
Combined Approach of GWAS and Phylogenetic Analyses to Identify New Candidate Genes That Participate in Arabidopsis thaliana Primary Root Development Using Cellular Measurements and Primary Root Length
 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results
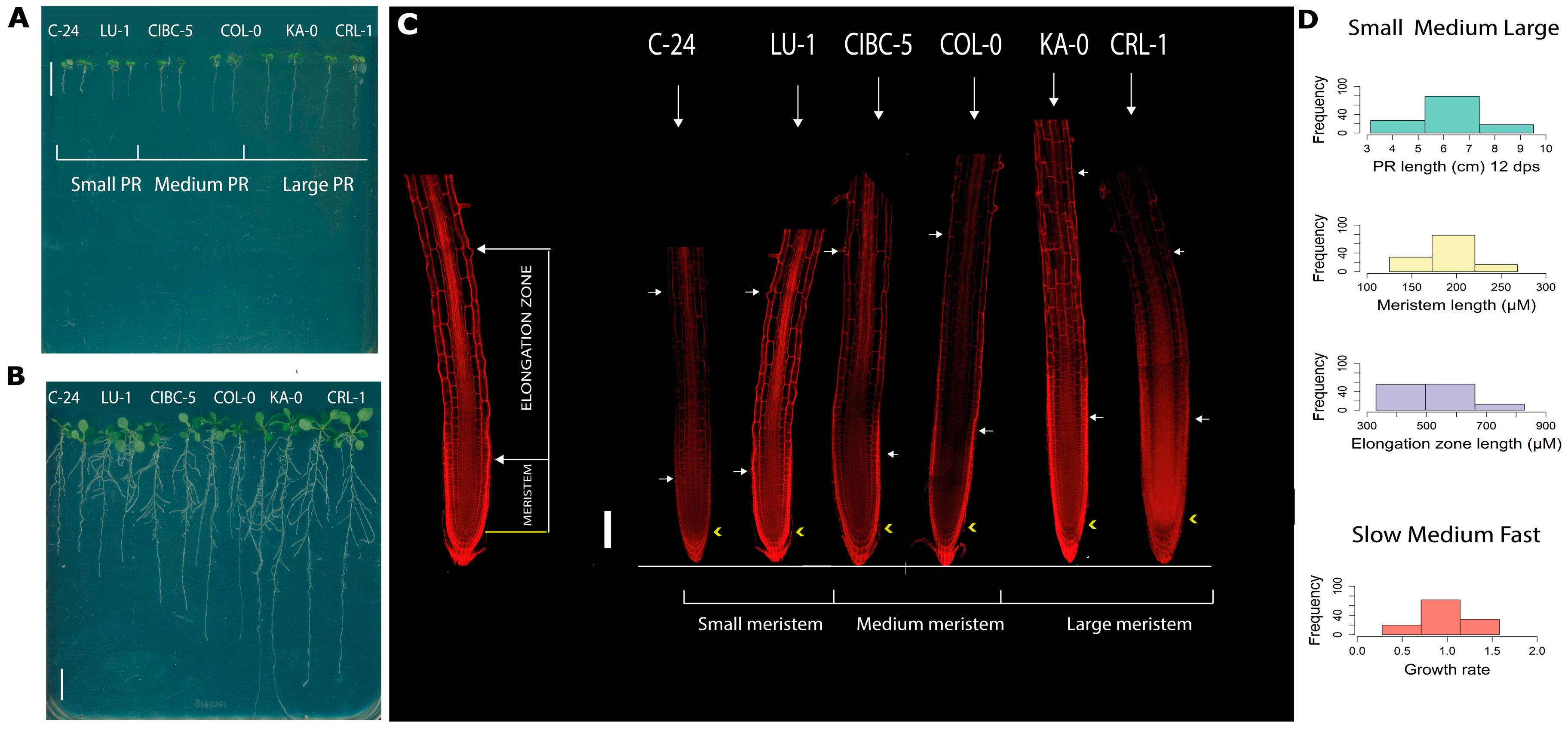
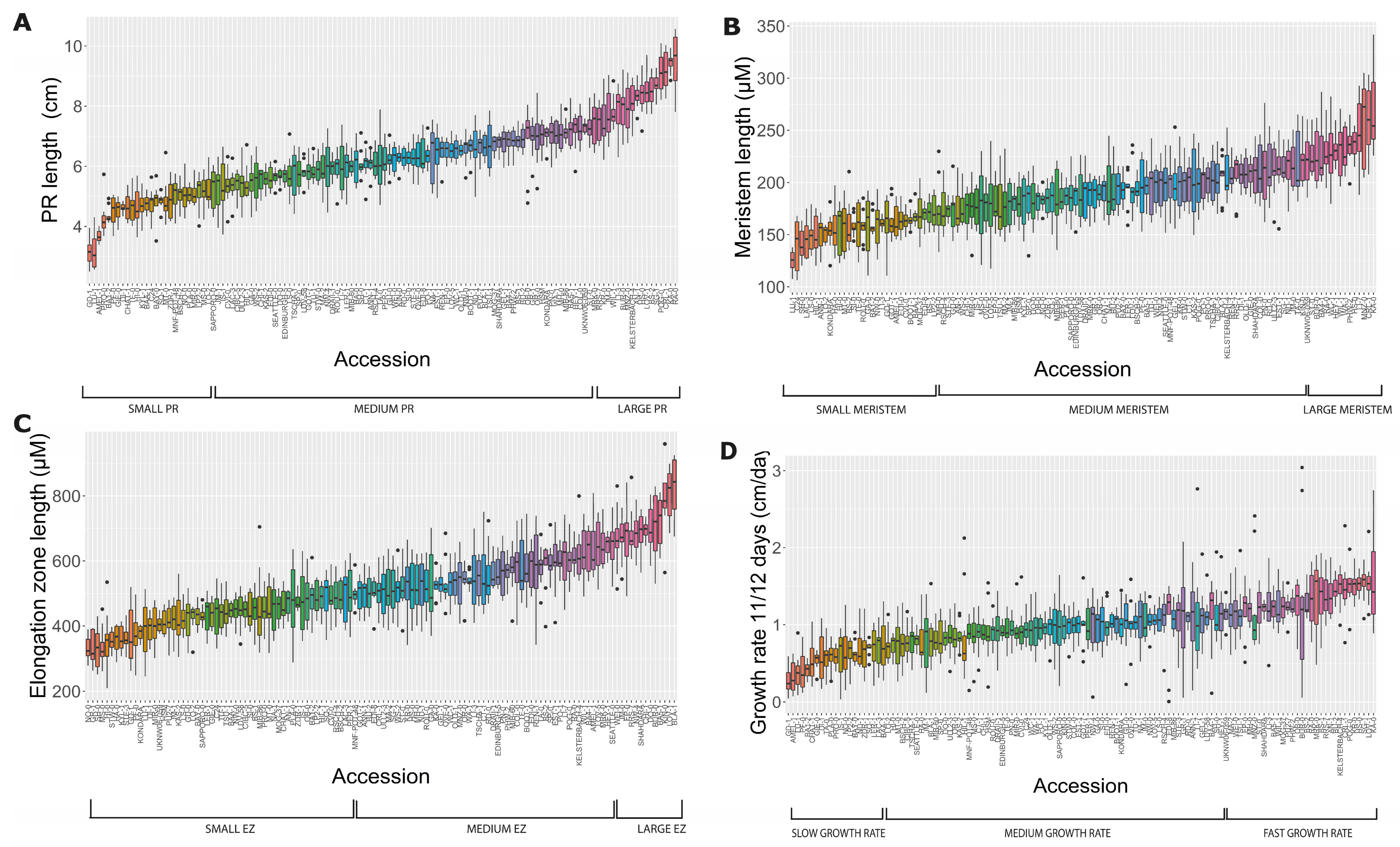
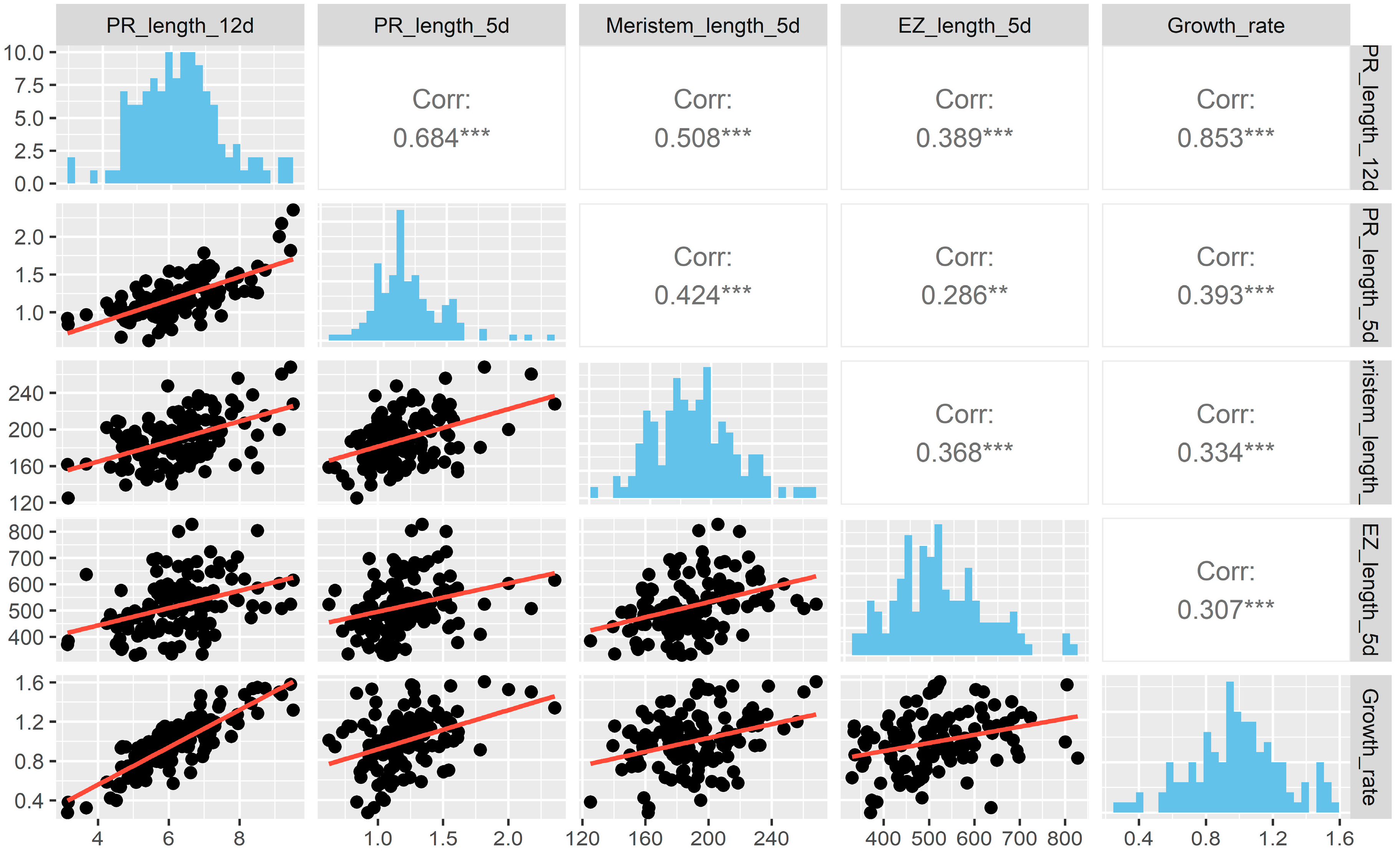
2.1. Natural Allelic Variation Determines Primary Root Development in Arabidopsis
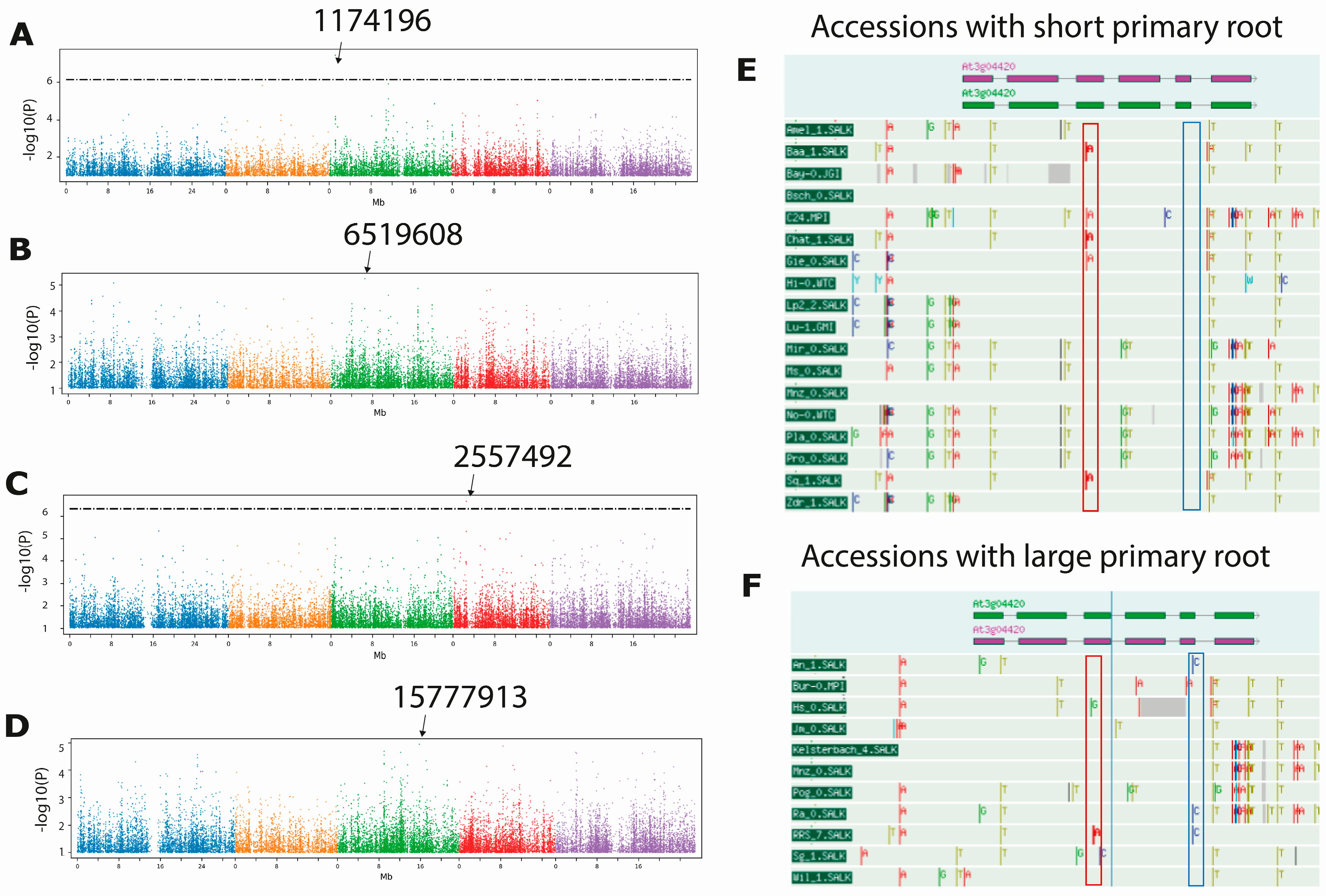
2.2. GWAS Reveal Genes That May Explain the Natural Variation in the Length of the Primary Root, the Meristem and the Elongation Zone
2.3. Phylogenetic Tree Reconstructions Reveal Additional Accessions for Further Studies
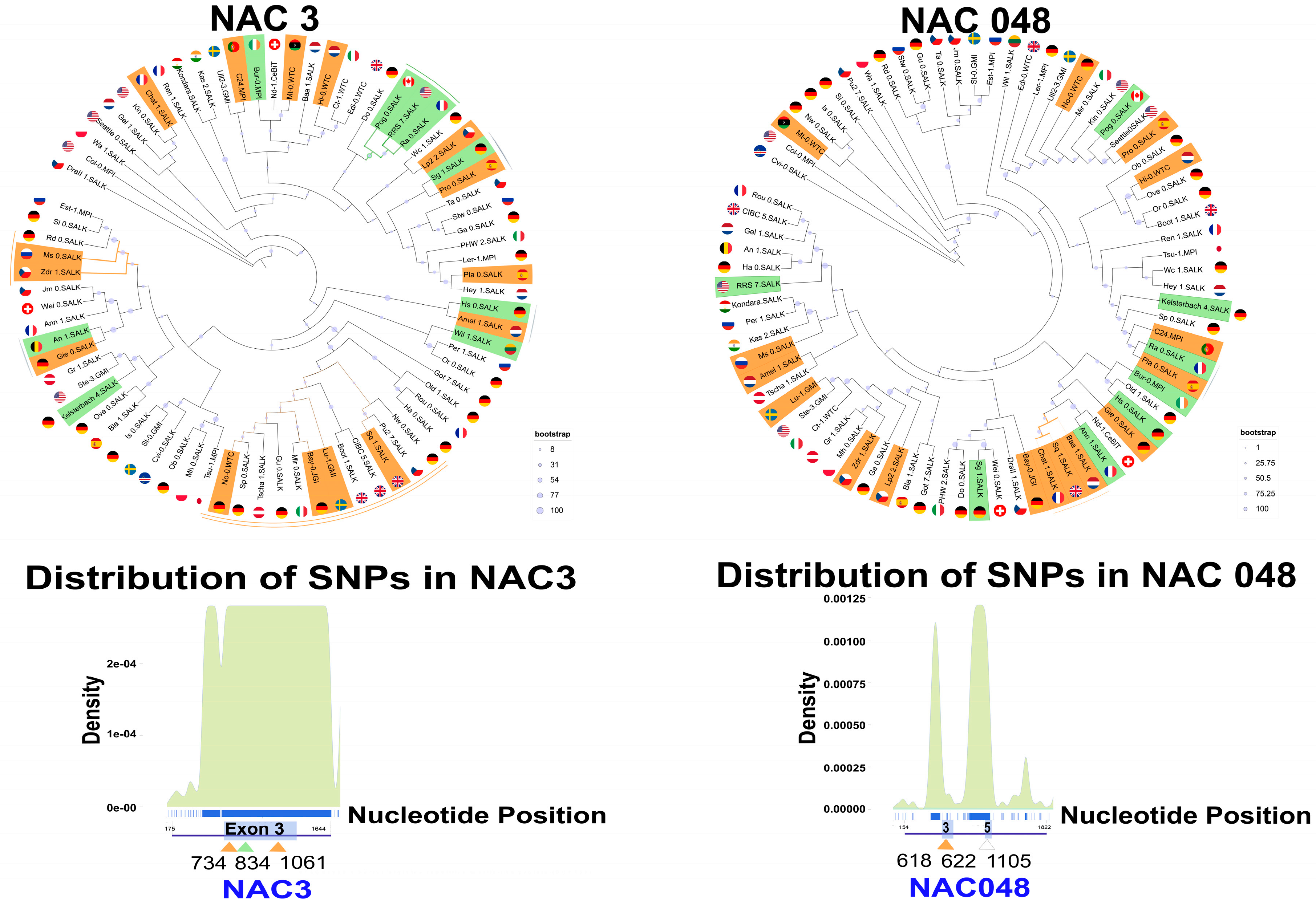
2.4. Complex Patterns of SNPs and Indels in NAC3 and NAC048
3. Discussion
3.1. Natural Populations of Arabidopsis Have Wide Variation of Primary Root Length, Growth Rate and Features at the Cellular Level
3.2. GWAS Uncover Novel Genes with the Potential to Explain the Natural Variation in the Length of the Primary Root, the Meristem, and the Elongation Zone
3.3. Phylogenetic Gene Tree Reconstruction Allowed for the Investigation of Evolutionary Trends and Discovery of Additional Accessions for Further Studies
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Pseudo-Schiff Assay
4.3. Microscopy Visualization
4.4. Quantitative Analysis
4.5. Plant Phenotype Studies
4.6. Genome-Wide Association Studies
4.7. Polymorphism Patterns in the Selected Genes in Accessions with Extreme Phenotypes
4.8. Phylogenetic Tree Construction and Analysis
4.9. In Silico Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-Wide Association Studies in Plants: The Missing Heritability Is in the Field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D. Natural Variation in Arabidopsis: From Molecular Genetics to Ecological Genomics. Plant Physiol. 2012, 158, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Deja-Muylle, A.; Parizot, B.; Motte, H.; Beeckman, T. Exploiting Natural Variation in Root System Architecture via Genome-Wide Association Studies. J. Exp. Bot. 2020, 71, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A. Root Decisions. Plant Cell Environ. 2009, 32, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Beemster, G.T.S.; Baskin, T.I. Stunted Plant 1 Mediates Effects of Cytokinin, but Not of Auxin, on Cell Division and Expansion in the Root of Arabidopsis. Plant Physiol. 2000, 124, 1718–1727. [Google Scholar] [CrossRef]
- Zluhan-Martínez, E.; López-Ruíz, B.A.; García-Gómez, M.L.; García-Ponce, B.; Sánchez, M.d.l.P.; Álvarez-Buylla, E.R.; Garay-Arroyo, A. Integrative Roles of Phytohormones on Cell Proliferation, Elongation and Differentiation in the Arabidopsis Thaliana Primary Root. Front. Plant Sci. 2021, 12, 659155. [Google Scholar] [CrossRef]
- Julkowska, M.M.; Koevoets, I.T.; Mol, S.; Hoefsloot, H.; Feron, R.; Tester, M.A.; Keurentjes, J.J.B.; Korte, A.; Haring, M.A.; de Boer, G.-J.; et al. Genetic Components of Root Architecture Remodeling in Response to Salt Stress. Plant Cell 2017, 29, 3198–3213. [Google Scholar] [CrossRef]
- Deolu-Ajayi, A.O.; Meyer, A.J.; Haring, M.A.; Julkowska, M.M.; Testerink, C. Genetic Loci Associated with Early Salt Stress Responses of Roots. iScience 2019, 21, 458–473. [Google Scholar] [CrossRef]
- Gifford, M.L.; Banta, J.A.; Katari, M.S.; Hulsmans, J.; Chen, L.; Ristova, D.; Tranchina, D.; Purugganan, M.D.; Coruzzi, G.M.; Birnbaum, K.D. Plasticity Regulators Modulate Specific Root Traits in Discrete Nitrogen Environments. PLoS Genet 2013, 9, e1003760. [Google Scholar] [CrossRef]
- Satbhai, S.B.; Setzer, C.; Freynschlag, F.; Slovak, R.; Kerdaffrec, E.; Busch, W. Natural Allelic Variation of FRO2 Modulates Arabidopsis Root Growth under Iron Deficiency. Nat. Commun. 2017, 8, 15603. [Google Scholar] [CrossRef]
- Bouain, N.; Satbhai, S.B.; Korte, A.; Saenchai, C.; Desbrosses, G.; Berthomieu, P.; Busch, W.; Rouached, H. Natural Allelic Variation of the AZI1 Gene Controls Root Growth under Zinc-Limiting Condition. PLoS Genet. 2018, 14, e1007304. [Google Scholar] [CrossRef]
- Bouain, N.; Korte, A.; Satbhai, S.B.; Nam, H.-I.; Rhee, S.Y.; Busch, W.; Rouached, H. Systems Genomics Approaches Provide New Insights into Arabidopsis Thaliana Root Growth Regulation under Combinatorial Mineral Nutrient Limitation. PLoS Genet. 2019, 15, e1008392. [Google Scholar] [CrossRef]
- Li, B.; Sun, L.; Huang, J.; Göschl, C.; Shi, W.; Chory, J.; Busch, W. GSNOR Provides Plant Tolerance to Iron Toxicity via Preventing Iron-Dependent Nitrosative and Oxidative Cytotoxicity. Nat. Commun. 2019, 10, 3896. [Google Scholar] [CrossRef]
- Ghimire, B.; Saminathan, T.; Bodunrin, A.; Abburi, V.L.; Kshetry, A.O.; Shinde, S.; Nimmakayala, P.; Reddy, U.K. Genome-Wide Association Study of Natural Variation in Arabidopsis Exposed to Acid Mine Drainage Toxicity and Validation of Associated Genes with Reverse Genetics. Plants 2021, 10, 191. [Google Scholar] [CrossRef]
- Justamante, M.S.; Ibáñez, S.; Peidró, A.; Pérez-Pérez, J.M. A Genome-Wide Association Study Identifies New Loci Involved in Wound-Induced Lateral Root Formation in Arabidopsis Thaliana. Front. Plant Sci. 2019, 10, 311. [Google Scholar] [CrossRef]
- Slovak, R.; Setzer, C.; Roiuk, M.; Bertels, J.; Göschl, C.; Jandrasits, K.; Beemster, G.T.S.; Busch, W. Ribosome Assembly Factor Adenylate Kinase 6 Maintains Cell Proliferation and Cell Size Homeostasis during Root Growth. New Phytol. 2020, 225, 2064–2076. [Google Scholar] [CrossRef]
- Lachowiec, J.; Shen, X.; Queitsch, C.; Carlborg, Ö. A Genome-Wide Association Analysis Reveals Epistatic Cancellation of Additive Genetic Variance for Root Length in Arabidopsis Thaliana. PLoS Genet. 2015, 11, e1005541. [Google Scholar] [CrossRef]
- Meijón, M.; Satbhai, S.B.; Tsuchimatsu, T.; Busch, W. Genome-Wide Association Study Using Cellular Traits Identifies a New Regulator of Root Development in Arabidopsis. Nat. Genet. 2014, 46, 77–81. [Google Scholar] [CrossRef]
- Baluska, F.; Volkmann, D.; Barlow, P.W. Specialized Zones of Development in Roots: View from the Cellular Level. Plant Physiol. 1996, 112, 3–4. [Google Scholar] [CrossRef]
- Verbelen, J.P.; De Cnodder, T.; Le, J.; Vissenberg, K.; Baluška, F. The Root Apex of Arabidopsis Thaliana Consists of Four Distinct Zones of Growth Activities: Meristematic Zone, Transition Zone, Fast Elongation Zone and Growth Terminating Zone; Taylor & Francis: Oxford, UK, 2006; Volume 1. [Google Scholar]
- Ivanov, V.B.; Dubrovsky, J.G. Longitudinal Zonation Pattern in Plant Roots: Conflicts and Solutions. Trends Plant Sci. 2013, 18, 237–243. [Google Scholar] [CrossRef]
- Salvi, E.; Rutten, J.P.; Di Mambro, R.; Polverari, L.; Licursi, V.; Negri, R.; Dello Ioio, R.; Sabatini, S.; Ten Tusscher, K. A Self-Organized PLT/Auxin/ARR-B Network Controls the Dynamics of Root Zonation Development in Arabidopsis Thaliana. Dev. Cell 2020, 53, 431–443.e23. [Google Scholar] [CrossRef] [PubMed]
- Dolan, L.; Janmaat, K.; Willemsen, V.; Linstead, P.; Poethig, S.; Roberts, K.; Scheres, B. Cellular Organisation of the Arabidopsis Thaliana Root. Development 1993, 119, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, H.; Umeda, M. Hormonal Control of Cell Division and Elongation along Differentiation Trajectories in Roots. J. Exp. Bot. 2014, 65, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Chaiwanon, J.; Wang, Z.-Y. Spatiotemporal Brassinosteroid Signaling and Antagonism with Auxin Pattern Stem Cell Dynamics in Arabidopsis Roots HHS Public Access Author Manuscript. Curr. Biol. 2015, 25, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Garay-Arroyo, A.; Ortiz-Moreno, E.; De La Paz Sánchez, M.; Murphy, A.S.; García-Ponce, B.; Marsch-Martínez, N.; De Folter, S.; Corvera-Poiré, A.; Jaimes-Miranda, F.; Pacheco-Escobedo, M.A.; et al. The MADS Transcription Factor XAL2/AGL14 Modulates Auxin Transport during Arabidopsis Root Development by Regulating PIN Expression. EMBO J. 2013, 32, 2884–2895. [Google Scholar] [CrossRef]
- Tapia-Ló Pez, R.; García-Ponce, B.; Dubrovsky, J.G.; Garay-Arroyo, A.; Pérez-Ruíz, R.V.; Kim, S.-H.; Acevedo, F.; Pelaz, S.; Alvarez-Buylla, E.R. An AGAMOUS-Related MADS-Box Gene, XAL1 (AGL12), Regulates Root Meristem Cell Proliferation and Flowering Transition in Arabidopsis. Plant Physiol. 2008, 146, 1182–1192. [Google Scholar] [CrossRef]
- Ubeda-Tomás, S.; Federici, F.; Casimiro, I.; Beemster, G.T.S.; Bhalerao, R.; Swarup, R.; Doerner, P.; Haseloff, J.; Bennett, M.J. Gibberellin Signaling in the Endodermis Controls Arabidopsis Root Meristem Size. Curr. Biol. 2009, 19, 1194–1199. [Google Scholar] [CrossRef]
- Cajero-Sanchez, W.; Aceves-Garcia, P.; Fernández-Marcos, M.; Gutiérrez, C.; Rosas, U.; García-Ponce, B.; Álvarez-Buylla, E.R.; Sánchez, M.d.l.P.; Garay-Arroyo, A. Natural Root Cellular Variation in Responses to Osmotic Stress in Arabidopsis Thaliana Accessions. Genes 2019, 10, 983. [Google Scholar] [CrossRef]
- Li, M.; Wang, M.; Lin, Q.; Wang, M.; Niu, X.; Cheng, J.; Xu, M.; Qin, Y.; Liao, X.; Xu, J.; et al. Symplastic Communication in the Root Cap Directs Auxin Distribution to Modulate Root Development. JIPB 2022, 64, 859–870. [Google Scholar] [CrossRef]
- Napsucialy-Mendivil, S.; Alvarez-Venegas, R.; Shishkova, S.; Dubrovsky, J.G. ARABIDOPSIS HOMOLOG of TRITHORAX1 (ATX1) Is Required for Cell Production, Patterning, and Morphogenesis in Root Development. J. Exp. Bot. 2014, 65, 6373–6384. [Google Scholar] [CrossRef]
- Street, I.H.; Aman, S.; Zubo, Y.; Ramzan, A.; Wang, X.; Shakeel, S.N.; Kieber, J.J.; Eric Schaller, G. Ethylene Inhibits Cell Proliferation of the Arabidopsis Root Meristem. Plant Physiol. 2015, 169, 338–350. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Sun, J.; Li, B.; Zhu, Q.; Chen, S.; Zhang, H. Arabidopsis Fatty Acid Desaturase FAD2 Is Required for Salt Tolerance during Seed Germination and Early Seedling Growth. PLoS ONE 2012, 7, e30355. [Google Scholar] [CrossRef]
- Ratner, B. The Correlation Coefficient: Its Values Range between +1/−1, or Do They? J. Target. Meas. Anal. Mark. 2009, 17, 139–142. [Google Scholar] [CrossRef]
- Horton, M.W.; Hancock, A.M.; Huang, Y.S.; Toomajian, C.; Atwell, S.; Auton, A.; Muliyati, N.W.; Platt, A.; Sperone, F.G.; Vilhjálmsson, B.J.; et al. Genome-Wide Patterns of Genetic Variation in Worldwide Arabidopsis Thaliana Accessions from the RegMap Panel. Nat. Genet. 2012, 44, 212–216. [Google Scholar] [CrossRef]
- Arouisse, B.; Korte, A.; Eeuwijk, F.; Kruijer, W. Imputation of 3 Million SNPs in the Arabidopsis Regional Mapping Population. Plant J. 2020, 102, 872–882. [Google Scholar] [CrossRef]
- LaRue, T.; Lindner, H.; Srinivas, A.; Exposito-Alonso, M.; Lobet, G.; Dinneny, J.R. Uncovering Natural Variation in Root System Architecture and Growth Dynamics Using a Robotics-Assisted Phenomics Platform. eLife 2022, 11, e76968. [Google Scholar] [CrossRef]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The Crosstalks Between Jasmonic Acid and Other Plant Hormone Signaling Highlight the Involvement of Jasmonic Acid as a Core Component in Plant Response to Biotic and Abiotic Stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef]
- Qiao, H.; Chang, K.N.; Yazaki, J.; Ecker, J.R. Interplay between Ethylene, ETP1/ETP2 F-Box Proteins, and Degradation of EIN2 Triggers Ethylene Responses in Arabidopsis. Genes Dev. 2009, 23, 512–521. [Google Scholar] [CrossRef]
- Balazadeh, S.; Kwasniewski, M.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Xue, G.-P.; Mueller-Roeber, B. ORS1, an H2O2-Responsive NAC Transcription Factor, Controls Senescence in Arabidopsis Thaliana. Mol. Plant 2011, 4, 346–360. [Google Scholar] [CrossRef]
- Yan, Z.; Jia, J.; Yan, X.; Shi, H.; Han, Y. Arabidopsis KHZ1 and KHZ2, Two Novel Non-Tandem CCCH Zinc-Finger and K-Homolog Domain Proteins, Have Redundant Roles in the Regulation of Flowering and Senescence. Plant Mol. Biol. 2017, 95, 549–565. [Google Scholar] [CrossRef]
- Lin, W.-D.; Liao, Y.-Y.; Yang, T.J.W.; Pan, C.-Y.; Buckhout, T.J.; Schmidt, W. Coexpression-Based Clustering of Arabidopsis Root Genes Predicts Functional Modules in Early Phosphate Deficiency Signaling. Plant Physiol. 2011, 155, 1383–1402. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Wang, H.; Tang, X. NAC Transcription Factors in Plant Multiple Abiotic Stress Responses: Progress and Prospects. Front. Plant Sci. 2015, 6, 902. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Blanco, C.; Andrade, J.; Becker, C.; Bemm, F.; Bergelson, J.; Borgwardt, K.M.; Cao, J.; Chae, E.; Dezwaan, T.M.; Ding, W.; et al. 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis Thaliana. Cell 2016, 166, 481–491. [Google Scholar] [CrossRef] [PubMed]
- European Union Chromosome 3 Arabidopsis Genome Sequencing Consortium; The Institute for Genomic Research; Kazusa DNA Research Institute Sequence and Analysis of Chromosome 3 of the Plant Arabidopsis Thaliana. Nature 2000, 408, 820–823. [CrossRef]
- Cajero Sánchez, W.; García-Ponce, B.; Sánchez, M. de la P.; Álvarez-Buylla, E.R.; Garay-Arroyo, A. Identifying the Transition to the Maturation Zone in Three Ecotypes of Arabidopsis Thaliana Roots. Commun. Integr. Biol. 2018, 11, e1395993. [Google Scholar] [CrossRef]
- Seren, Ü.; Vilhjálmsson, B.J.; Horton, M.W.; Meng, D.; Forai, P.; Huang, Y.S.; Long, Q.; Segura, V.; Nordborg, M. GWAPP: A Web Application for Genome-Wide Association Mapping in Arabidopsis. Plant Cell 2013, 24, 4793–4805. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Statist. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. ALeaves Facilitates On-Demand Exploration of Metazoan Gene Family Trees on MAFFT Sequence Alignment Server with Enhanced Interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New Tools for the Analysis of Genome-Wide SNP Data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | SNP Position | Maximum Score | MAF | Genes 10 Kb Window | Trait |
|---|---|---|---|---|---|
| 3 | 1174196 | 7.444 * | 0.235 | AT3G04420.1 NAC domain-containing protein 48 (NAC048) AT3G04410.1 NAC domain transcriptional regulator superfamily protein AT3G04430.1 NAC domain-containing protein 49 AT3G04440.1 Plasma-membrane choline transporter family protein AT3G04443.1 hypothetical protein | PR length 4 dps PR length 5 dps PR length 6 dps |
| 3 | 3866768 | 6.815 * | 0.056 | AT3G12120.1 fatty acid desaturase 2 (FAD2) AT3G12130.1 KH domain-containing protein/zinc finger (CCCH type) family protein. AT3G12140.1 Emsy N Terminus (ENT)/plant Tudor-like domain-containing protein | PR length 2 dps |
| 3 | 11031439 | 6.777 * | 0.073 | AT3G29034.1 Transmembrane protein AT3G29035.1 NAC domain-containing protein 3 | PR length 6 dps PR length 8 dps PR length 9 dps PR length 10 dps PR length 11 dps PR length 12 dps |
| 4 | 2557492 | 6.650 * | 0.057 | AT4G04990.1 Serine/arginine repetitive matrix-like protein (DUF761) | EZ length |
| 3 | 23146095 | 6.664 * | 0.081 | AT3G62570.1 Tetratricopeptide repeat (TPR)-like superfamily protein AT3G62580.1 Late embryogenesis abundant protein (LEA) family protein AT3G62590.1 alpha/beta-Hydrolases superfamily protein AT3G62600.1 DNAJ heat shock family protein | PR length 2 dps PR length 3 dps |
| 3 | 6519608 | 5.246 | 0.317 | AT3G18900.1 Ternary complex factor MIP1 leucine-zipper protein AT3G18905.1 F-box/associated interaction domain protein AT3G18910.1 EIN2 targeting protein 2 AT3G18930.1 RING/U-box superfamily protein | Growth rate 12 dps |
| 3 | 15777913 | 4.954 | 0.081 | AT3G43960.1 Cysteine proteinases superfamily protein AT3G43970.1 Hypothetical protein AT3G43980.1 Ribosomal protein S14p/S29e family protein AT3G43990.1 Bromo-adjacent homology (BAH) domain-containing protein | Meristem length |
| Gene | Trait | CDS Position | Nucleotide Change | Amino Acid Change | Trait Classification | Percentage of Accessions |
|---|---|---|---|---|---|---|
| AT3G04420.1 NAC domain containing protein 48 | PR length 4 dps | 452 Third exon | Missense variant gGt/gAt | G/D | Short | 16.6% |
| Large | 8.3% | |||||
| 814 Fifth exon | Missense variant Gtg/Ctg | V/L | Short | 0% | ||
| Large | 33.3% | |||||
| AT3G12120.1 Fatty acid desaturase 2 | PR length 2 dps | 1059 First exon | Synonymous variant gtA/gtG | V | Short | 38% |
| Large | 45% | |||||
| AT3G29035.1 NAC domain containing protein 3 | PR length 12 dps | 847 Third exón | Missense variant Ttt/Gtt | F/V | Short | 33.3% |
| Large | 9% | |||||
| AT3G43960.1 Cysteine proteinases superfamily protein | EZ | 224 First exón | Missense variant gAt/gCt | D/A | Short | 14.2% |
| Large | 17.6% | |||||
| AT3G43970.1 hypothetical protein | Meristem length | 45 Second exon | Synonymous variant gcT/gcG | A | Short | 0% |
| Large | 50% | |||||
| AT3G18900.1 ternary complex factor MIP1 leucine-zipper protein | Growth rate | 87 Fourth exon | Missense variant Gga/Cga | G/R | Slow | 21.4% |
| Fast | 55.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Ruiz, B.A.; Quezada-Rodríguez, E.H.; Piñeyro-Nelson, A.; Tovar, H.; García-Ponce, B.; Sánchez, M.d.l.P.; Álvarez-Buylla, E.R.; Garay-Arroyo, A. Combined Approach of GWAS and Phylogenetic Analyses to Identify New Candidate Genes That Participate in Arabidopsis thaliana Primary Root Development Using Cellular Measurements and Primary Root Length. Plants 2022, 11, 3162. https://doi.org/10.3390/plants11223162
López-Ruiz BA, Quezada-Rodríguez EH, Piñeyro-Nelson A, Tovar H, García-Ponce B, Sánchez MdlP, Álvarez-Buylla ER, Garay-Arroyo A. Combined Approach of GWAS and Phylogenetic Analyses to Identify New Candidate Genes That Participate in Arabidopsis thaliana Primary Root Development Using Cellular Measurements and Primary Root Length. Plants. 2022; 11(22):3162. https://doi.org/10.3390/plants11223162
Chicago/Turabian StyleLópez-Ruiz, Brenda Anabel, Elsa H. Quezada-Rodríguez, Alma Piñeyro-Nelson, Hugo Tovar, Berenice García-Ponce, María de la Paz Sánchez, Elena R. Álvarez-Buylla, and Adriana Garay-Arroyo. 2022. "Combined Approach of GWAS and Phylogenetic Analyses to Identify New Candidate Genes That Participate in Arabidopsis thaliana Primary Root Development Using Cellular Measurements and Primary Root Length" Plants 11, no. 22: 3162. https://doi.org/10.3390/plants11223162