

Dynamic Alteration of Microbial Communities of Duckweeds from Nature to Nutrient-Deficient Condition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
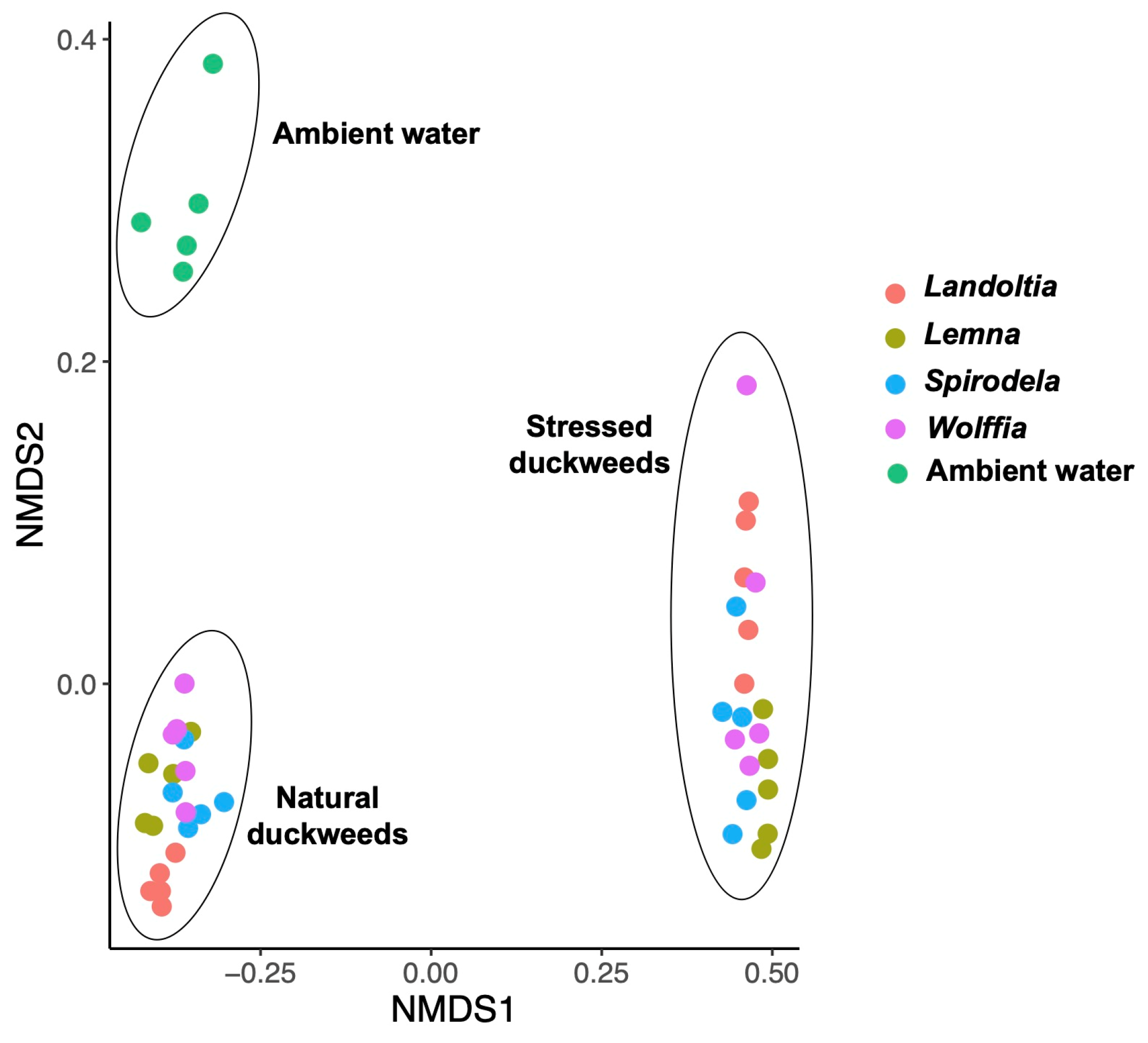
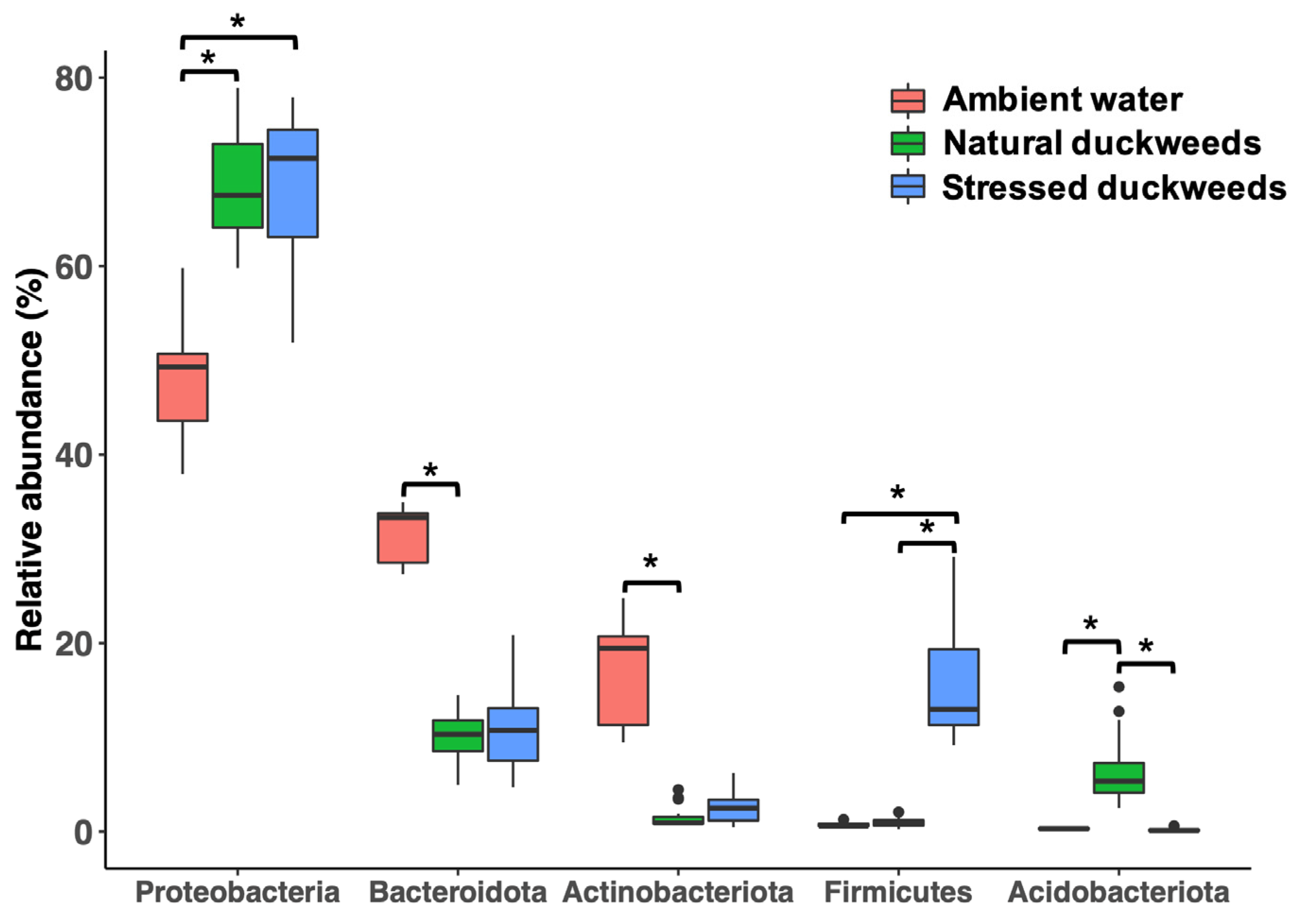
2. Results
2.1. Microbial Diversity and Composition
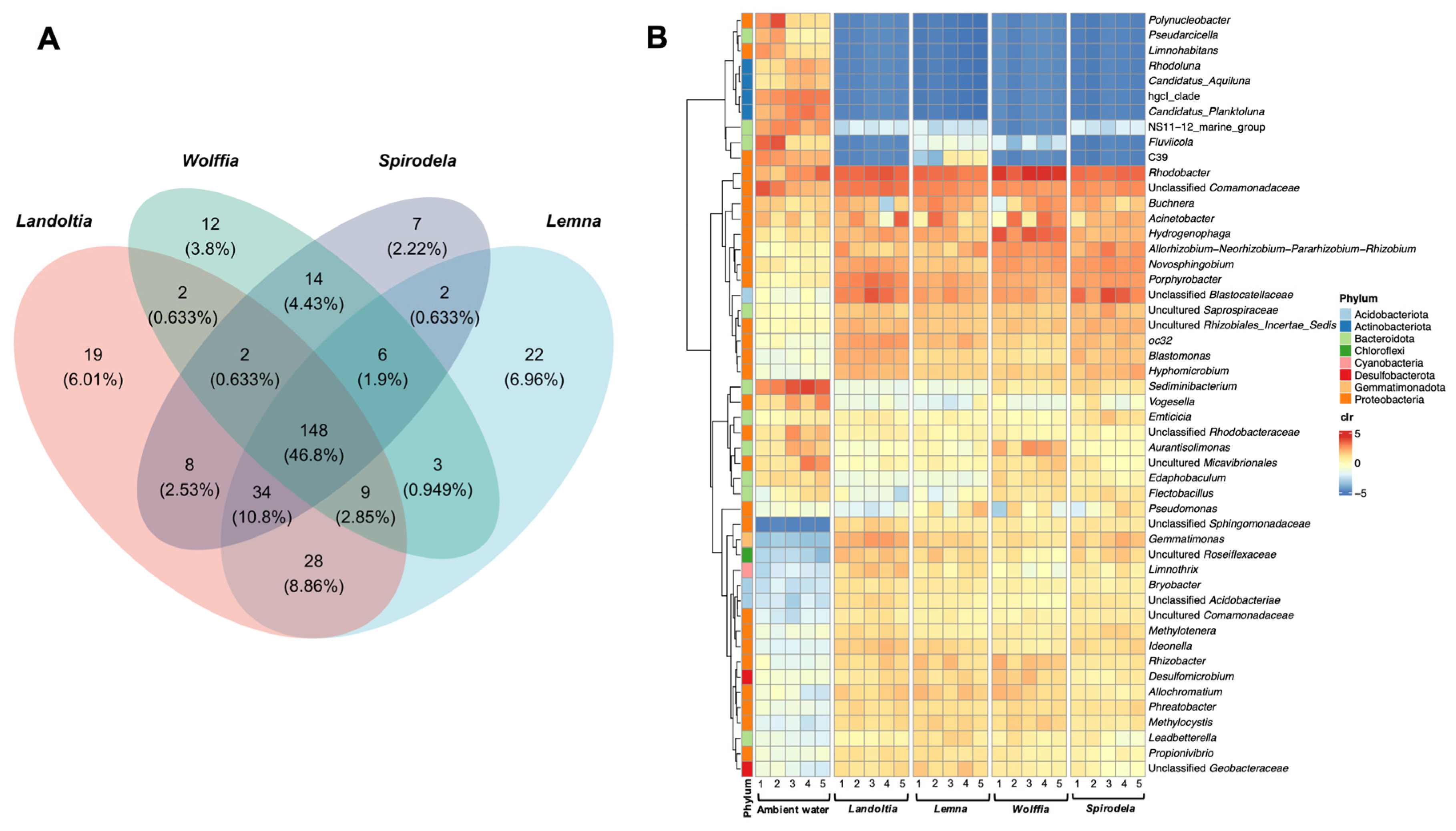
2.2. Core Microbiomes of Natural Duckweed
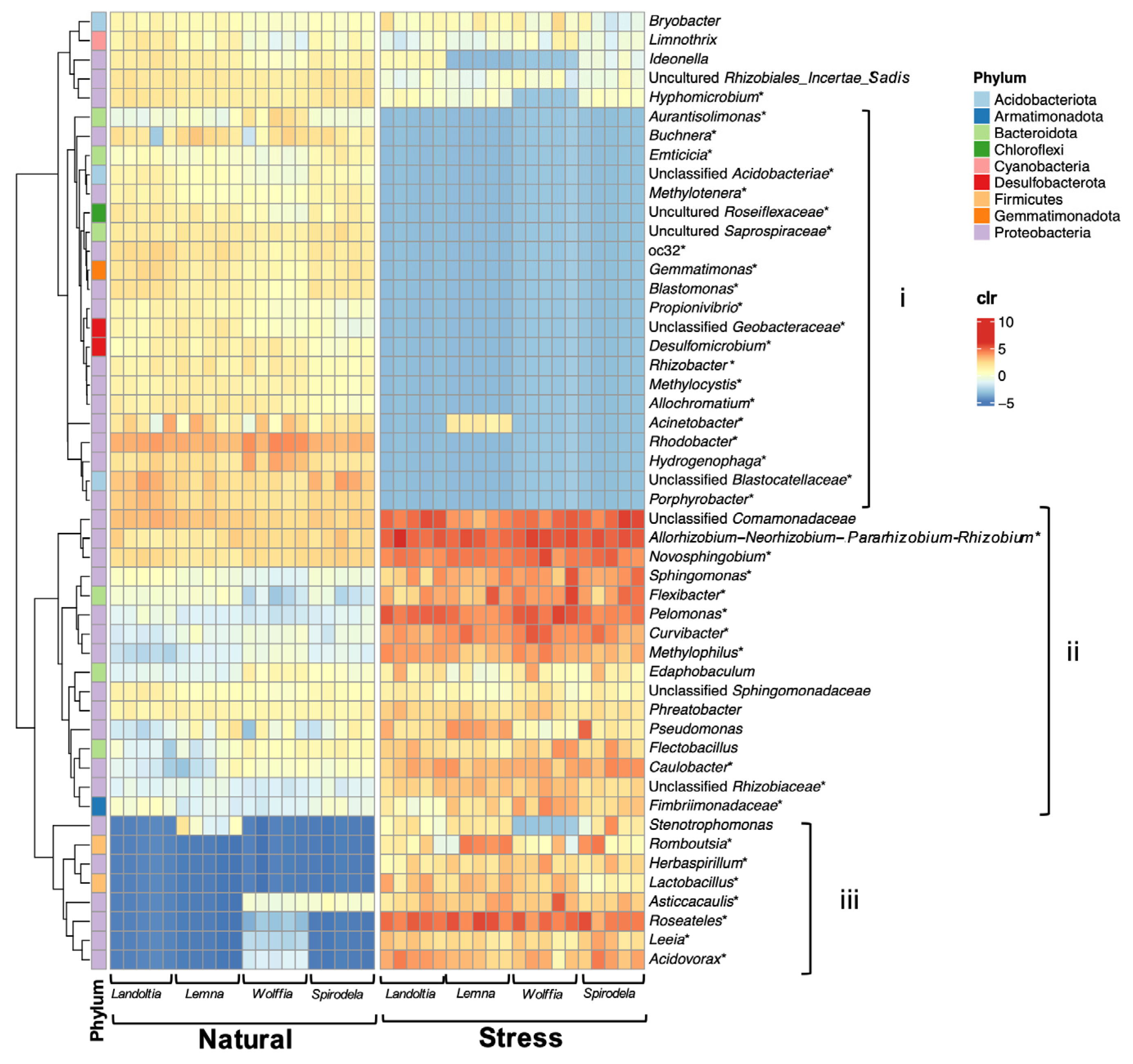
2.3. Nutrient-Deficient Condition Altered Duckweed Core Microbiomes
2.4. Functional Prediction of Microbial Communities of Duckweeds
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Experimental Design for Nutrient-Deficient Condition
4.3. Duckweed Genotyping
4.4. DNA Extraction and Sequencing
4.5. Data Processing and Metagenome Analysis
4.6. Functional Metagenome Prediction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bog, M.; Appenroth, K.-J.; Sree, K.S. Duckweed (Lemnaceae): Its molecular taxonomy. Front. Sustain. Food Syst. 2019, 3, 117. [Google Scholar] [CrossRef]
- Van der Spiegel, M.; Noordam, M.Y.; van der Fels-Klerx, H.J. Safety of novel protein sources (insects, microalgae, seaweed, duckweed, and rapeseed) and legislative aspects for their application in food and feed production. Compr. Rev. Food Sci. Food Saf. 2013, 12, 662–678. [Google Scholar] [CrossRef]
- Cui, W.; Cheng, J.J. Growing duckweed for biofuel production: A review. Plant Biol. 2015, 17 (Suppl S1), 16–23. [Google Scholar] [CrossRef] [PubMed]
- Appenroth, K.J.; Sree, K.S.; Bog, M.; Ecker, J.; Seeliger, C.; Bohm, V.; Lorkowski, S.; Sommer, K.; Vetter, W.; Tolzin-Banasch, K.; et al. Nutritional value of the duckweed species of the genus Wolffia (Lemnaceae) as human food. Front. Chem. 2018, 6, 483. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, J.; Fang, Y.; Zhao, Y.; Tian, X.; Jin, Y.; Zhao, H. Microbial community succession and pollutants removal of a novel carriers enhanced duckweed treatment system for rural wastewater in Dianchi Lake basin. Bioresour. Technol. 2019, 276, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Acosta, K.; Appenroth, K.J.; Borisjuk, L.; Edelman, M.; Heinig, U.; Jansen, M.A.K.; Oyama, T.; Pasaribu, B.; Schubert, I.; Sorrels, S.; et al. Return of the Lemnaceae: Duckweed as a model plant system in the genomics and postgenomics era. Plant Cell 2021, 33, 3207–3234. [Google Scholar] [CrossRef]
- Yamaga, F.; Washio, K.; Morikawa, M. Sustainable biodegradation of phenol by Acinetobacter calcoaceticus P23 isolated from the rhizosphere of duckweed Lemna aoukikusa. Environ. Sci. Technol. 2010, 44, 6470–6474. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.; Xu, J.; Acosta, K.; Poulev, A.; Lebeis, S.; Lam, E. Bacterial production of indole related compounds reveals their role in association between duckweeds and endophytes. Front. Chem. 2018, 6, 265. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, W.; Sugawara, M.; Miwa, K.; Morikawa, M. Plant growth-promoting bacterium Acinetobacter calcoaceticus P23 increases the chlorophyll content of the monocot Lemna minor (duckweed) and the dicot Lactuca sativa (lettuce). J. Biosci. Bioeng. 2014, 118, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, Y.; Jog, R.; Morikawa, M. Effects of co-inoculation of two different plant growth-promoting bacteria on duckweed. Plant Growth Regul. 2018, 86, 287–296. [Google Scholar] [CrossRef]
- Khairina, Y.; Jog, R.; Boonmak, C.; Toyama, T.; Oyama, T.; Morikawa, M. Indigenous bacteria, an excellent reservoir of functional plant growth promoters for enhancing duckweed biomass yield on site. Chemosphere 2021, 268, 129247. [Google Scholar] [CrossRef]
- Acosta, K.; Xu, J.; Gilbert, S.; Denison, E.; Brinkman, T.; Lebeis, S.; Lam, E. Duckweed hosts a taxonomically similar bacterial assemblage as the terrestrial leaf microbiome. PLoS ONE 2020, 15, e0228560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizawa, H.; Kuroda, M.; Inoue, D.; Morikawa, M.; Ike, M. Community dynamics of duckweed-associated bacteria upon inoculation of plant growth-promoting bacteria. FEMS Microbiol. Ecol. 2020, 96, fiaa101. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.M.; Yu, Z.H.; Luo, D.Y.; Laurich, J.; Passeport, E.; Frederickson, M.E. Resilience to multiple stressors in an aquatic plant and its microbiome. Am. J. Bot. 2020, 107, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, O.M.; Castrillo, G.; Herrera Paredes, S.; Salas Gonzalez, I.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, T.; Tanaka, Y.; Tamaki, H.; Yoneda, Y.; Makino, A.; Tateno, Y.; Li, Y.; Toyama, T.; Kamagata, Y.; Mori, K. Comparative analysis of microbial communities in fronds and roots of three duckweed species: Spirodela polyrhiza, Lemna minor, and Lemna aequinoctialis. Microbes Environ. 2020, 35, ME20081. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Hiroshima, N.; Ishizawa, H.; Ike, M. Whole structures, core taxa, and functional properties of duckweed microbiomes. Bioresour. Technol. Rep. 2022, 18, 101060. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Sheldrake, M.; Rosenstock, N.P.; Mangan, S.; Revillini, D.; Sayer, E.J.; Olsson, P.A.; Verbruggen, E.; Tanner, E.V.J.; Turner, B.L.; Wright, S.J. Responses of arbuscular mycorrhizal fungi to long-term inorganic and organic nutrient addition in a lowland tropical forest. ISME J. 2018, 12, 2433–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Gilbert, S.; Poulev, A.; Acosta, K.; Lebeis, S.; Long, C.; Lam, E. Host-specific and tissue-dependent orchestration of microbiome community structure in traditional rice paddy ecosystems. Plant Soil 2020, 452, 379–395. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Herrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2017, 50, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E4284–E4293. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Coleman-Derr, D. Causes and consequences of a conserved bacterial root microbiome response to drought stress. Curr. Opin. Microbiol. 2019, 49, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.K.; Mohammed, M.; Ibny, F.Y.I.; Dakora, F.D. Rhizobia as a source of plant growth-promoting molecules: Potential applications and possible operational mechanisms. Front. Sustain. Food Syst. 2021, 4, 619676. [Google Scholar] [CrossRef]
- Khan, A.L.; Waqas, M.; Kang, S.M.; Al-Harrasi, A.; Hussain, J.; Al-Rawahi, A.; Al-Khiziri, S.; Ullah, I.; Ali, L.; Jung, H.Y.; et al. Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J. Microbiol. 2014, 52, 689–695. [Google Scholar] [CrossRef]
- Rangjaroen, C.; Rerkasem, B.; Teaumroong, N.; Noisangiam, R.; Lumyong, S. Promoting plant growth in a commercial rice cultivar by endophytic diazotrophic bacteria isolated from rice landraces. Ann. Microbiol. 2015, 65, 253–266. [Google Scholar] [CrossRef]
- Banik, A.; Mukhopadhaya, S.K.; Dangar, T.K. Characterization of N2-fixing plant growth promoting endophytic and epiphytic bacterial community of Indian cultivated and wild rice (Oryza spp.) genotypes. Planta 2016, 243, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Gamal-Eldin, H.; Elbanna, K. Field evidence for the potential of Rhodobacter capsulatus as biofertilizer for flooded rice. Curr. Microbiol. 2011, 62, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Iwamoto, Y.; Hirakawa, Y.; Mori, K.; Yamada, N.; Maki, T.; Yamamoto, S.; Miyasaka, H. Plant-growth-promoting effect by cell components of purple non-sulfur photosynthetic bacteria. Microorganisms 2022, 10, 771. [Google Scholar] [CrossRef]
- Staudinger, C.; Mehmeti-Tershani, V.; Gil-Quintana, E.; Gonzalez, E.M.; Hofhansl, F.; Bachmann, G.; Wienkoop, S. Evidence for a rhizobia-induced drought stress response strategy in Medicago truncatula. J. Proteom. 2016, 136, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igiehon, N.O.; Babalola, O.O.; Aremu, B.R. Genomic insights into plant growth promoting rhizobia capable of enhancing soybean germination under drought stress. BMC Microbiol. 2019, 19, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulumello, J.; Chabert, N.; Rodriguez, J.; Long, J.; Nalin, R.; Achouak, W.; Heulin, T. Rhizobium alamii improves water stress tolerance in a non-legume. Sci. Total Environ. 2021, 797, 148895. [Google Scholar] [CrossRef] [PubMed]
- Vives-Peris, V.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Salt stress alleviation in citrus plants by plant growth-promoting rhizobacteria Pseudomonas putida and Novosphingobium sp. Plant Cell Rep. 2018, 37, 1557–1569. [Google Scholar] [CrossRef]
- Makino, A.; Nakai, R.; Yoneda, Y.; Toyama, T.; Tanaka, Y.; Meng, X.-Y.; Mori, K.; Ike, M.; Morikawa, M.; Kamagata, Y.; et al. Isolation of aquatic plant growth-promoting bacteria for the floating plant duckweed (Lemna minor). Microorganisms 2022, 10, 1564. [Google Scholar] [CrossRef]
- Aylward, F.O.; McDonald, B.R.; Adams, S.M.; Valenzuela, A.; Schmidt, R.A.; Goodwin, L.A.; Woyke, T.; Currie, C.R.; Suen, G.; Poulsen, M. Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl. Environ. Microbiol. 2013, 79, 3724–3733. [Google Scholar] [CrossRef] [Green Version]
- Lamont, J.R.; Wilkins, O.; Bywater-Ekegärd, M.; Smith, D.L. From yogurt to yield: Potential applications of lactic acid bacteria in plant production. Soil Biol. Biochem. 2017, 111, 1–9. [Google Scholar] [CrossRef]
- Quattrini, M.; Bernardi, C.; Stuknyte, M.; Masotti, F.; Passera, A.; Ricci, G.; Vallone, L.; De Noni, I.; Brasca, M.; Fortina, M.G. Functional characterization of Lactobacillus plantarum ITEM 17215: A potential biocontrol agent of fungi with plant growth promoting traits, able to enhance the nutritional value of cereal products. Food Res. Int. 2018, 106, 936–944. [Google Scholar] [CrossRef]
- Shah, A.A.; Eguchi, T.; Mayumi, D.; Kato, S.; Shintani, N.; Kamini, N.R.; Nakajima-Kambe, T. Purification and properties of novel aliphatic-aromatic co-polyesters degrading enzymes from newly isolated Roseateles depolymerans strain TB-87. Polym. Degrad. Stab. 2013, 98, 609–618. [Google Scholar] [CrossRef]
- Ishizawa, H.; Kuroda, M.; Inoue, D.; Ike, M. Genome-wide identification of bacterial colonization and fitness determinants on the floating macrophyte, duckweed. Commun. Biol. 2022, 5, 68. [Google Scholar] [CrossRef]
- Allard-Massicotte, R.; Tessier, L.; Lécuyer, F.; Lakshmanan, V.; Lucier, J.-F.; Garneau, D.; Caudwell, L.; Vlamakis, H.; Bais, H.P.; Beauregard, P.B. Bacillus subtilis early colonization of Arabidopsis thaliana roots involves multiple chemotaxis receptors. mBio 2016, 7, e01664-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Sun, D.; Zhu, J.; Liu, W. Two-component signal transduction systems: A major strategy for connecting input stimuli to biofilm formation. Front. Microbiol. 2019, 9, 3279. [Google Scholar] [CrossRef]
- Espinosa-Urgel, M.; Salido, A.; Ramos, J.L. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J. Bacteriol. 2000, 182, 2363–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Gil, M.; Yousef-Coronado, F.; Espinosa-Urgel, M. LapF, the second largest Pseudomonas putida protein, contributes to plant root colonization and determines biofilm architecture. Mol. Microbiol. 2010, 77, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Y.; Yan, Y.; Ermakova, M.; Kerstetter, R.; Messing, J. DNA barcoding of the Lemnaceae, a family of aquatic monocots. BMC Plant Biol. 2010, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Robeson, M.S., 2nd; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- McKnight, D.T.; Huerlimann, R.; Bower, D.S.; Schwarzkopf, L.; Alford, R.A.; Zenger, K.R. microDecon: A highly accurate read-subtraction tool for the post-sequencing removal of contamination in metabarcoding studies. Environ. DNA 2019, 1, 14–25. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Sorensen, T.A. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content and its application to analyses of the vegetation on Danish commons. Biol. Skar. 1948, 5, 1–34. [Google Scholar]
- Anderson, M.J. Permutational multivariate analysis of variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Wiley: Hoboken, NJ, USA, 2017; pp. 1–5. [Google Scholar] [CrossRef]
- Wickham, H. Package ‘ggplot2’: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Volume 10, pp. 970–978. [Google Scholar]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual comparisons by ranking methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunyoo, C.; Roongsattham, P.; Khumwan, S.; Phonmakham, J.; Wonnapinij, P.; Thamchaipenet, A. Dynamic Alteration of Microbial Communities of Duckweeds from Nature to Nutrient-Deficient Condition. Plants 2022, 11, 2915. https://doi.org/10.3390/plants11212915
Bunyoo C, Roongsattham P, Khumwan S, Phonmakham J, Wonnapinij P, Thamchaipenet A. Dynamic Alteration of Microbial Communities of Duckweeds from Nature to Nutrient-Deficient Condition. Plants. 2022; 11(21):2915. https://doi.org/10.3390/plants11212915
Chicago/Turabian StyleBunyoo, Chakrit, Peerapat Roongsattham, Sirikorn Khumwan, Juthaporn Phonmakham, Passorn Wonnapinij, and Arinthip Thamchaipenet. 2022. "Dynamic Alteration of Microbial Communities of Duckweeds from Nature to Nutrient-Deficient Condition" Plants 11, no. 21: 2915. https://doi.org/10.3390/plants11212915