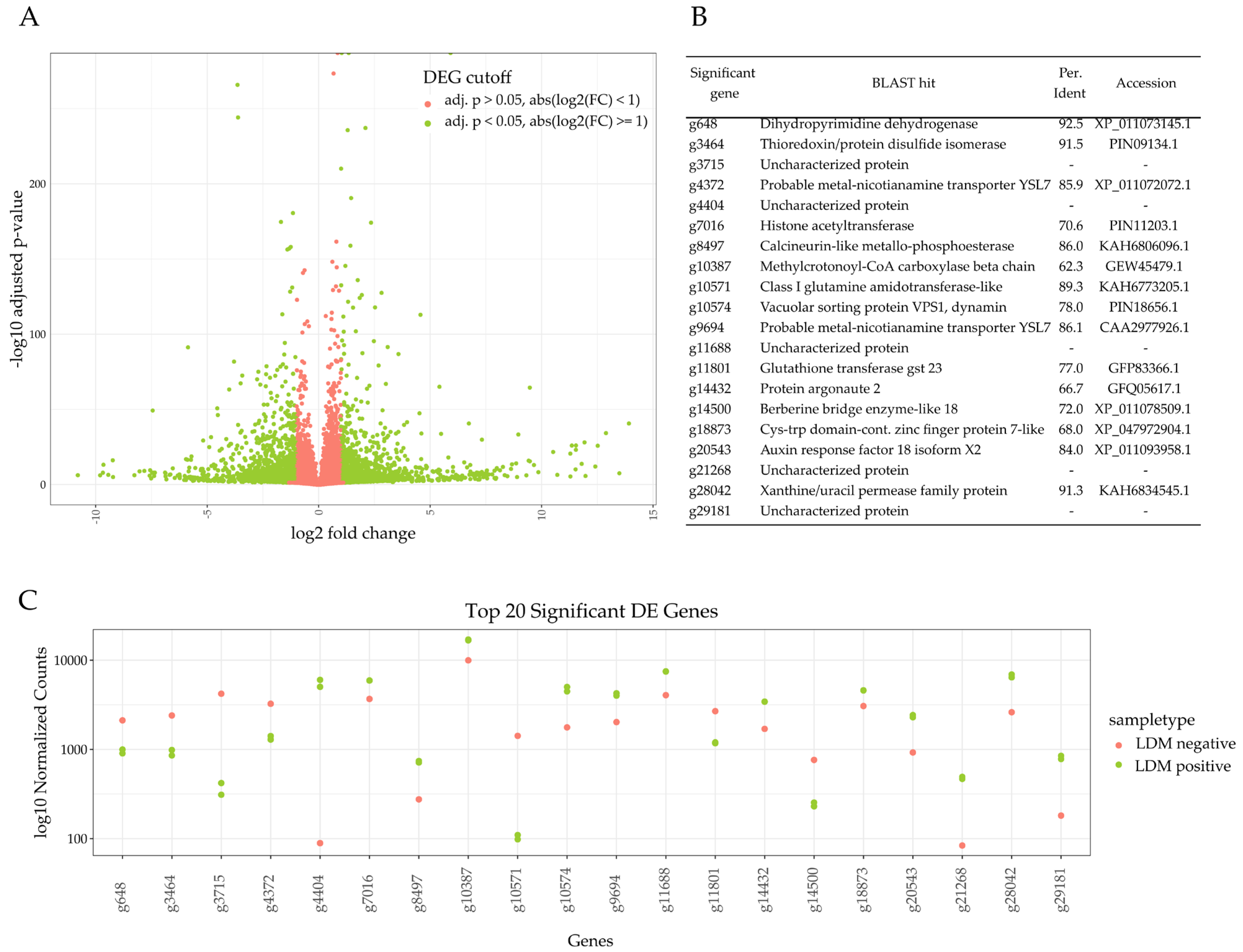
2.2. Identification of DEG
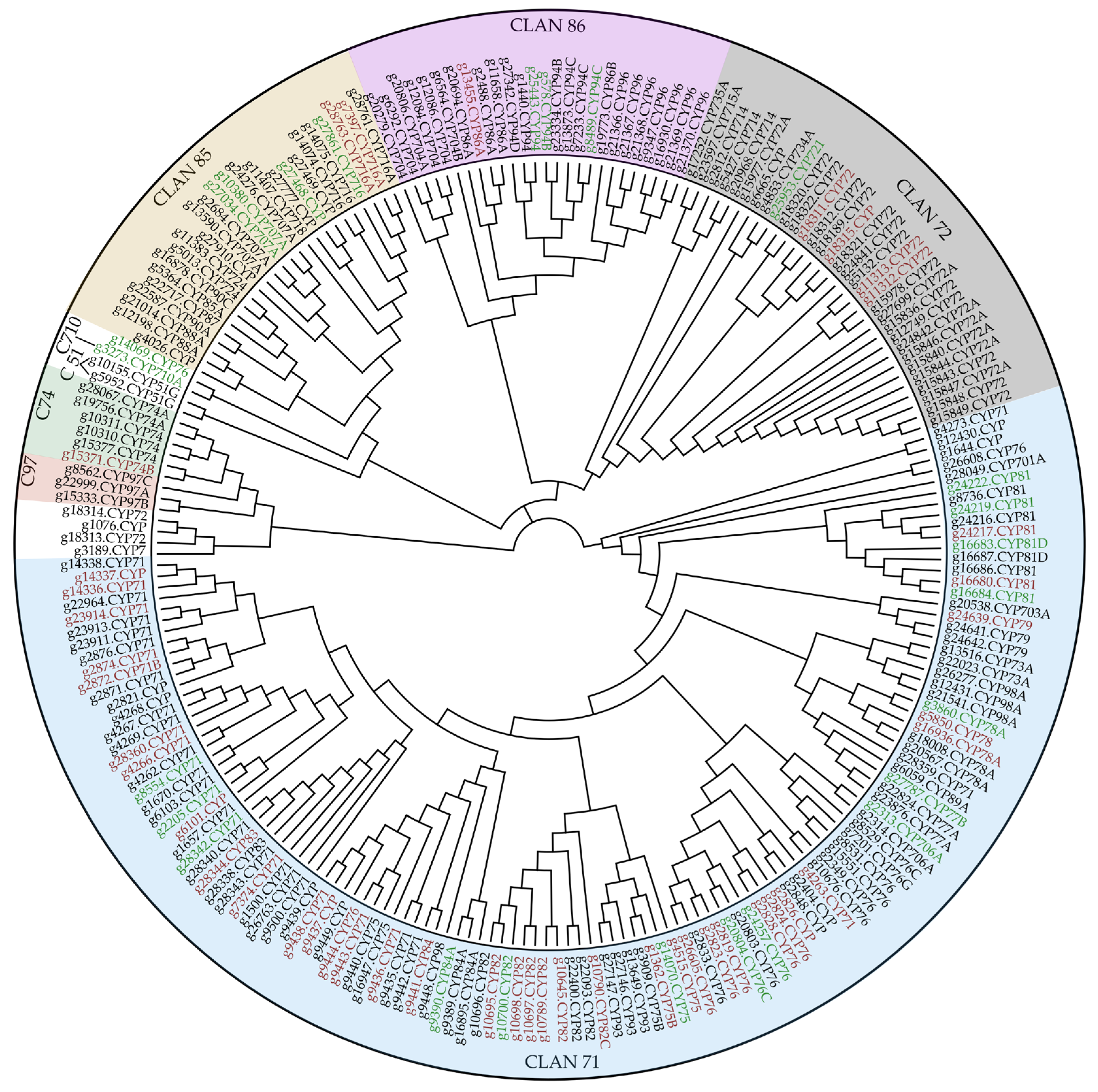
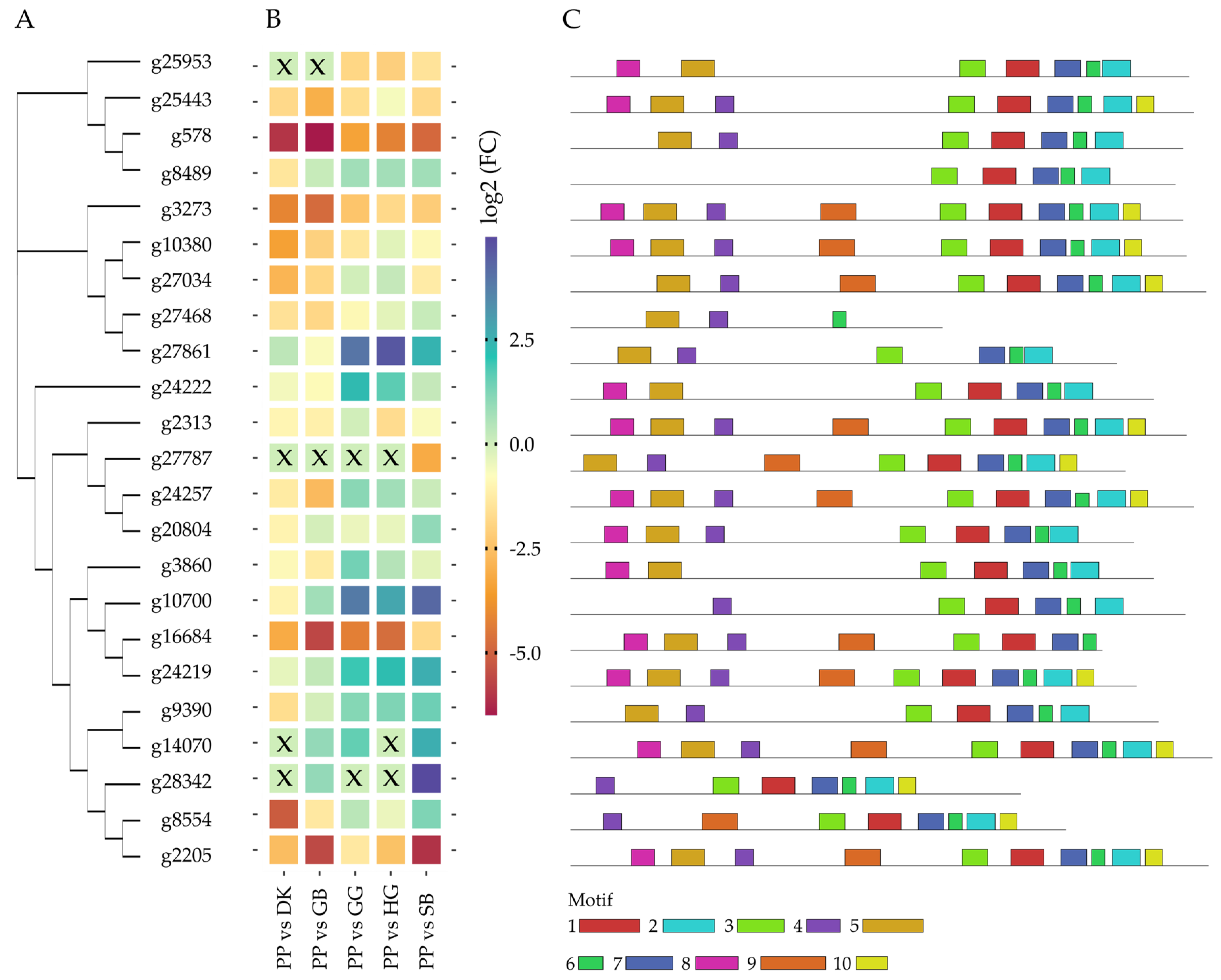
2.3. Terpene Tailoring through CYPs between Plant Cultivars
3. Materials and Methods
3.1. Plant Material
3.2. Genomic Resource
3.3. RNA Preparation and Short Read Sequencing
3.4. Mapping and Annotation of Aligned Reads
3.5. Evaluation of Differential Gene Expression between Aerial Plant Parts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritz, M.; Ahmad, N.; Brueck, T.; Mehlmer, N. Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids. Plants 2023, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, S.; Jacquemond-Collet, I.; Fabre, N.; Stanislas, E.; Moulis, C. Iridoid keto-glycosides from Caryopteris × Clandonensis. Phytochemistry 1999, 51, 767–769. [Google Scholar] [CrossRef]
- Matsumoto, T.; Mayer, C.; Eugster, C.H. α-Caryopteron, ein neues Pyrano-juglon aus Caryopteris clandonensis. Helv. Chim. Acta 1969, 52, 808–812. [Google Scholar] [CrossRef]
- Blythe, E.K.; Tabanca, N.; Demirci, B.; Bernier, U.R.; Agramonte, N.M.; Ali, A.; Baser, H.C.; Khan, I.A. Composition of the essential oil of Pink ChablisTM bluebeard (Caryopteris ×clandonensis ’Durio’) and its biological activity against the yellow fever mosquito Aedes aegypti. Nat. Volatiles Essent. Oils 2015, 2, 11–21. [Google Scholar]
- Abdelaty, N.A.; Attia, E.Z.; Hamed, A.N.E.; Desoukey, S.Y. A review on various classes of secondary metabolites and biological activities of Lamiaceae (Labiatae) (2002–2018). J. Adv. Biomed. Pharm. Sci. 2021, 4, 16–31. [Google Scholar] [CrossRef]
- Siciliano, T.; Bader, A.; Vassallo, A.; Braca, A.; Morelli, I.; Pizza, C.; De Tommasi, N. Secondary metabolites from Ballota undulata (Lamiaceae). Biochem. Syst. Ecol. 2005, 33, 341–351. [Google Scholar] [CrossRef]
- Mimica-Dukic, N.; Bozin, B.; Mentha, L. Species (Lamiaceae) as Promising Sources of Bioactive Secondary Metabolites. Curr. Pharm. Des. 2008, 14, 3141–3150. [Google Scholar] [CrossRef]
- Kliebenstein, D.J. Secondary metabolites and plant/environment interactions: A view through Arabidopsis thaliana tinged glasses. Plant. Cell Environ. 2004, 27, 675–684. [Google Scholar] [CrossRef]
- Boncan, D.A.T.; Tsang, S.S.K.; Li, C.; Lee, I.H.T.; Lam, H.M.; Chan, T.F.; Hui, J.H.L. Terpenes and Terpenoids in Plants: Interactions with Environment and Insects. Int. J. Mol. Sci. 2020, 21, 7382. [Google Scholar] [CrossRef]
- Holopainen, J.K.; Himanen, S.J.; Yuan, J.S.; Chen, F.; Stewart, C.N. Ecological functions of terpenoids in changing climates. Nat. Prod. 2013, 1, 2913–2940. [Google Scholar]
- Wang, T.; Li, L.; Zhuang, W.; Zhang, F.; Shu, X.; Wang, N.; Wang, Z. Recent Research Progress in Taxol Biosynthetic Pathway and Acylation Reactions Mediated by Taxus Acyltransferases. Molecules 2021, 26, 2855. [Google Scholar] [CrossRef] [PubMed]
- Kamatou, G.P.P.; Vermaak, I.; Viljoen, A.M.; Lawrence, B.M. Menthol: A simple monoterpene with remarkable biological properties. Phytochemistry 2013, 96, 15–25. [Google Scholar] [CrossRef]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, B.; Kim, D.W.; Huh, G.; Lee, H.; Kang, K.; Lee, J.W. Synthesis and biological evaluation of isoliquiritigenin derivatives as a neuroprotective agent against glutamate mediated neurotoxicity in HT22 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127058. [Google Scholar] [CrossRef]
- Mazimba, O. Umbelliferone: Sources, chemistry and bioactivities review. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 223–232. [Google Scholar] [CrossRef]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lešnik, S.; Furlan, V.; Bren, U. Rosemary (Rosmarinus officinalis L.): Extraction techniques, analytical methods and health-promoting biological effects. Phytochem. Rev. 2021, 20, 1273–1328. [Google Scholar] [CrossRef]
- Furlan, V.; Bren, U. Helichrysum italicum: From Extraction, Distillation, and Encapsulation Techniques to Beneficial Health Effects. Foods 2023, 12, 802. [Google Scholar] [CrossRef]
- Fadilah, N.Q.; Jittmittraphap, A.; Leaungwutiwong, P.; Pripdeevech, P.; Dhanushka, D.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Virucidal Activity of Essential Oils From Citrus x aurantium L. Against Influenza A Virus H1N1: Limonene as a Potential Household Disinfectant Against Virus. Nat. Prod. Commun. 2022, 17, 1934578X211072713. [Google Scholar] [CrossRef]
- Chassagne, F.; Samarakoon, T.; Porras, G.; Lyles, J.T.; Dettweiler, M.; Marquez, L.; Salam, A.M.; Shabih, S.; Farrokhi, D.R.; Quave, C.L. A Systematic Review of Plants With Antibacterial Activities: A Taxonomic and Phylogenetic Perspective. Front. Pharmacol. 2021, 11, 2069. [Google Scholar] [CrossRef]
- Islam, A.K.M.M.; Suttiyut, T.; Anwar, M.P.; Juraimi, A.S.; Kato-Noguchi, H. Allelopathic Properties of Lamiaceae Species: Prospects and Challenges to Use in Agriculture. Plants 2022, 11, 1478. [Google Scholar] [CrossRef]
- Byers, K.J.R.P.; Bradshaw, H.D.; Riffell, J.A. Three floral volatiles contribute to differential pollinator attraction in monkeyflowers (Mimulus). J. Exp. Biol. 2014, 217, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, R.; Schmidt, A.; Peters, R.J. Isoprenyl diphosphate synthases: The chain length determining step in terpene biosynthesis. Planta 2018, 249, 9–20. [Google Scholar] [CrossRef]
- Dickschat, J.S. Bacterial Diterpene Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 15964–15976. [Google Scholar] [CrossRef]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant terpenoid synthases: Molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Karunanithi, P.S.; Zerbe, P. Terpene Synthases as Metabolic Gatekeepers in the Evolution of Plant Terpenoid Chemical Diversity. Front. Plant Sci. 2019, 10, 1166. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhu, X.; Wang, H.; Liu, T.; Cheng, J.; Jiang, H. Discovery and modification of cytochrome P450 for plant natural products biosynthesis. Synth. Syst. Biotechnol. 2020, 5, 187. [Google Scholar] [CrossRef]
- Foti, R.S.; Honaker, M.; Nath, A.; Pearson, J.T.; Buttrick, B.; Isoherranen, N.; Atkins, W.M. Catalytic vs. Inhibitory Promiscuity in Cytochrome P450s: Implications for Evolution of New Function. Biochemistry 2011, 50, 2387. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Knoll, M.; Sirim, D.; Wagner, F.; Funke, S.; Pleiss, J.; Bateman, A. The Cytochrome P450 Engineering Database: A navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 2007, 23, 2015–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R. The Cytochrome P450 Homepage. Hum. Genom. 2009, 4, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R. Cytochrome P450 nomenclature, 2004. Methods Mol. Biol. 2006, 320, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Mohamed, R. Plant cytochrome P450s: Nomenclature and involvement in natural product biosynthesis. Protoplasma 2015, 253, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.T.; Liao, P.; Crocoll, C.; Boachon, B.; Förster, C.; Leidecker, F.; Wiese, N.; Zhao, D.; Wood, J.C.; Buell, C.R.; et al. The biosynthesis of thymol, carvacrol, and thymohydroquinone in Lamiaceae proceeds via cytochrome P450s and a short-chain dehydrogenase. Proc. Natl. Acad. Sci. USA 2021, 118, e2110092118. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Geniza, M.; Naithani, S.; Phillips, J.L.; Haq, E.; Jaiswal, P. Chia (Salvia hispanica) Gene Expression Atlas Elucidates Dynamic Spatio-Temporal Changes Associated With Plant Growth and Development. Front. Plant Sci. 2021, 12, 667678. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Dong, Y.; Hao, H.; Ling, Z.; Bai, H.; Wang, H.; Cui, H.; Shi, L. Time-series transcriptome provides insights into the gene regulation network involved in the volatile terpenoid metabolism during the flower development of lavender. BMC Plant Biol. 2019, 19, 313. [Google Scholar] [CrossRef] [Green Version]
- Lichman, B.R.; Godden, G.T.; Buell, C.R. Gene and genome duplications in the evolution of chemodiversity: Perspectives from studies of Lamiaceae. Curr. Opin. Plant Biol. 2020, 55, 74–83. [Google Scholar] [CrossRef]
- Bak, S.; Beisson, F.; Bishop, G.; Hamberger, B.; Höfer, R.; Paquette, S.; Werck-Reichhart, D. Cytochromes P450. Arab. Book 2011, 9, e0144. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Ye, S.; Wang, Y.; Xu, L.; Zhu, X.; Yang, J.; Feng, H.; Yu, R.; Karanja, B.; Gong, Y.; et al. Transcriptome-based gene profiling provides novel insights into the characteristics of radish root response to Cr stress with next-generation sequencing. Front. Plant Sci. 2015, 6, 202. [Google Scholar] [CrossRef]
- Manzano, A.; Carnero-Diaz, E.; Herranz, R.; Medina, F.J. Recent transcriptomic studies to elucidate the plant adaptive response to spaceflight and to simulated space environments. iScience 2022, 25, 104687. [Google Scholar] [CrossRef]
- Howlader, J.; Robin, A.H.K.; Natarajan, S.; Biswas, M.K.; Sumi, K.R.; Song, C.Y.; Park, J.-I.; Nou, I.-S. Transcriptome Analysis by RNA–Seq Reveals Genes Related to Plant Height in Two Sets of Parent-hybrid Combinations in Easter lily (Lilium longiflorum). Sci. Rep. 2020, 10, 9082. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, S.; Khokhar, W.; Jabre, I.; Reddy, A.S.N.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Alternative splicing and protein diversity: Plants versus animals. Front. Plant Sci. 2019, 10, 708. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309. [Google Scholar] [CrossRef] [Green Version]
- Curie, C.; Cassin, G.; Couch, D.; Divol, F.; Higuchi, K.; Le Jean, M.; Misson, J.; Schikora, A.; Czernic, P.; Mari, S. Metal movement within the plant: Contribution of nicotianamine and yellow stripe 1-like transporters. Ann. Bot. 2009, 103, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, Y.; Masuda, H.; Bashir, K.; Inoue, H.; Tsukamoto, T.; Takahashi, M.; Nakanishi, H.; Aoki, N.; Hirose, T.; Ohsugi, R.; et al. Rice metal-nicotianamine transporter, OsYSL2, is required for the long-distance transport of iron and manganese. Plant J. 2010, 62, 379–390. [Google Scholar] [CrossRef]
- Li, Z.; Li, W.; Guo, M.; Liu, S.; Liu, L.; Yu, Y.; Mo, B.; Chen, X.; Gao, L. Origin, evolution and diversification of plant ARGONAUTE proteins. Plant J. 2022, 109, 1086–1097. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Li, R.; Yuan, L.; Dai, Y.; Wang, X. Identification and functional analysis of a protein disulfide isomerase (AtPDI1) in Arabidopsis thaliana. Front. Plant Sci. 2018, 9, 913. [Google Scholar] [CrossRef]
- Finkelstein, R. Abscisic Acid Synthesis and Response. Arab. Book 2013, 11, e0166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Zhao, S.; Zhang, M.; Dong, K.; Chen, Y.; Fu, C.; Yu, L. Transcriptome assembly and systematic identification of novel cytochrome P450s in taxus chinensis. Front. Plant Sci. 2017, 8, 1468. [Google Scholar] [CrossRef] [Green Version]
- Degtyarenko, K.N. Structural domains of P450-containing monooxygenase systems. Protein Eng. Des. Sel. 1995, 8, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasav, A.P.; Barvkar, V.T. Phylogenomic analysis of cytochrome P450 multigene family and their differential expression analysis in Solanum lycopersicum L. suggested tissue specific promoters. BMC Genom. 2019, 20, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, G.; Schachner, M.; Gabbiani, G.; Minerdi, D.; Savoi, S.; Sabbatini, P. Role of Cytochrome P450 Enzyme in Plant Microorganisms’ Communication: A Focus on Grapevine. Int. J. Mol. Sci. 2023, 24, 4695. [Google Scholar] [CrossRef]
- Bathe, U.; Tissier, A. Cytochrome P450 enzymes: A driving force of plant diterpene diversity. Phytochemistry 2019, 161, 149–162. [Google Scholar] [CrossRef]
- Hansen, C.C.; Nelson, D.R.; Møller, B.L.; Werck-Reichhart, D. Plant cytochrome P450 plasticity and evolution. Mol. Plant 2021, 14, 1244–1265. [Google Scholar] [CrossRef]
- Haudenschild, C.; Schalk, M.; Karp, F.; Croteau, R. Functional Expression of Regiospecific Cytochrome P450 Limonene Hydroxylases from Mint (Mentha spp.) in Escherichia coli and Saccharomyces cerevisiae. Arch. Biochem. Biophys. 2000, 379, 127–136. [Google Scholar] [CrossRef]
- Lupien, S.; Karp, F.; Wildung, M.; Croteau, R. Regiospecific cytochrome P450 limonene hydroxylases from mint (Mentha) species: cDNA isolation, characterization, and functional expression of (-)-4S-limonene-3-hydroxylase and (-)-4S-limonene-6-hydroxylase. Arch. Biochem. Biophys. 1999, 368, 181–192. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, C.; Too, H.P. Multienzyme Biosynthesis of Dihydroartemisinic Acid. Molecules 2017, 22, 1422. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hillwig, M.L.; Wang, Q.; Peters, R.J. Parsing a multifunctional biosynthetic gene cluster from rice: Biochemical characterization of CYP71Z6 & 7. FEBS Lett. 2011, 585, 3446. [Google Scholar] [CrossRef] [Green Version]
- Sawai, S.; Saito, K. Triterpenoid Biosynthesis and Engineering in Plants. Front. Plant Sci. 2011, 585, 3446–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Qi, X.; Yu, X.; Zheng, Y.; Liu, Z.; Fang, H.; Li, L.; Bai, Y.; Liang, C.; Li, W. Genome-Wide Analysis of Terpene Synthase Gene Family in Mentha longifolia and Catalytic Activity Analysis of a Single Terpene Synthase. Genes 2021, 12, 518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, J.; Cao, S.; Bai, F.; Yang, Y.; Huang, S.; Yao, L.; Anzai, Y.; Kato, F.; et al. New reactions and products resulting from alternative interactions between the P450 enzyme and redox partners. J. Am. Chem. Soc. 2014, 136, 3640–3646. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ortega, A.; Vinaixa, M.; Zebec, Z.; Takano, E.; Scrutton, N.S. A Toolbox for Diverse Oxyfunctionalisation of Monoterpenes OPEN. Sci. Rep. 2018, 8, 14396. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-Genome Annotation with BRAKER. Methods Mol. Biol. 2019, 1962, 65. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgis, H.Z. Red: An intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinform. 2015, 16, 227. [Google Scholar] [CrossRef] [Green Version]
- Grüning, B.; Yusuf, D.; Houwaart, T.; Anika; Miladi, M.; Gu, Q.; Batut, B.; Soranzo, N.; Gamaleldin, H.; Von Kuster, G.; et al. Bgruening/Galaxytools: September Release 2019; Zenodo: Geneva, Switzerland, 2018. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 March 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Batut, B.; Freeberg, M.; Heydarian, M.; Erxleben, A.; Videm, P.; Blank, C.; Doyle, M.; Soranzo, N.; van Heusden, P.; Delisle, L. Reference-Based RNA-Seq Data Analysis (Galaxy Training Materials). Available online: https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/ref-based/tutorial.html#citing-this-tutorial (accessed on 25 March 2023).
- Batut, B.; Hiltemann, S.; Bagnacani, A.; Baker, D.; Bhardwaj, V.; Blank, C.; Bretaudeau, A.; Brillet-Guéguen, L.; Čech, M.; Chilton, J.; et al. Community-Driven Data Analysis Training for Biology. Cell Syst. 2018, 6, 752–758.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kweon, O.; Kim, S.J.; Kim, J.H.; Nho, S.W.; Bae, D.; Chon, J.; Hart, M.; Baek, D.H.; Kim, Y.C.; Wang, W.; et al. CYPminer: An automated cytochrome P450 identification, classification, and data analysis tool for genome data sets across kingdoms. BMC Bioinform. 2020, 21, 160. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioinformatics Training at the Harvard Chan Bioinformatics Core. Available online: https://hbctraining.github.io/main/ (accessed on 19 April 2023).
- Pantiora, P.; Furlan, V.; Matiadis, D.; Mavroidi, B.; Perperopoulou, F.; Papageorgiou, A.C.; Sagnou, M.; Bren, U.; Pelecanou, M.; Labrou, N.E. Monocarbonyl Curcumin Analogues as Potent Inhibitors against Human Glutathione Transferase P1-1. Antioxidants 2023, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Kores, K.; Kolenc, Z.; Furlan, V.; Bren, U. Inverse Molecular Docking Elucidating the Anticarcinogenic Potential of the Hop Natural Product Xanthohumol and Its Metabolites. Foods 2022, 11, 1253. [Google Scholar] [CrossRef]
- Wen, W.; Yu, R. Artemisinin biosynthesis and its regulatory enzymes: Progress and perspective. Pharmacogn. Rev. 2011, 5, 189. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Xu, Z.; Song, C.; Chen, S. Herbgenomics: Decipher molecular genetics of medicinal plants. Innovation 2022, 3, 100322. [Google Scholar] [CrossRef]
- Alami, M.M.; Ouyang, Z.; Zhang, Y.; Shu, S.; Yang, G.; Mei, Z.; Wang, X. The Current Developments in Medicinal Plant Genomics Enabled the Diversification of Secondary Metabolites’ Biosynthesis. Int. J. Mol. Sci. 2022, 23, 15932. [Google Scholar] [CrossRef]
- Cheng, Q.Q.; Ouyang, Y.; Tang, Z.Y.; Lao, C.C.; Zhang, Y.Y.; Cheng, C.S.; Zhou, H. Review on the Development and Applications of Medicinal Plant Genomes. Front. Plant Sci. 2021, 12, 2981. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Caryopteris × clandonensis Cultivar | Raw Reads in Bases | Q20 in % | Q30 in % | Clean Reads in Bases | Q20 in % | Q30 in % | Totally Mapped in % | Uniquely Mapped in % | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unique | Duplicate | Unique | Duplicate | ||||||||
| Dark Knight | R1 | 24,501,785 | 19,238,555 | 99.95 | 94.76 | 13,072,273 | 29,945,355 | 99.99 | 95.08 | 87.8 | 79.3 |
| R2 | 26,380,719 | 17,359,621 | 99.25 | 87.90 | 16,204,470 | 26,813,158 | 99.46 | 88.25 | |||
| Grand Bleu | R1 | 17,917,215 | 51,971,129 | 99.85 | 93.75 | 11,552,426 | 57,659,626 | 99.98 | 94.21 | 85.8 | 76.8 |
| R2 | 18,808,258 | 51,080,086 | 99.51 | 92.12 | 13,260,446 | 55,951,606 | 99.68 | 92.42 | |||
| Good as Gold | R1 | 22,797,327 | 27,074,692 | 99.60 | 94.52 | 13,160,322 | 31,359,084 | 99.95 | 95.08 | 86.7 | 75.4 |
| R2 | 25,142,438 | 24,729,581 | 99.35 | 89.41 | 16,112,061 | 28,407,345 | 99.54 | 89.76 | |||
| Hint of Gold | R1 | 20,547,645 | 20,953,229 | 99.89 | 94.67 | 15,165,071 | 33,935,373 | 99.98 | 95.06 | 86.4 | 77.2 |
| R2 | 23,044,700 | 18,456,174 | 99.38 | 88.40 | 18,084,814 | 31,015,630 | 99.56 | 88.81 | |||
| Sunny Blue | R1 | 20,535,582 | 25,022,034 | 99.96 | 94.96 | 13,908,152 | 27,181,140 | 99.99 | 95.35 | 87.0 | 80.5 |
| R2 | 22,771,085 | 22,786,531 | 99.39 | 88.30 | 16,573,745 | 24,515,547 | 99.56 | 88.60 | |||
| Pink Perfection | R1 | 25,751,312 | 28,610,858 | 99.96 | 94.23 | 12,295,625 | 26,046,846 | 99.99 | 94.60 | 87.7 | 82.0 |
| R2 | 29,512,685 | 24,849,485 | 99.40 | 90.42 | 14,649,539 | 23,692,932 | 99.58 | 90.70 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritz, M.; Ahmad, N.; Brueck, T.; Mehlmer, N. Differential RNA-Seq Analysis Predicts Genes Related to Terpene Tailoring in Caryopteris × clandonensis. Plants 2023, 12, 2305. https://doi.org/10.3390/plants12122305
Ritz M, Ahmad N, Brueck T, Mehlmer N. Differential RNA-Seq Analysis Predicts Genes Related to Terpene Tailoring in Caryopteris × clandonensis. Plants. 2023; 12(12):2305. https://doi.org/10.3390/plants12122305
Chicago/Turabian StyleRitz, Manfred, Nadim Ahmad, Thomas Brueck, and Norbert Mehlmer. 2023. "Differential RNA-Seq Analysis Predicts Genes Related to Terpene Tailoring in Caryopteris × clandonensis" Plants 12, no. 12: 2305. https://doi.org/10.3390/plants12122305