
Evaluating the Rhizosphere and Endophytic Microbiomes of a Bamboo Plant in Response to the Long-Term Application of Heavy Organic Amendment

Abstract
:1. Introduction
2. Results
2.1. Soil Physiochemical Characteristics and C:N:P Stoichiometry
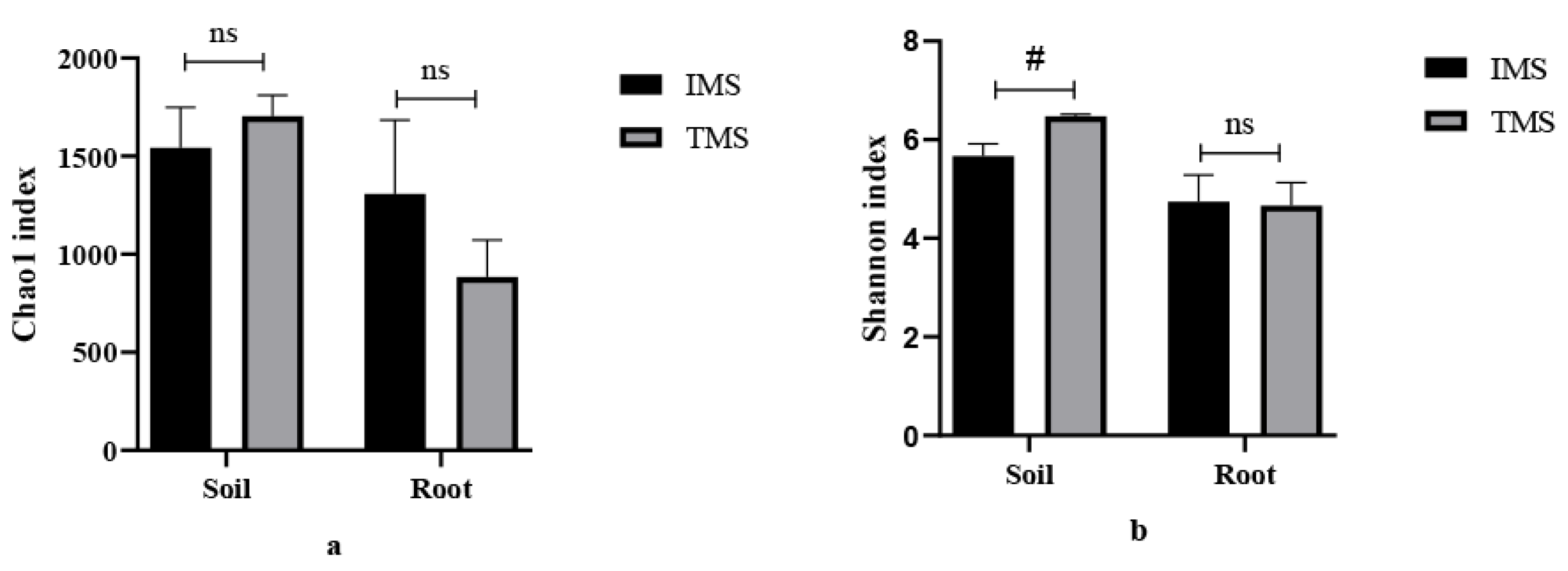
2.2. α-Diversity of Bacterial Communities
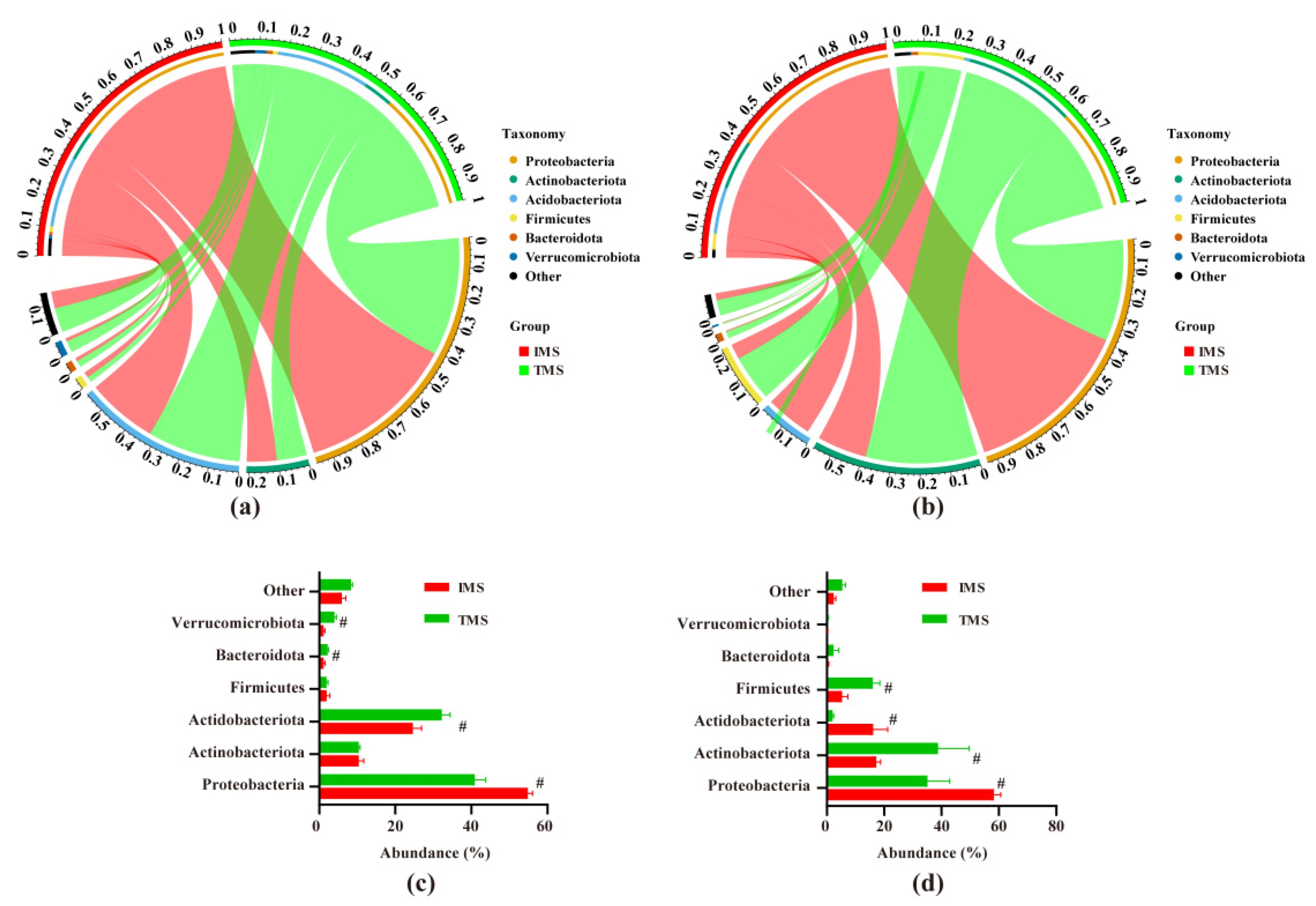
2.3. Compositions of Bacterial Communities
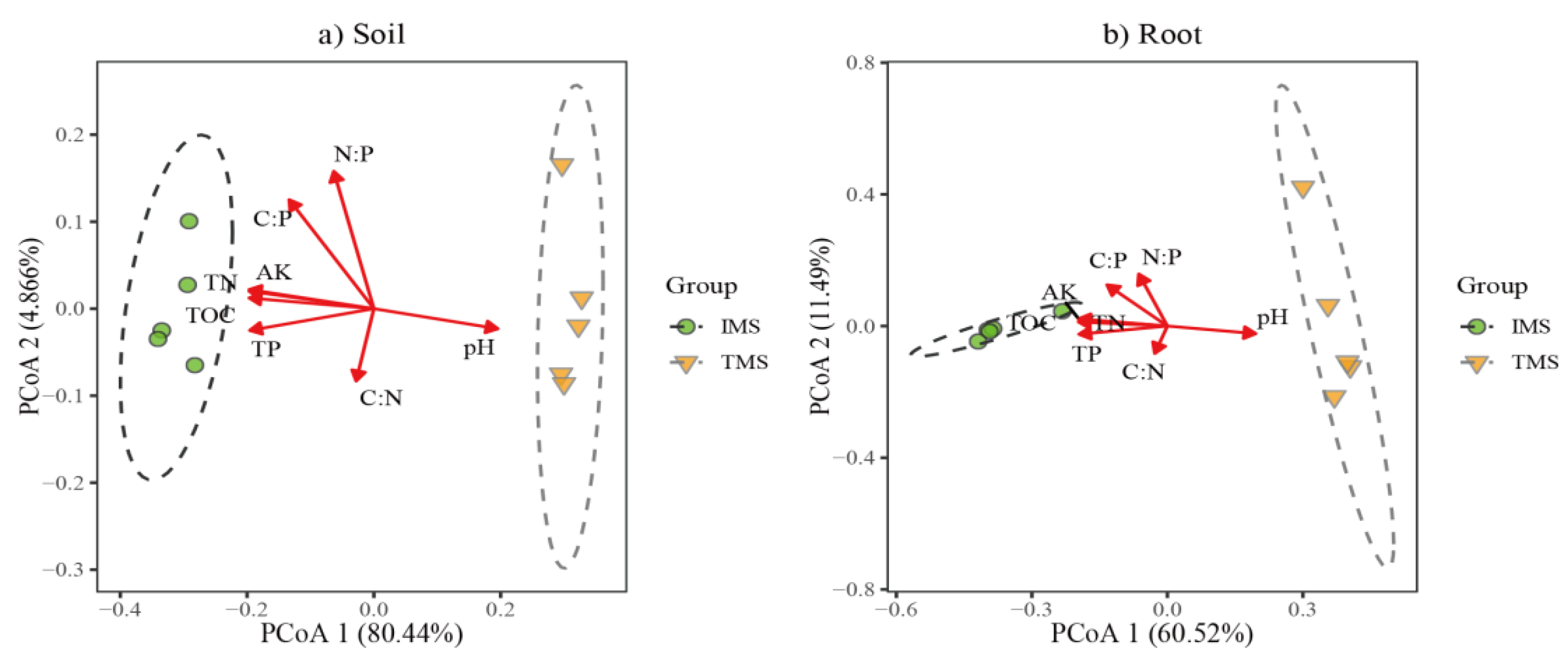
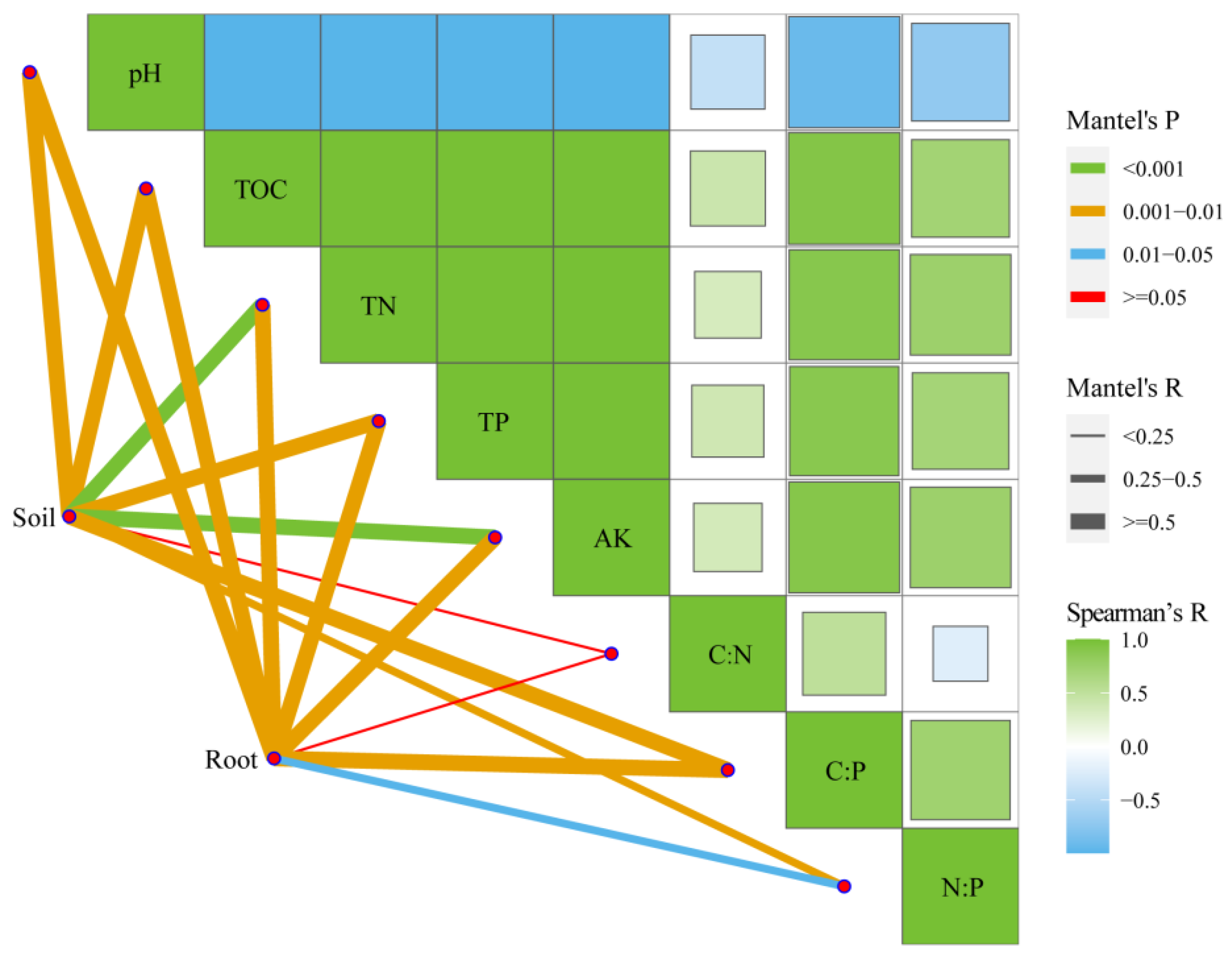
2.4. Factors Driving the Bacterial Communities
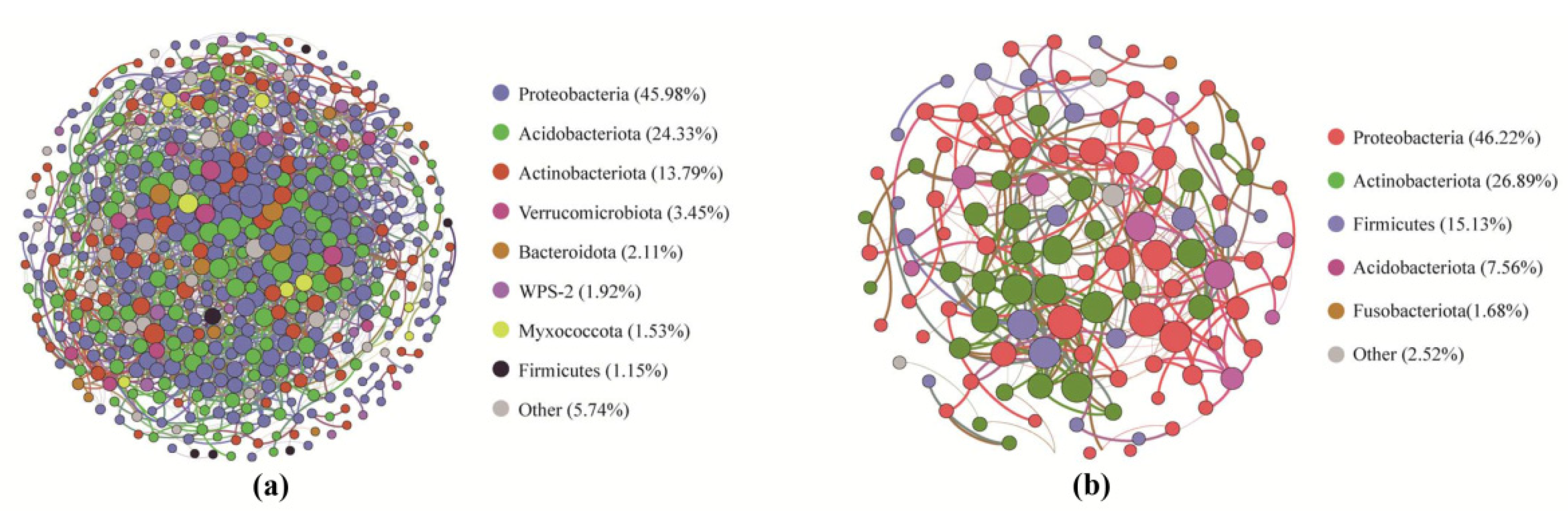
2.5. Properties of Microbial Co-Occurrence Networks
3. Discussion
3.1. Influences of IMS on Soil Physicochemical Properties
3.2. Influences of IMS on the Bamboo Rhizosphere Soil Bacterial Communities Compositions
3.3. Influences of IMS on the Root Endophytic Bacterial Communities Compositions
4. Materials and Methods
4.1. Sample Collection
4.2. Analysis of Soil Physicochemical Properties and Enzyme Activities
4.3. DNA Extraction and Sequencing
4.4. Analysis of Sequencing Data
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, X.; Zhou, G.; Jiang, H.; Yu, S.; Fu, J.; Li, W.; Wang, W.; Ma, Z.; Peng, C. Carbon sequestration by Chinese bamboo forests and their ecological benefits: Assessment of potential, problems, and future challenges. Environ. Rev. 2011, 19, 418–428. [Google Scholar] [CrossRef]
- Fang, W.; He, J.; Lu, X.; Chen, J. Cultivation techniques of early shooting and high yielding for Lei bamboo sprout. J. Zhejiang A F Univ. 1994, 11, 121–128. [Google Scholar]
- Li, Y.; Jiang, P.; Chang, S.X.; Wu, J.; Lin, L. Organic mulch and fertilization affect soil carbon pools and forms under intensively managed bamboo (Phyllostachys praecox) forests in southeast China. J. Soils Sediment. 2010, 10, 739–747. [Google Scholar] [CrossRef]
- Zhai, W.; Zhong, Z.; Gao, G.; Yang, H. Influence of mulching management on soil bacterial structure and diversity in Phyllostachys praecox stands. Sci. Silvae Sin. 2017, 53, 133–142. [Google Scholar]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef]
- Lynch, J. Root architecture and plant productivity. Plant Physiol. 1995, 109, 7–13. [Google Scholar] [CrossRef]
- Castrillo, G.; Teixeira, P.J.P.L.; Paredes, S.H.; Law, T.F.; de Lorenzo, L.; Feltcher, M.E.; Finkel, O.M.; Breakfield, N.W.; Mieczkowski, P.; Jones, C.D.; et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef]
- Qin, Y.; Druzhinina, I.S.; Pan, X.; Yuan, Z. Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 2016, 34, 1245–1259. [Google Scholar] [CrossRef]
- Puri, R.R.; Dangi, S.R.; Dhungana, S.A.; Itoh, K. Diversity and plant growth promoting ability of culturable endophytic bacteria in nepalese sweet potato. Adv. Appl. Microbiol. 2018, 8, 734–761. [Google Scholar] [CrossRef]
- Puri, R.R.; Adachi, F.; Omichi, M.; Saeki, Y.; Yamamoto, A.; Hayashi, S.; Ali, M.A.; Itoh, K. Metagenomic study of endophytic bacterial community of sweet potato (Ipomoea batatas) cultivated in different soil and climatic conditions. World J. Microbiol. Biotechnol. 2019, 35, 176. [Google Scholar] [CrossRef]
- Longley, R.; Noel, Z.A.; Benucci, G.M.N.; Chilvers, M.I.; Trail, F.; Bonito, G. Crop management impacts the soybean (Glycine max) microbiome. Front. Microbiol. 2020, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.-C.; Schlaeppi, K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Fabiańska, I.; Gerlach, N.; Almario, J.; Bucher, M. Plant-mediated effects of soil phosphorus on the root-associated fungal microbiota in Arabidopsis thaliana. New Phytol. 2019, 221, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.d.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ge, Y.; Song, J.; Rensing, C. Assembly of root-associated microbial community of typical rice cultivars in different soil types. Biol. Fertil. Soils 2020, 56, 249–260. [Google Scholar] [CrossRef]
- Edwards, J.A.; Santos-Medellín, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef]
- Cai, R.; Huang, F.; Sun, D.; Qin, H.; Huang, F.; Zhuang, S.; Zhou, G.; Cao, Z. Temporal and spatial variation of soil organic matters in Phyllostachys praecox stands with intensive cultivation management. J. Zhejiang A F Univ. 2007, 24, 450–455. [Google Scholar]
- Wu, G.; Qu, P.; Sun, E.; Chang, Z.; Xu, Y.; Huang, H. Physical, chemical, and rheological properties of rice husks treated by composting process. BioResources 2015, 10, 227–239. [Google Scholar] [CrossRef]
- Cleveland, C.C.; Liptzin, D. C:N:P stoichiometry in soil: Is there a “Redfield ratio” for the microbial biomass? Biogeochemistry 2007, 85, 235–252. [Google Scholar] [CrossRef]
- Tang, C.; Rengel, Z. Role of plant cation/anion uptake ratio in soil acidification. In Handbook of Soil Acidity; CRC Press: Boca Raton, FL, USA, 2003; pp. 71–96. [Google Scholar]
- Ajwa, H.A.; Tabatabai, M.A. Decomposition of different organic materials in soils. Biol. Fertil. Soils 1994, 18, 175–182. [Google Scholar] [CrossRef]
- Tian, H.; Chen, G.; Zhang, C.; Melillo, J.M.; Hall, C.A.S. Pattern and variation of C:N:P ratios in China’s soils: A synthesis of observational data. Biogeochemistry 2010, 98, 139–151. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Q.; Li, S.; Ge, J.; Liang, C.; Qin, H.; Xu, Q.; Fuhrmann, J.J. Diversity and function of soil bacterial communities in response to long-term intensive management in a subtropical bamboo forest. Geoderma 2019, 354, 113894. [Google Scholar] [CrossRef]
- Shi, S.; Tian, L.; Nasir, F.; Bahadur, A.; Batool, A.; Luo, S.; Yang, F.; Wang, Z.; Tian, C. Response of microbial communities and enzyme activities to amendments in saline-alkaline soils. Appl. Soil Ecol. 2019, 135, 16–24. [Google Scholar] [CrossRef]
- Zverev, A.O.; Pershina, E.V.; Shapkin, V.M.; Kichko, A.K.; Mitrofanova, O.P.; Kobylyanskii, V.D.; Yuzikhin, O.S.; Belimov, A.A.; Andronov, E.E. Molecular analysis of the rhizosphere microbial communities from gramineous plants grown on contrasting soils. Microbiology 2020, 89, 231–241. [Google Scholar] [CrossRef]
- Jangid, K.; Williams, M.A.; Franzluebbers, A.J.; Sanderlin, J.S.; Reeves, J.H.; Jenkins, M.B.; Endale, D.M.; Coleman, D.C.; Whitman, W.B. Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol. Biochem. 2008, 40, 2843–2853. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef]
- Van Der Heijden, M.G.A.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Dai, Y.; Tian, H.; Zhou, W.; Lv, J. Soil organic carbon transformation and dynamics of microorganisms under different organic amendments. Sci. Total Environ. 2021, 750, 141719. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, H.; Gao, C. Differential responses of soil bacterial taxa to long-term P, N, and organic manure application. J. Soils Sediments 2016, 16, 1046–1058. [Google Scholar] [CrossRef]
- Sichert, A.; Corzett, C.H.; Schechter, M.S.; Unfried, F.; Markert, S.; Becher, D.; Fernandez-Guerra, A.; Liebeke, M.; Schweder, T.; Polz, M.F.; et al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 2020, 5, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Yeves, P.J.; Ghai, R.; Mehrshad, M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Reconstruction of diverse verrucomicrobial genomes from metagenome datasets of freshwater reservoirs. Front. Microbiol. 2017, 8, 2131. [Google Scholar] [CrossRef]
- Kopáček, J.; Cosby, B.J.; Evans, C.D.; Hruška, J.; Moldan, F.; Oulehle, F.; Šantrůčková, H.; Tahovská, K.; Wright, R.F. Nitrogen, organic carbon and sulphur cycling in terrestrial ecosystems: Linking nitrogen saturation to carbon limitation of soil microbial processes. Biogeochemistry 2013, 115, 33–51. [Google Scholar] [CrossRef]
- Khadem, A.F.; Pol, A.; Jetten, M.S.M.; Op den Camp, H.J.M. Nitrogen fixation by the verrucomicrobial methanotroph methylaidiphilum fumariolicum SolV. Microbiology 2010, 156, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Pol, A.; Jetten, M.; Op den Camp, H.J.M. Ammonia oxidation and nitrite reduction in the verrucomicrobial methanotroph Methylacidiphilum fumariolicum SolV. Front. Microbiol. 2017, 8, 1901. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, G.; Wu, Z.; Wen, X.; Zhong, H.; Zhong, Z.; Yang, C.; Bian, F.; Gai, X. Responses of soil nutrients and microbial communities to intercropping medicinal plants in Moso bamboo plantations in subtropical China. Environ. Sci. Pollut. Res. 2020, 27, 2301–2310. [Google Scholar] [CrossRef]
- Qi, D.; Wieneke, X.; Tao, J.; Zhou, X.; Desilva, U. Soil ph is the primary factor correlating with soil microbiome in karst rocky desertification regions in the Wushan County, Chongqing, China. Front. Microbiol. 2018, 9, 1027. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Y.; Yang, T.; Chu, H. Threshold effects of soil pH on microbial co-occurrence structure in acidic and alkaline arable lands. Sci. Total Environ. 2021, 800, 149592. [Google Scholar] [CrossRef]
- Morriën, E.; Hannula, S.; Snoek, L.; Helmsing, N.; Zweers, A.j.; Hollander, M.; Lujan Soto, R.; Bouffaud, M.-L.; Buée, M.; Dimmers, W.J.; et al. Soil networks become more connected and take up more carbon as nature restoration progresses. Nat. Commun. 2017, 8, 14349. [Google Scholar] [CrossRef]
- Fan, K.; Weisenhorn, P.; Jack, G.; Shi, Y.; Bai, Y.; Chu, H. Soil ph correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Soil Biol. Biochem. 2018, 121, 185–192. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, Z.; Gai, X.; Du, X.; Bian, F.; Yang, C.; Gao, G.; Wen, X. Changes of root endophytic bacterial community along a chronosequence of intensively managed Lei bamboo (Phyllostachys praecox) forests in subtropical China. Microorganisms 2019, 7, 616. [Google Scholar] [CrossRef]
- Tang, M.; Liu, J.; Hou, W.; Stubbendieck, R.M.; Xiong, H.; Jin, J.; Gong, J.; Cheng, C.; Tang, X.; Liu, Y.; et al. Structural variability in the bulk soil, rhizosphere, and root endophyte fungal communities of Themeda japonica plants under different grades of karst rocky desertification. Plant Soil 2021, 475, 105–122. [Google Scholar] [CrossRef]
- Gomes, T.; Pereira, J.A.; Benhadi, J.; Lino-Neto, T.; Baptista, P. Endophytic and epiphytic phyllosphere fungal communities are shaped by different environmental factors in a mediterranean ecosystem. Microb. Ecol. 2018, 76, 668–679. [Google Scholar] [CrossRef]
- Estendorfer, J.; Stempfhuber, B.; Haury, P.; Vestergaard, G.; Rillig, M.C.; Joshi, J.; Schröder, P.; Schloter, M. The influence of land use intensity on the plant-associated microbiome of Dactylis glomerata L. Front. Plant Sci. 2017, 8, 930. [Google Scholar] [CrossRef]
- Zhang, X.; Gai, X.; Yang, C.; Ying, J.; Li, W.; Du, X.; Zhong, Z.; Shao, Q.; Bian, F. Effects of chicken farming on soil properties and root-associated bacterial communities in a bamboo (Phyllostachys praecox) ecosystem. Appl. Soil Ecol. 2021, 157, 103725. [Google Scholar] [CrossRef]
- Rahimlou, S.; Bahram, M.; Tedersoo, L. Phylogenomics reveals the evolution of root nodulating α- and β-proteobacteria (rhizobia). Microbiol. Res. 2021, 250, 126788. [Google Scholar] [CrossRef]
- del Barrio-Duque, A.; Samad, A.; Nybroe, O.; Antonielli, L.; Sessitsch, A.; Compant, S. Interaction between endophytic proteobacteria strains and Serendipita indica enhances biocontrol activity against fungal pathogens. Plant Soil 2020, 451, 277–305. [Google Scholar] [CrossRef]
- Solaiyappan Mani, S.; Reinhold-Hurek, B. RNA-seq provides new insights into the gene expression changes in Azoarcus olearius BH72 under nitrogen-deficient and replete conditions beyond the nitrogen fixation process. Microorganisms 2021, 9, 1888. [Google Scholar] [CrossRef]
- Williams Kelly, P.; Sobral Bruno, W.; Dickerman Allan, W. A robust species tree for the alphaproteobacteria. J. Bacteriol. 2007, 189, 4578–4586. [Google Scholar] [CrossRef]
- Yoneda, Y.; Yamamoto, K.; Makino, A.; Tanaka, Y.; Meng, X.-Y.; Hashimoto, J.; Shin-ya, K.; Satoh, N.; Fujie, M.; Toyama, T.; et al. Novel plant-associated acidobacteria promotes growth of common floating aquatic plants, duckweeds. Microorganisms 2021, 9, 1133. [Google Scholar] [CrossRef]
- Kielak, A.M.; Cipriano, M.A.P.; Kuramae, E.E. Acidobacteria strains from subdivision 1 act as plant growth-promoting bacteria. Arch. Microbiol. 2016, 198, 987–993. [Google Scholar] [CrossRef]
- Ranjeet, K.T.; Janice, L.S.; Carina, M.J.; Don, L.C.; Michelle, H.S.; Lee, A.D.; Bailey, J.F.; Morra, M.J. Novel plant-microbe rhizosphere interaction involving Streptomyces lydicus WYEC108 and the pea plant (Pisum sativum). Appl. Environ. Microbiol. 2002, 68, 2161–2171. [Google Scholar]
- Musa, Z.; Ma, J.; Egamberdieva, D.; Abdelshafy Mohamad, O.A.; Abaydulla, G.; Liu, Y.; Li, W.-J.; Li, L. Diversity and antimicrobial potential of cultivable endophytic actinobacteria associated with the medicinal plant Thymus roseus. Front. Microbiol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Shimizu, M.; Yazawa, S.; Ushijima, Y. A promising strain of endophytic Streptomyces sp. for biological control of cucumber anthracnose. J. Gen. Plant Pathol. 2009, 75, 27–36. [Google Scholar] [CrossRef]
- Velloso, C.C.V.; Ribeiro, V.P.; de Carvalho, C.G.; de Oliveira, C.A.; de Paula Lana, U.G.; Marriel, I.E.; de Sousa, S.M.; Gomes, E.A. Tropical endophytic bacillus species enhance plant growth and nutrient uptake in cereals. In Endophytes: Mineral Nutrient Management, Volume 3; Maheshwari, D.K., Dheeman, S., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 157–180. [Google Scholar]
- Bokhari, A.; Essack, M.; Lafi, F.F.; Andres-Barrao, C.; Jalal, R.; Alamoudi, S.; Razali, R.; Alzubaidy, H.; Shah, K.H.; Siddique, S.; et al. Bioprospecting desert plant Bacillus endophytic strains for their potential to enhance plant stress tolerance. Sci. Rep. 2019, 9, 18154. [Google Scholar] [CrossRef]
- Chen, S. Ecological Adaptation of Senescence Mechanism of the Major Organs of Phyllostachys Violascens to Mulching Management; Chinese Academy of Forestry: Beijing, China, 2014. [Google Scholar]
- Santolini, M.; Barabási, A.-L. Predicting perturbation patterns from the topology of biological networks. Proc. Natl. Acad. Sci. USA 2018, 115, E6375–E6383. [Google Scholar] [CrossRef]
- Lu, R.K. Soil and Agro-Chemical Analytical Methods; China Agricultural Science and Technology Press: Beijing, China, 1999; pp. 146–195. [Google Scholar]
- Beckers, B.; Op De Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using qiime 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Dixon, P. Vegan, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Huang, H.Y.; Zhou, L.; Chen, J.; Wei, T. Ggcor: Extended Tools for Correlation Analysis and Visualization; R Package Version: 0.7.6; R Core Team: Vienna, Austria, 2020; p. 7. [Google Scholar]
- Langfelder, P.; Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media (ICWSM-09), San Jose, CA, USA, 17–20 May 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMS | TMS | |
|---|---|---|
| pH | 4.47 ± 0.01 b | 4.74 ± 0.01 a |
| TOC (g/kg) | 87.54 ± 2.96 a | 36.92 ± 0.92 b |
| TN (g/kg) | 6.22 ± 0.17 a | 2.73 ± 0.13 b |
| TP (g/kg) | 0.95 ± 0.03 a | 0.46 ± 0.02 b |
| AK (mg/kg) | 296.82 ± 6.06 a | 79.16 ± 5.21 b |
| C:N | 14.09 ± 0.51 a | 13.55 ± 0.92 a |
| C:P | 92.01 ± 3.24 a | 80.42 ± 2.67 b |
| N:P | 6.53 ± 0.14 a | 5.96 ± 0.41 b |
| Soil | Root | |||
|---|---|---|---|---|
| IMS | TMS | IMS | TMS | |
| Nodes | 1064 | 1232 | 381 | 222 |
| Edges | 6419 | 8225 | 1643 | 462 |
| Average degree | 12.066 | 13.352 | 8.625 | 4.162 |
| Modularity | 1.389 | 1.375 | 1.354 | 3.631 |
| Graph density | 0.011 | 0.011 | 0.023 | 0.019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Huang, Z.; Zhong, Z.; Li, Q.; Bian, F.; Gao, G.; Yang, C.; Wen, X. Evaluating the Rhizosphere and Endophytic Microbiomes of a Bamboo Plant in Response to the Long-Term Application of Heavy Organic Amendment. Plants 2022, 11, 2129. https://doi.org/10.3390/plants11162129
Zhang X, Huang Z, Zhong Z, Li Q, Bian F, Gao G, Yang C, Wen X. Evaluating the Rhizosphere and Endophytic Microbiomes of a Bamboo Plant in Response to the Long-Term Application of Heavy Organic Amendment. Plants. 2022; 11(16):2129. https://doi.org/10.3390/plants11162129
Chicago/Turabian StyleZhang, Xiaoping, Zhiyuan Huang, Zheke Zhong, Qiaoling Li, Fangyuan Bian, Guibin Gao, Chuanbao Yang, and Xing Wen. 2022. "Evaluating the Rhizosphere and Endophytic Microbiomes of a Bamboo Plant in Response to the Long-Term Application of Heavy Organic Amendment" Plants 11, no. 16: 2129. https://doi.org/10.3390/plants11162129