
Synthesis, Properties and Spatial Structure of 4-[(3,5-Dimethyl-1,2-oxazol-4-yl)sulfonyl]cytisine

 , , ,
, , ,
Abstract
:1. Introduction
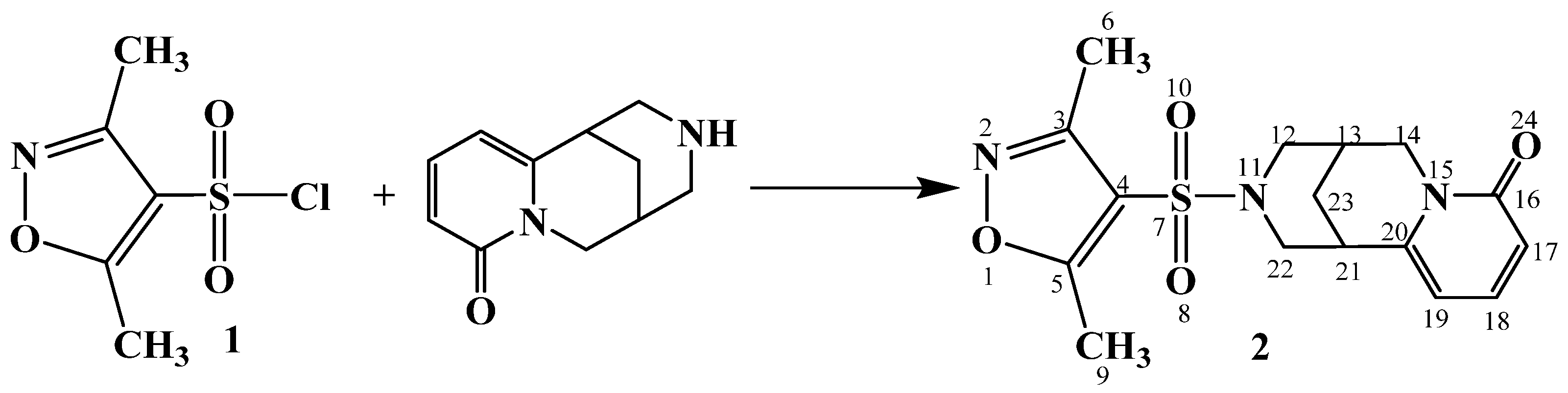
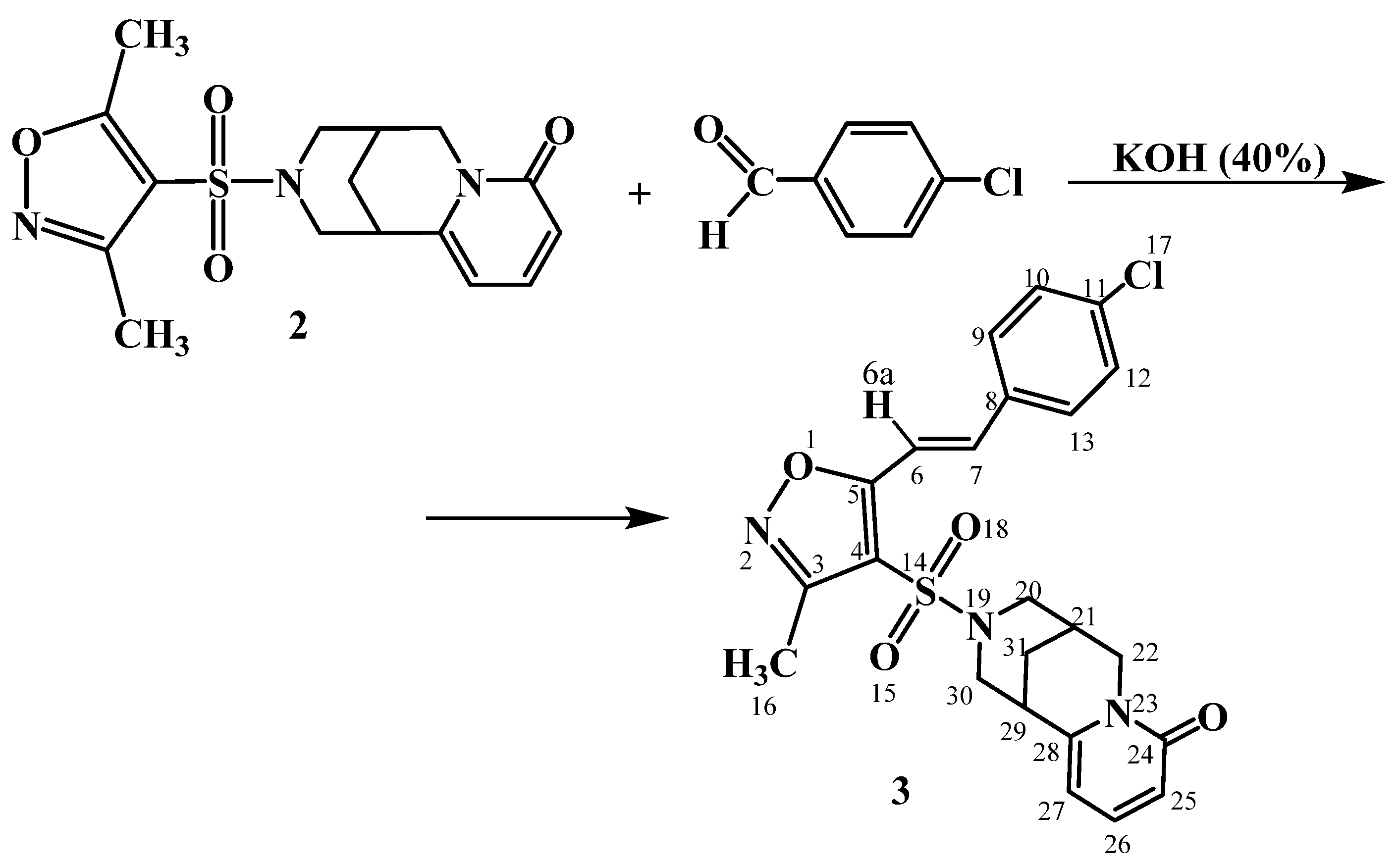
2. Results and Discussion
3. Conclusions
- Structures of compounds 2 and 3 have been determined with 1H, 13C NMR spectroscopy.
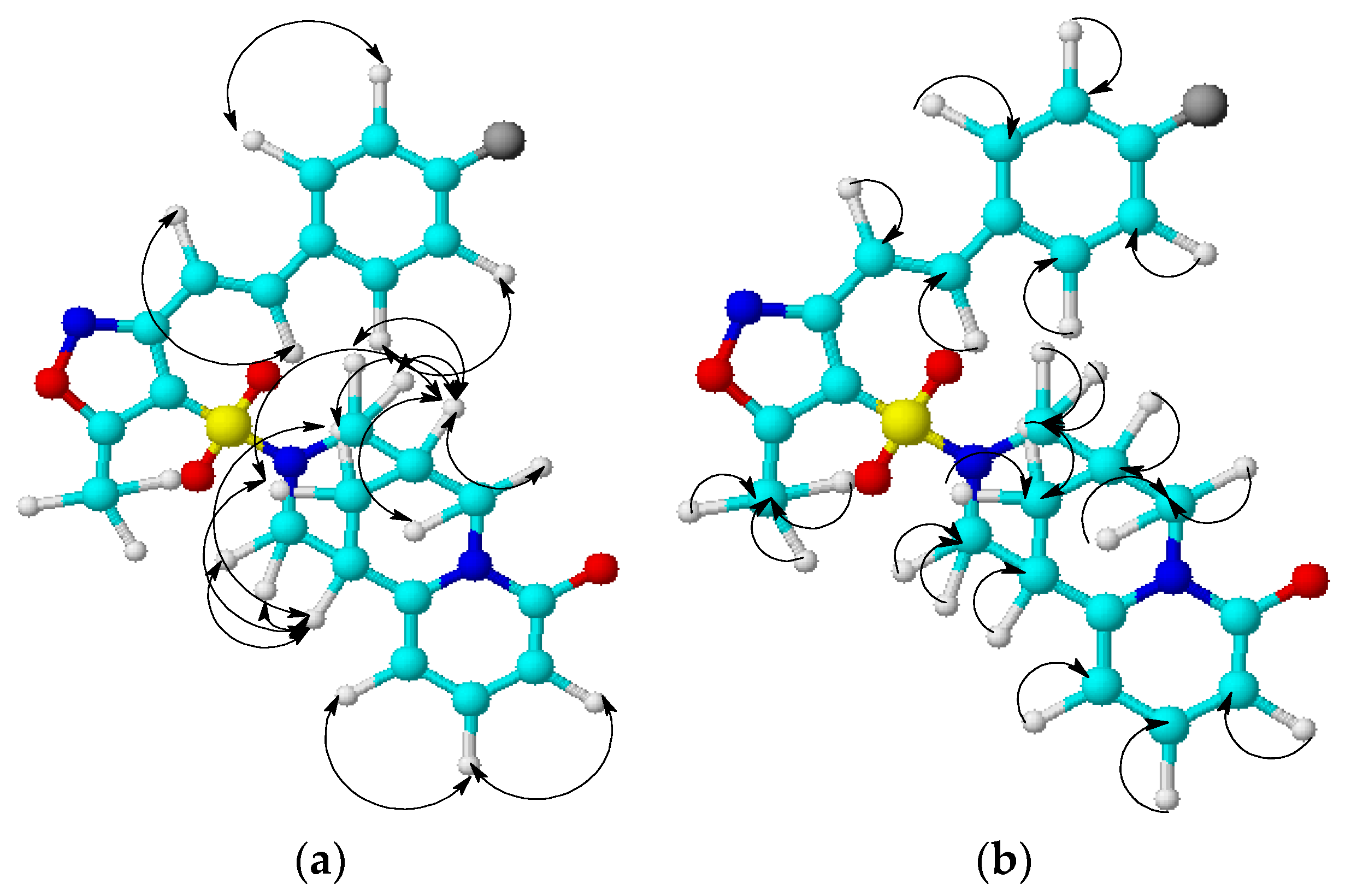
- 2D NMR spectroscopy of COSY (1H-1H), HMQC (1H-13C) and HMBC (1H-13C) has been used to study the mutual influence of atoms inside molecules of 2 and 3.
- The X-ray diffraction analysis has established the spatial structure of compound 2. All the parameters of a crystal structure and its structural features have been determined.
- The reaction with p-chlorobenzaldehyde has defined that a methyl group at the 5-position of the 4-sulfamidisoxazole cycle has been more active in the condensation reaction with the substituted aromatic aldehydes.
- The biological screening has shown a high hemorheological activity of 4-[(3,5-dimethyl-1,2-oxazol-4-yl)sulfonyl]cytisine 2. It has been as good as the well-known angioprotector pentoxifylline.
4. Experimental
Experimental Procedures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ragavendra, B.; Divya, K.G.; Padmaja, A.; Padmavathi, V. Synthesis and antimicrobial activity of bisazolylsulfonyl amines. Indian J. Chem. 2016, 55B, 1376–1383. [Google Scholar]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011, 10, 655–683. [Google Scholar] [PubMed]
- Linton, A.L.; Chatfield, W.R. A clinical trial of sulphamethoxazole in pyelonephritis with simplified methods of assessment. Br. J. Urol. 1965, 37, 515–517. [Google Scholar] [CrossRef]
- Shefter, E.; Chmielewicz, Z.F.; Blount, J.F.; Brennan, B.F.; Sackman, P. Biological implications of molecular and crystal structures of sulfadimethoxine, sulfadoxine, and sulfisoxazole. J. Pharm. Sci. 1972, 61, 872–877. [Google Scholar] [CrossRef]
- Delfs, F.M.; Loop, W. Solubility and acidity of 2-sulfanilamido-4,5-dimethyloxazole. Arzneimittel-Forschung 1961, 11, 402–403. [Google Scholar] [PubMed]
- Browne, S.G. Trial of a long-acting sulfonamide sulfaphenazole (Orisul, Ciba), in the treatment of leprosy. Int. J. Lepr. 1961, 29, 502–505. [Google Scholar] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2002, 23, 146–189. [Google Scholar] [CrossRef]
- Husain, A.; Madhesia, D. Heterocyclic compounds as carbonic anhydrase inhibitor. J. Enzyme Inhib. Med. Chem. 2012, 27, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Fitzmaurice, R.J.; Caddick, S. 3,5-Isoxazoles from α-bromo-pentafluorophenyl vinylsulfonates: Synthesis of sulfonates and sulfonamides. Org. Biomol. Chem. 2009, 7, 4349–4351. [Google Scholar] [CrossRef]
- Rapposelli, S.; Lapucci, A.; Minutolo, F.; Orlandini El Ortore, G.; Pinza, M.; Balsamo, A. Synthesis and COX-2 inhibitory properties of N-phenyl- and N-benzylsubstituted amides of 2-(4-metylsulfonylphenyl)cyclopent-1-ene-1- carboxylic acid and of their pyrazole, thiophene and isoxazole analogs. Farmaco 2004, 59, 25–31. [Google Scholar] [CrossRef]
- Barril, X.; Borgognoni, J.; Brough, P.A. 4,5-Diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef]
- Talley, J.J.; Brown, D.L.; Carter, J.S. 4-[5-Methyl-3-phenylisoxazol]4-yl]-benzenesulfonamide, valdecoxib: A potent and selective inhibitor of COX-2. J. Med. Chem. 2000, 43, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S.; et al. 3,5-Dimethylisoxazoles Act As Acetyl-lysine-mimetic Bromodomain. J. Med. Chem. 2011, 54, 6761–6770. [Google Scholar] [CrossRef] [PubMed]
- Freer, A.A.; Robins, D.J.; Sheldrake, G.N. Structures of (−)-cytisine and (−)-N-methylcytisine: Tricyclic quinolizidine alkaloids. Acta Crystallogr. 1987, C43, 1119–1122. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Smol’yakov, A.F.; Karnoukhova, V.A.; Osintseva, S.V.; Petrova, P.R.; Koval’skaya, A.V.; Tsypysheva, I.P. Crystal and Molecular Structures of Methylcytisine Nitro-Derivatives. Pharm. Chem. J. 2017, 50, 826–832. [Google Scholar] [CrossRef]
- Owczarzak, A.; Grzeskiewicz, A.M.; Kubicki, M. Experimental studies of charge density distribution in the crystals of cytisine and N-methylcytisine. Inside the Fake Tobacco. Struct. Chem. 2017, 28, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- Cambridge Crystallographic Database, Release 2021; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2021.
- Kulakov, I.V.; Nurkenov, O.A.; Ainabaev, A.A.; Turdybekov, D.M.; Turdybekov, K.M. Synthesis of Dithiocarbamine Derivatives on the Matrix of Cytisine, Anabasine and d-Pseudoephedrine Alkaloids. Crystalline Structure of N-Cytisine Dithiocarbamate Ammonium Salt. Russ. J. Gen. Chem. 2009, 79, 1716–1719. [Google Scholar] [CrossRef]
- Turdybekov, K.M.; Kulakov, I.V.; Turdybekov, D.M.; Mahmutova, A.S. Conformational states and crystal structure of N-formylcytisine. Russ. J. Gen. Chem. 2017, 87, 2493–2496. [Google Scholar] [CrossRef]
- Korsakov, M.K. Sulfonamide derivatives of binuclear azole-containing systems: Synthesis and properties. Diss. Doc. Chem. Sci. 2018, 453. Available online: https://www.dissercat.com/content/sulfonamidnye-proizvodnye-dvuyadernykh-azolsoderzhashchikh-sistem-sintez-i-svoistva (accessed on 8 December 2022). (In Russian).
- Plotnikov, M.B.; Koltunov, A.A.; Aliev, O.I. A method for selecting drugs that affect the rheological properties of the blood in vitro. Eksperimental’naia I Klin. Farmakol. 1996, 59, 54–55. [Google Scholar]
- SMART V5.051 and SAINT V5.00. Madison, Bruker AXS Inc.: Billerica, MA, USA, 2005.
- Sheldrick, G.N. SADABS. Madison, Bruker AXS Inc.: Billerica, MA, USA, 2008.
- Sheldrick, G.N. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.N. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | d | Bond | d |
|---|---|---|---|
| S(1)-O(3) | 1.424(2) | C(9)-C(13) | 1.523(4) |
| S(1)-O(2) | 1.427(2) | C(9)-H(9A) | 0.9800 |
| S(1)-N(12) | 1.634(2) | C(10)-H(10A) | 0.9700 |
| S(1)-C(4′) | 1.749(3) | C(10)-H(10B) | 0.9700 |
| O(1)-C(2) | 1.231(3) | C(11)-N(12) | 1.472(4) |
| N(1)-C(6) | 1.372(3) | C(11)-H(11A) | 0.9700 |
| N(1)-C(2) | 1.400(3) | C(11)-H(11B) | 0.9700 |
| N(1)-C(10) | 1.487(3) | N(12)-C(13) | 1.484(3) |
| C(2)-C(3) | 1.427(4) | C(13)-H(13A) | 0.9700 |
| C(3)-C(4) | 1.348(5) | C(13)-H(13B) | 0.9700 |
| C(3)-H(3B) | 0.9300 | O(1′)-C(5′) | 1.334(4) |
| C(4)-C(5) | 1.396(4) | O(1′)-N(2′) | 1.418(4) |
| C(4)-H(4A) | 0.9300 | N(2′)-C(3′) | 1.304(4) |
| C(5)-C(6) | 1.362(4) | C(3′)-C(4′) | 1.412(4) |
| C(5)-H(5A) | 0.9300 | C(3′)-C(6′) | 1.475(6) |
| C(6)-C(7) | 1.500(4) | C(4′)-C(5′) | 1.372(4) |
| C(7)-C(8) | 1.526(4) | C(5′)-C(7′) | 1.480(5) |
| C(7)-C(11) | 1.532(4) | C(6′)-H(6′A) | 0.9600 |
| C(7)-H(7A) | 0.9800 | C(6′)-H(6′B) | 0.9600 |
| C(8)-C(9) | 1.515(5) | C(6′)-H(6′C) | 0.9600 |
| C(8)-H(8A) | 0.9700 | C(7′)-H(7′A) | 0.9600 |
| C(8)-H(8B) | 0.9700 | C(7′)-H(7′B) | 0.9600 |
| C(9)-C(10) | 1.512(4) | C(7′)-H(7′C) | 0.9600 |
| Angle | ω | Angle | ω |
|---|---|---|---|
| O(3)-S(1)-O(2) | 120.01(14) | N(1)-C(10)-H(10A) | 108.6 |
| O(3)-S(1)-N(12) | 107.74(13) | C(9)-C(10)-H(10A) | 108.6 |
| O(2)-S(1)-N(12) | 107.29(13) | N(1)-C(10)-H(10B) | 108.6 |
| O(3)-S(1)-C(4′) | 106.65(14) | C(9)-C(10)-H(10B) | 108.6 |
| O(2)-S(1)-C(4′) | 107.25(14) | H(10A)-C(10)-H(10B) | 107.6 |
| N(12)-S(1)-C(4′) | 107.31(12) | N(12)-C(11)-C(7) | 108.9(2) |
| C(6)-N(1)-C(2) | 123.0(2) | N(12)-C(11)-H(11A) | 109.9 |
| C(6)-N(1)-C(10) | 123.5(2) | C(7)-C(11)-H(11A) | 109.9 |
| C(2)-N(1)-C(10) | 113.4(2) | N(12)-C(11)-H(11B) | 109.9 |
| O(1)-C(2)-N(1) | 119.6(3) | C(7)-C(11)-H(11B) | 109.9 |
| O(1)-C(2)-C(3) | 124.8(3) | H(11A)-C(11)-H(11B) | 108.3 |
| N(1)-C(2)-C(3) | 115.6(3) | C(11)-N(12)-C(13) | 113.4(2) |
| C(4)-C(3)-C(2) | 121.2(3) | C(11)-N(12)-S(1) | 117.54(17) |
| C(4)-C(3)-H(3B) | 119.4 | C(13)-N(12)-S(1) | 115.02(19) |
| C(2)-C(3)-H(3B) | 119.4 | N(12)-C(13)-C(9) | 109.5(2) |
| C(3)-C(4)-C(5) | 120.8(3) | N(12)-C(13)-H(13A) | 109.8 |
| C(3)-C(4)-H(4A) | 119.6 | C(9)-C(13)-H(13A) | 109.8 |
| C(5)-C(4)-H(4A) | 119.6 | N(12)-C(13)-H(13B) | 109.8 |
| C(6)-C(5)-C(4) | 120.1(3) | C(9)-C(13)-H(13B) | 109.8 |
| C(6)-C(5)-H(5A) | 120.0 | H(13A)-C(13)-H(13B) | 108.2 |
| C(4)-C(5)-H(5A) | 120.0 | C(5′)-O(1′)-N(2′) | 108.7(2) |
| C(5)-C(6)-N(1) | 119.3(2) | C(3′)-N(2′)-O(1′) | 106.7(3) |
| C(5)-C(6)-C(7) | 122.0(2) | N(2′)-C(3′)-C(4′) | 110.1(3) |
| N(1)-C(6)-C(7) | 118.7(2) | N(2′)-C(3′)-C(6′) | 118.5(3) |
| C(6)-C(7)-C(8) | 110.9(2) | C(4′)-C(3′)-C(6′) | 131.4(3) |
| C(6)-C(7)-C(11) | 111.1(2) | C(5′)-C(4′)-C(3′) | 105.9(3) |
| C(8)-C(7)-C(11) | 109.8(2) | C(5′)-C(4′)-S(1) | 126.7(2) |
| C(6)-C(7)-H(7A) | 108.3 | C(3′)-C(4′)-S(1) | 127.4(2) |
| C(8)-C(7)-H(7A) | 108.3 | O(1′)-C(5′)-C(4′) | 108.6(3) |
| C(11)-C(7)-H(7A) | 108.3 | O(1′)-C(5′)-C(7′) | 117.0(3) |
| C(9)-C(8)-C(7) | 106.5(2) | C(4′)-C(5′)-C(7′) | 134.4(3) |
| C(9)-C(8)-H(8A) | 110.4 | C(3′)-C(6′)-H(6′A) | 109.5 |
| C(7)-C(8)-H(8A) | 110.4 | C(3′)-C(6′)-H(6′B) | 109.5 |
| C(9)-C(8)-H(8B) | 110.4 | H(6′A)-C(6′)-H(6′B) | 109.5 |
| C(7)-C(8)-H(8B) | 110.4 | C(3′)-C(6′)-H(6′C) | 109.5 |
| H(8A)-C(8)-H(8B) | 108.6 | H(6′A)-C(6′)-H(6′C) | 109.5 |
| C(10)-C(9)-C(8) | 111.1(2) | H(6′B)-C(6′)-H(6′C) | 109.5 |
| C(10)-C(9)-C(13) | 112.2(2) | C(5′)-C(7′)-H(7′A) | 109.5 |
| C(8)-C(9)-C(13) | 110.4(2) | C(5′)-C(7′)-H(7′B) | 109.5 |
| C(10)-C(9)-H(9A) | 107.6 | H(7′A)-C(7′)-H(7′B) | 109.5 |
| C(8)-C(9)-H(9A) | 107.6 | C(5′)-C(7′)-H(7′C) | 109.5 |
| C(13)-C(9)-H(9A) | 107.6 | H(7′A)-C(7′)-H(7′C) | 109.5 |
| N(1)-C(10)-C(9) | 114.4(2) | H(7′B)-C(7′)-H(7′C) | 109.5 |
| Angle | Τ | Angle | τ |
|---|---|---|---|
| C(6)-N(1)-C(2)-O(1) | 179.4(2) | O(3)-S(1)-N(12)-C(11) | 168.7(2) |
| C(10)-N(1)-C(2)-O(1) | 1.6(4) | O(2)-S(1)-N(12)-C(11) | 38.1(2) |
| C(6)-N(1)-C(2)-C(3) | −0.9(4) | C(4′)-S(1)-N(12)-C(11) | −76.8(2) |
| C(10)-N(1)-C(2)-C(3) | −178.7(2) | O(3)-S(1)-N(12)-C(13) | −53.9(2) |
| O(1)-C(2)-C(3)-C(4) | −178.3(3) | O(2)-S(1)-N(12)-C(13) | 175.6(2) |
| N(1)-C(2)-C(3)-C(4) | 2.0(4) | C(4′)-S(1)-N(12)-C(13) | 60.6(2) |
| C(2)-C(3)-C(4)-C(5) | −1.2(5) | C(11)-N(12)-C(13)-C(9) | −56.2(3) |
| C(3)-C(4)-C(5)-C(6) | −0.9(5) | S(1)-N(12)-C(13)-C(9) | 164.5(2) |
| C(4)-C(5)-C(6)-N(1) | 1.9(4) | C(10)-C(9)-C(13)-N(12) | −66.1(3) |
| C(4)-C(5)-C(6)-C(7) | −177.7(3) | C(8)-C(9)-C(13)-N(12) | 58.4(3) |
| C(2)-N(1)-C(6)-C(5) | −1.1(4) | C(5′)-O(1′)-N(2′)-C(3′) | −0.2(4) |
| C(10)-N(1)-C(6)-C(5) | 176.5(3) | O(1′)-N(2′)-C(3′)-C(4′) | 0.4(4) |
| C(2)-N(1)-C(6)-C(7) | 178.6(2) | O(1′)-N(2′)-C(3′)-C(6′) | 178.5(4) |
| C(10)-N(1)-C(6)-C(7) | −3.8(3) | N(2′)-C(3′)-C(4′)-C(5′) | −0.4(4) |
| C(5)-C(6)-C(7)-C(8) | −148.2(3) | C(6′)-C(3′)-C(4′)-C(5′) | −178.2(4) |
| N(1)-C(6)-C(7)-C(8) | 32.1(3) | N(2′)-C(3′)-C(4′)-S(1) | 178.4(2) |
| C(5)-C(6)-C(7)-C(11) | 89.3(3) | C(6′)-C(3′)-C(4′)-S(1) | 0.6(6) |
| N(1)-C(6)-C(7)-C(11) | −90.3(3) | O(3)-S(1)-C(4′)-C(5′) | 29.7(3) |
| C(6)-C(7)-C(8)-C(9) | −60.9(3) | O(2)-S(1)-C(4′)-C(5′) | 159.4(3) |
| C(11)-C(7)-C(8)-C(9) | 62.3(3) | N(12)-S(1)-C(4′)-C(5′) | −85.6(3) |
| C(7)-C(8)-C(9)-C(10) | 63.5(3) | O(3)-S(1)-C(4′)-C(3′) | −148.9(3) |
| C(7)-C(8)-C(9)-C(13) | −61.6(3) | O(2)-S(1)-C(4′)-C(3′) | −19.1(3) |
| C(6)-N(1)-C(10)-C(9) | 5.7(4) | N(12)-S(1)-C(4′)-C(3′) | 95.9(3) |
| C(2)-N(1)-C(10)-C(9) | −176.5(2) | N(2′)-O(1′)-C(5′)-C(4′) | 0.0(3) |
| C(8)-C(9)-C(10)-N(1) | −36.3(3) | N(2′)-O(1′)-C(5′)-C(7′) | −179.3(3) |
| C(13)-C(9)-C(10)-N(1) | 87.8(3) | C(3′)-C(4′)-C(5′)-O(1′) | 0.3(3) |
| C(6)-C(7)-C(11)-N(12) | 63.2(3) | S(1)-C(4′)-C(5′)-O(1′) | −178.6(2) |
| C(8)-C(7)-C(11)-N(12) | −59.9(3) | C(3′)-C(4′)-C(5′)-C(7′) | 179.3(4) |
| C(7)-C(11)-N(12)-C(13) | 56.9(3) | S(1)-C(4′)-C(5′)-C(7′) | 0.5(5) |
| C(7)-C(11)-N(12)-S(1) | −164.97(18) |
| Atom | x | y | z |
|---|---|---|---|
| S(1) | 1570(1) | 6334(1) | 2084(1) |
| O(1) | 3784(4) | 4085(3) | 6024(2) |
| O(2) | −479(3) | 5822(3) | 1848(2) |
| O(3) | 2015(4) | 7585(3) | 2713(2) |
| N(1) | 3553(3) | 3144(2) | 4497(2) |
| C(2) | 2863(4) | 3270(3) | 5396(2) |
| C(3) | 1088(5) | 2419(4) | 5507(2) |
| C(4) | 195(5) | 1522(4) | 4792(3) |
| C(5) | 965(4) | 1421(4) | 3914(2) |
| C(6) | 2621(4) | 2250(3) | 3765(2) |
| C(7) | 3456(4) | 2222(3) | 2821(2) |
| C(8) | 5760(4) | 2453(4) | 2978(2) |
| C(9) | 6137(4) | 3981(4) | 3445(2) |
| C(10) | 5433(4) | 4025(4) | 4428(2) |
| C(11) | 2442(4) | 3421(3) | 2127(2) |
| N(12) | 2927(3) | 4914(3) | 2551(2) |
| C(13) | 5147(4) | 5205(4) | 2775(2) |
| O(1′) | 4206(5) | 7819(3) | −55(2) |
| N(2′) | 2656(6) | 6928(3) | −579(2) |
| C(3′) | 1633(5) | 6326(4) | 55(2) |
| C(4′) | 2446(4) | 6796(3) | 997(2) |
| C(5′) | 4050(5) | 7722(3) | 885(2) |
| C(6′) | −125(9) | 5360(5) | −294(3) |
| C(7′) | 5530(5) | 8596(4) | 1557(3) |
| Tested Parameter | Blood Viscosity (mPa × s) at Various Spindle Speeds, rpm | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 4 | 6 | 8 | 12 | 20 | 40 | 60 | |
| Reference viscosity, n = 2 | 2.71 ± 0.05 | 2.25 ± 0.02 | 2.05 ± 0.01 | 1.80 ± 0.04 | 1.68 ± 0.04 | 1.45 ± 0.02 | 1.30 ± 0.6 | 1.27 ± 0.05 |
| Blood viscosity after 1 h of incubation at 43 °C in a control, n = 4 | 8.75 ± 0.26 | 7.05 ± 0.14 | 5.30 ± 0.07 | 4.46 ± 0.10 | 3.65 ± 0.29 | 3.33 ± 0.42 | 3.01 ± 0.37 | 2.85 ± 0.41 |
| p1 = 0.0001 | p1 = 0.00002 | p1 = 0.00001 | p1 = 0.0001 | p1 = 0.0113 | p1 = 0.0420 | p1 = 0.0368 | p1 = 0.0627 | |
| Blood viscosity after 1 h of incubation at 43 °C, samples with compound 2, n = 4 | 6.83 ± 0.10 | 5.21 ± 0.06 | 4.74 ± 0.08 | 3.81 ± 0.08 | 3.01 ± 0.6 | 2.67 ± 0.9 | 2.32 ± 0.15 | 2.17 ± 0.10 |
| p1 = 0.00001 p2 = 0.0005 | p1 = 0.00001 p2 = 0.00002 | p1 = 0.00003 p2 = 0.0021 | p1 = 0.0001 p2 = 0.0018 | p1 = 0.0261 p2 = 0.1511 | p1 = 0.0118 p2 = 0.2044 | p1 = 0.0109 p2 = 0.1332 | p1 = 0.0040 p2 = 0.1580 | |
| Tested Parameter | Blood Viscosity (mPa × s) at Various Spindle Speeds, rpm | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 4 | 6 | 8 | 12 | 20 | 40 | 60 | |
| Reference viscosity, n = 2 | 5.94 ± 0.59 | 4.90 ± 0.43 | 4.10 ± 0.38 | 3.87 ± 0.34 | 3.40 ± 0.29 | 2.69 ± 0.26 | 2.32 ± 0.12 | 2.21 ± 0.12 |
| Blood viscosity after 1 h of incubation at 43 °C in a control, n = 4 | 7.53 ± 0.45 | 6.36 ± 0.40 | 5.79 ± 0.44 | 5.19 ± 0.31 | 4.37 ± 0.13 | 3.56 ± 0.15 | 2.76 ± 0.09 | 2.53 ± 0.07 |
| p1 = 0.0519 | p1 = 0.0364 | p1 = 0.0250 | p1 = 0.0184 | p1 = 0.0026 | p1 = 0.0065 | p1 = 0.0098 | p1 = 0.0218 | |
| Blood viscosity after 1 h of incubation at 43 °C, samples with pentoxifylline, n = 4 | 7.03 ± 0.43 | 5.81 ± 0.30 | 5.00 ± 0.21 | 4.56 ± 0.16 | 4.05 ± 0.10 | 3.24 ± 0.14 | 2.56 ± 0.08 | 2.39 ± 0.07 |
| p1 = 0.1584 p2 = 0.4306 | p1 = 0.1009 p2 = 0.2800 | p1 = 0.0357 p2 = 0.1205 | p1 = 0.0532 p2 = 0.0855 | p1 = 0.0171 p2 = 0.0631 | p1 = 0.0563 p2 = 0.1353 | p1 = 0.0960 p2 = 0.0999 | p1 = 0.1887 p2 = 0.1590 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrayev, M.K.; Nurkenov, O.A.; Rakhimberlinova, Z.B.; Takibayeva, A.T.; Turdybekov, D.M.; Seilkhanov, T.M.; Issabayeva, M.B.; Kelmyalene, A.A.; Kezdikbayeva, A.T.; Mendibayeva, A.Z. Synthesis, Properties and Spatial Structure of 4-[(3,5-Dimethyl-1,2-oxazol-4-yl)sulfonyl]cytisine. Plants 2023, 12, 137. https://doi.org/10.3390/plants12010137
Ibrayev MK, Nurkenov OA, Rakhimberlinova ZB, Takibayeva AT, Turdybekov DM, Seilkhanov TM, Issabayeva MB, Kelmyalene AA, Kezdikbayeva AT, Mendibayeva AZ. Synthesis, Properties and Spatial Structure of 4-[(3,5-Dimethyl-1,2-oxazol-4-yl)sulfonyl]cytisine. Plants. 2023; 12(1):137. https://doi.org/10.3390/plants12010137
Chicago/Turabian StyleIbrayev, Marat K., Oralgazy A. Nurkenov, Zhanara B. Rakhimberlinova, Altynaray T. Takibayeva, Dastan M. Turdybekov, Tulegen M. Seilkhanov, Meruyert B. Issabayeva, Assel A. Kelmyalene, Assel T. Kezdikbayeva, and Anel Z. Mendibayeva. 2023. "Synthesis, Properties and Spatial Structure of 4-[(3,5-Dimethyl-1,2-oxazol-4-yl)sulfonyl]cytisine" Plants 12, no. 1: 137. https://doi.org/10.3390/plants12010137