
The Hidden Potential of High-Throughput RNA-Seq Re-Analysis, a Case Study for DHDPS, Key Enzyme of the Aspartate-Derived Lysine Biosynthesis Pathway and Its Role in Abiotic and Biotic Stress Responses in Soybean



Abstract
:1. Introduction
2. Results
2.1. Phylogenetic Analysis of DHDPS in Plants
2.2. RNA-Seq Re-Analysis Method Validation
2.3. Novel DHDPS Expression Atlas Data by RNA-Seq Re-Analysis
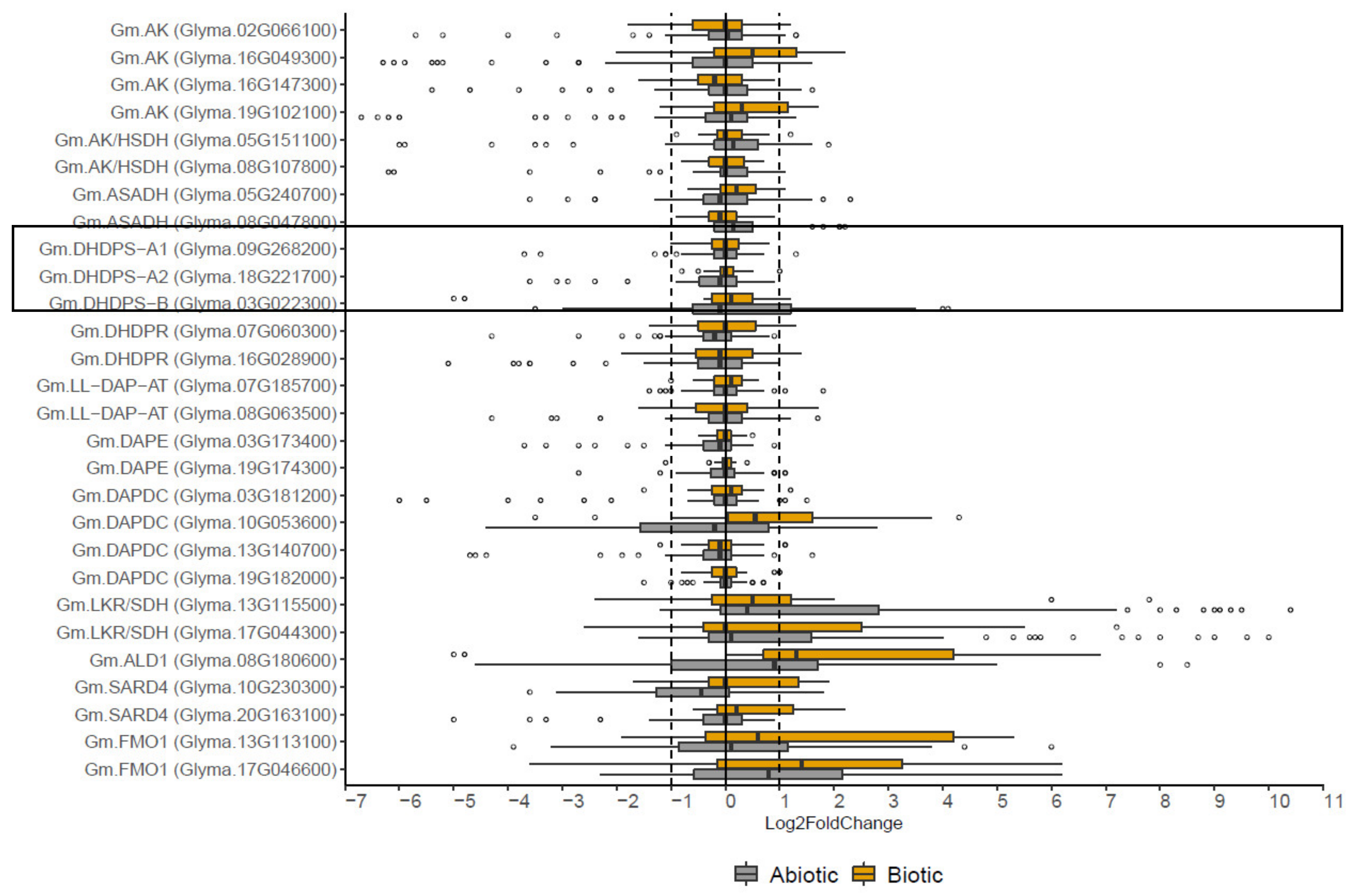
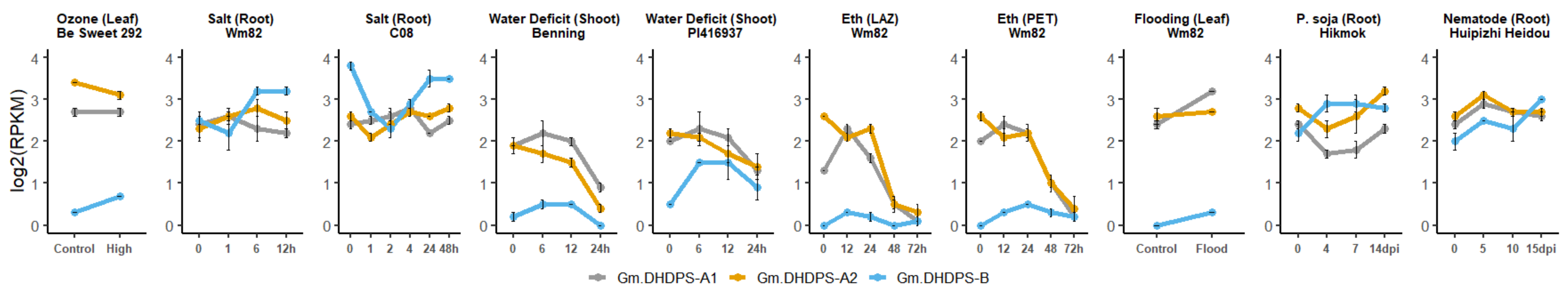
2.4. The Effect of Biotic and Abiotic Stress on the Expression of DHDPS and Other Genes Involved in the Aspartate-Derived Lys Biosynthesis and Catabolic Pathway
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Analysis of DHDPS
4.2. Identification of Enzymes Involved in Lysine Biosynthesis and Catabolism Other than DHDPS
4.3. RNA-Seq Data Re-Analysis and Differential Gene Expression Analysis
4.4. Statistical Analysis and Graphic Representations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jander, G.; Joshi, V. Recent Progress in Deciphering the Biosynthesis of Aspartate-Derived Amino Acids in Plants. Mol. Plant 2010, 3, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Blickling, S.; Knäblein, J. Feedback inhibition of dihydrodipicolinate synthase enzymes by L-lysine. Biol. Chem. 1997, 378, 207–210. [Google Scholar] [PubMed]
- Galili, G. New insights into the regulation and functional significance of lysine metabolism in plants. Annu. Rev. Plant Biol. 2002, 53, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Arruda, P.; Kemper, E.L.; Papes, F.; Leite, A. Regulation of lysine catabolism in higher plants. Trends Plant Sci. 2000, 5, 324–330. [Google Scholar] [CrossRef]
- Galili, G.; Tang, G.; Zhu, X.; Gakiere, B. Lysine catabolism: A stress and development super-regulated metabolic pathway. Curr. Opin. Plant Biol. 2001, 4, 261–266. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, Y. The Emergence of a Mobile Signal for Systemic Acquired Resistance. Plant Cell 2019, 31, 1414–1415. [Google Scholar] [CrossRef] [Green Version]
- Song, J.T. Divergent Roles in Arabidopsis thaliana Development and Defense of Two Homologous Genes, ABERRANT GROWTH AND DEATH2 and AGD2-LIKE DEFENSE RESPONSE PROTEIN1, Encoding Novel Aminotransferases. Plant Cell 2004, 16, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Ding, P.; Rekhter, D.; Ding, Y.; Feussner, K.; Busta, L.; Haroth, S.; Xu, S.; Li, X.; Jetter, R.; Feussner, I.; et al. Characterization of a Pipecolic Acid Biosynthesis Pathway Required for Systemic Acquired Resistance. Plant Cell 2016, 28, 2603–2615. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Zeier, J. l-lysine metabolism to N-hydroxypipecolic acid: An integral immune-activating pathway in plants. Plant J. 2018, 96, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Bernsdorff, F.; Döring, A.-C.; Gruner, K.; Schuck, S.; Bräutigam, A.; Zeier, J. Pipecolic Acid Orchestrates Plant Systemic Acquired Resistance and Defense Priming via Salicylic Acid-Dependent and -Independent Pathways. Plant Cell 2016, 28, 102–129. [Google Scholar] [CrossRef] [Green Version]
- Cheshire, R.M.; Miflin, B.J. The control of lysine biosynthesis in maize. Phytochemistry 1975, 14, 695–698. [Google Scholar] [CrossRef]
- Mazelis, M.; Whatley, F.R.; Whatley, J. The enzymology of lysine biosynthesis in higher plants The occurrence, characterization and some regulatory properties of dihydrodipicolinate synthase. FEBS Lett. 1977, 84, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Dereppe, C.; Bold, G.; Ghisalba, O.; Ebert, E.; Schär, H.-P. Purification and Characterization of Dihydrodipicolinate Synthase from Pea. Plant Physiol. 1992, 98, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Silk, G.W.; Matthews, B.F.; Somers, D.A.; Gengenbach, B.G. Cloning and expression of the soybean dapa gene encoding dihydrodipicolinate synthase. Plant Mol. Biol. 1994, 26, 989–993. [Google Scholar] [CrossRef]
- Vauterin, M.; Jacobs, M. Isolation of a poplar and an Arabidopsis thaliana dihydrodipicolinate synthase cDNA clone. Plant Mol. Biol. 1994, 25, 545–550. [Google Scholar] [CrossRef]
- Dante, R.A.; Neto, G.C.; Leite, A.; Yunes, J.A.; Arruda, P. The DapA gene encoding the lysine biosynthetic enzyme dihydrodipicolinate synthase from Coix lacryma-jobi: Cloning, characterization, and expression analysis. Plant Mol. Biol. 1999, 41, 551–561. [Google Scholar] [CrossRef]
- Kong, F.N.; Jiang, S.M.; Meng, X.B.; Song, C.L.; Shi, J.F.; Jin, D.M.; Jiang, S.L.; Wang, B. Cloning and Characterization of the DHDPS Gene Encoding the Lysine Biosynthetic Enzyme Dihydrodipocolinate Synthase from Zizania latifolia (Griseb). Plant Mol. Biol. Report. 2009, 27, 199–208. [Google Scholar] [CrossRef]
- Atkinson, S.C.; Dogovski, C.; Newman, J.; Dobson, R.C.; Perugini, M.A. Cloning, expression, purification and crystallization of dihydrodipicolinate synthase from the grapevine Vitis vinifera. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1537–1541. [Google Scholar] [CrossRef] [Green Version]
- Erzeel, E.; Van Bochaute, P.; Thu, T.T.; Angenon, G. Medicago truncatula dihydrodipicolinate synthase (DHDPS) enzymes display novel regulatory properties. Plant Mol. Biol. 2013, 81, 401–415. [Google Scholar] [CrossRef]
- Blickling, S.; Beisel, H.G.; Bozic, D.; Knäblein, J.; Laber, B.; Huber, R. Structure of dihydrodipicolinate synthase of Nicotiana sylvestris reveals novel quaternary structure. J. Mol. Biol. 1997, 274, 608–621. [Google Scholar] [CrossRef]
- Atkinson, S.C.; Dogovski, C.; Downton, M.T.; Czabotar, P.E.; Dobson, R.C.J.; Gerrard, J.A.; Wagner, J.; Perugini, M.A. Structural, kinetic and computational investigation of Vitis vinifera DHDPS reveals new insight into the mechanism of lysine-mediated allosteric inhibition. Plant Mol. Biol. 2013, 81, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.D.; Billakanti, J.M.; Wason, A.; Keller, S.; Mertens, H.D.; Atkinson, S.C.; Dobson, R.C.; Perugini, M.A.; Gerrard, J.A.; Pearce, F.G. Characterisation of the first enzymes committed to lysine biosynthesis in Arabidopsis thaliana. PLoS ONE 2012, 7, e40318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.J.; Lee, M.; Boarder, M.P.; Mangion, A.M.; Gendall, A.R.; Panjikar, S.; Perugini, M.A.; Soares Da Costa, T.P. Differential Lysine-Mediated Allosteric Regulation of Plant Dihydrodipicolinate Synthase Isoforms. FEBS J. 2021, 288, 4973–4986. [Google Scholar] [CrossRef] [PubMed]
- Soares da Costa, T.P.; Muscroft-Taylor, A.C.; Dobson, R.C.; Devenish, S.R.; Jameson, G.B.; Gerrard, J.A. How essential is the ‘essential’ active-site lysine in dihydrodipicolinate synthase? Biochimie 2010, 92, 837–845. [Google Scholar] [CrossRef]
- Da Costa, T.P.S.; Desbois, S.; Dogovski, C.; Gorman, M.A.; Ketaren, N.E.; Paxman, J.J.; Siddiqui, T.; Zammit, L.M.; Abbott, B.M.; Robins-Browne, R.M.; et al. Structural Determinants Defining the Allosteric Inhibition of an Essential Antibiotic Target. Structure 2016, 24, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Stuttmann, J.; Hubberten, H.M.; Rietz, S.; Kaur, J.; Muskett, P.; Guerois, R.; Bednarek, P.; Hoefgen, R.; Parker, J.E. Perturbation of Arabidopsis amino acid metabolism causes incompatibility with the adapted biotrophic pathogen Hyaloperonospora arabidopsidis. Plant Cell 2011, 23, 2788–2803. [Google Scholar] [CrossRef] [Green Version]
- Uppalapati, S.R.; Marek, S.M.; Lee, H.-K.; Nakashima, J.; Tang, Y.; Sledge, M.K.; Dixon, R.A.; Mysore, K.S. Global Gene Expression Profiling During Medicago truncatula–Phymatotrichopsis omnivora Interaction Reveals a Role for Jasmonic Acid, Ethylene, and the Flavonoid Pathway in Disease Development. Mol. Plant-Microbe Interact. 2009, 22, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Mah, K.M.; Uppalapati, S.R.; Tang, Y.; Allen, S.; Shuai, B. Gene expression profiling of Macrophomina phaseolina infected Medicago truncatula roots reveals a role for auxin in plant tolerance against the charcoal rot pathogen. Physiol. Mol. Plant Pathol. 2012, 79, 21–30. [Google Scholar] [CrossRef]
- He, J.; Benedito, V.A.; Wang, M.; Murray, J.D.; Zhao, P.X.; Tang, Y.; Udvardi, M.K. The Medicago truncatula gene expression atlas web server. BMC Bioinform. 2009, 10, 441. [Google Scholar] [CrossRef] [Green Version]
- Procter, J.B.; Thompson, J.; Letunic, I.; Creevey, C.; Jossinet, F.; Barton, G.J. Visualization of multiple alignments, phylogenies and gene family evolution. Nat. Methods 2010, 7, S16–S25. [Google Scholar] [CrossRef]
- Severin, A.J.; Woody, J.L.; Bolon, Y.-T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.-A.; Peng, D.-L.; et al. Global Dissection of Alternative Splicing in Paleopolyploid Soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Sundaresan, S.; Philosoph-Hadas, S.; Yang, R.; Meir, S.; Tucker, M.L. Examination of the Abscission-Associated Transcriptomes for Soybean, Tomato, and Arabidopsis Highlights the Conserved Biosynthesis of an Extensible Extracellular Matrix and Boundary Layer. Front. Plant Sci. 2015, 6, 1109. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Xiao, Z.; Li, M.W.; Wong, F.L.; Yung, W.S.; Ku, Y.S.; Wang, Q.; Wang, X.; Xie, M.; Yim, A.K.; et al. Transcriptomic reprogramming in soybean seedlings under salt stress. Plant Cell Environ. 2019, 42, 98–114. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Vaughn, J.N.; Abdel-Haleem, H.; Chavarro, C.; Abernathy, B.; Kim, K.; Jackson, S.A.; Li, Z. Transcriptomic changes due to water deficit define a general soybean response and accession-specific pathways for drought avoidance. BMC Plant Biol. 2015, 15, 26. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yao, Q.; Patil, G.B.; Agarwal, G.; Deshmukh, R.K.; Lin, L.; Wang, B.; Wang, Y.; Prince, S.J.; Song, L.; et al. Identification and Comparative Analysis of Differential Gene Expression in Soybean Leaf Tissue under Drought and Flooding Stress Revealed by RNA-Seq. Front. Plant Sci. 2016, 7, 1044. [Google Scholar] [CrossRef] [Green Version]
- Yendrek, C.R.; Koester, R.P.; Ainsworth, E.A. A comparative analysis of transcriptomic, biochemical, and physiological responses to elevated ozone identifies species-specific mechanisms of resilience in legume crops. J. Exp. Bot. 2015, 66, 7101–7112. [Google Scholar] [CrossRef] [Green Version]
- Belamkar, V.; Weeks, N.T.; Bharti, A.K.; Farmer, A.D.; Graham, M.A.; Cannon, S.B. Comprehensive characterization and RNA-Seq profiling of the HD-Zip transcription factor family in soybean (Glycine max) during dehydration and salt stress. BMC Genom. 2014, 15, 950. [Google Scholar] [CrossRef] [Green Version]
- Rasoolizadeh, A.; Labbe, C.; Sonah, H.; Deshmukh, R.K.; Belzile, F.; Menzies, J.G.; Belanger, R.R. Silicon protects soybean plants against Phytophthora sojae by interfering with effector-receptor expression. BMC Plant Biol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Zhu, X.; Wang, Y.; Jung, K.H.; Chen, L.; Xuan, Y.; Duan, Y. The transcriptomic changes of Huipizhi Heidou (Glycine max), a nematode-resistant black soybean during Heterodera glycines race 3 infection. J. Plant Physiol. 2018, 220, 96–104. [Google Scholar] [CrossRef]
- Craciun, A.; Jacobs, M.; Vauterin, M. Arabidopsis loss-of-function mutant in the lysine pathway points out complex regulation mechanisms. FEBS Lett. 2000, 487, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Hashimoto, T.; Kumpaisal, R.; Yamada, Y. Molecular cloning of wheat dihydrodipicolinate synthase. J. Biol. Chem. 1990, 265, 17451–17455. [Google Scholar] [CrossRef]
- Zheng, F.; Wu, H.; Zhang, R.; Li, S.; He, W.; Wong, F.-L.; Li, G.; Zhao, S.; Lam, H.-M. Molecular phylogeny and dynamic evolution of disease resistance genes in the legume family. BMC Genom. 2016, 17, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Engström, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; Rätsch, G.; Goldman, N.; Hubbard, T.J.; Harrow, J.; Guigó, R.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief. Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Vauterin, M.; Frankard, V.; Jacobs, M. The Arabidopsis thaliana dhdps gene encoding dihydrodipicolinate synthase, key enzyme of lysine biosynthesis, is expressed in a cell-specific manner. Plant Mol. Biol. 1999, 39, 695–708. [Google Scholar] [CrossRef]
- Sarrobert, C.; Thibaud, M.C.; Contard-David, P.; Gineste, S.; Bechtold, N.; Robaglia, C.; Nussaume, L. Identification of an Arabidopsis thaliana mutant accumulating threonine resulting from mutation in a new dihydrodipicolinate synthase gene. Plant J. 2000, 24, 357–367. [Google Scholar] [CrossRef]
- Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D.; Penin, A.A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016, 88, 1058–1070. [Google Scholar] [CrossRef]
- Carrere, S.; Verdier, J.; Gamas, P. MtExpress, a Comprehensive and Curated RNAseq-based Gene Expression Atlas for the Model Legume Medicago truncatula. Plant Cell Physiol. 2021, 62, 1494–1500. [Google Scholar] [CrossRef]
- Arruda, P.; Barreto, P. Lysine Catabolism Through the Saccharopine Pathway: Enzymes and Intermediates Involved in Plant Responses to Abiotic and Biotic Stress. Front. Plant Sci. 2020, 11, 587. [Google Scholar] [CrossRef]
- Kong, Y.; Xu, P.; Jing, X.; Chen, L.; Li, L.; Li, X. Decipher the ancestry of the plant-specific LBD gene family. BMC Genom. 2017, 18, 951. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Schläpfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-Wide Prediction of Metabolic Enzymes, Pathways, and Gene Clusters in Plants. Plant Physiol. 2017, 173, 2041–2059. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Leisner, C.P.; Ming, R.; Ainsworth, E.A. Distinct transcriptional profiles of ozone stress in soybean (Glycine max) flowers and pods. BMC Plant Biol. 2014, 14, 335. [Google Scholar] [CrossRef] [Green Version]
- Moran Lauter, A.N.; Peiffer, G.A.; Yin, T.; Whitham, S.A.; Cook, D.; Shoemaker, R.C.; Graham, M.A. Identification of candidate genes involved in early iron deficiency chlorosis signaling in soybean (Glycine max) roots and leaves. BMC Genom. 2014, 15, 702. [Google Scholar] [CrossRef] [Green Version]
- Leisner, C.P.; Yendrek, C.R.; Ainsworth, E.A. Physiological and transcriptomic responses in the seed coat of field-grown soybean (Glycine max L. Merr.) to abiotic stress. BMC Plant Biol. 2017, 17, 242. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, F.A.; Fuganti-Pagliarini, R.; Marcolino-Gomes, J.; Nakayama, T.J.; Molinari, H.B.C.; Lobo, F.P.; Harmon, F.G.; Nepomuceno, A.L. Daytime soybean transcriptome fluctuations during water deficit stress. BMC Genom. 2015, 16, 505. [Google Scholar] [CrossRef] [Green Version]
- Bencke-Malato, M.; De Souza, A.P.; Ribeiro-Alves, M.; Schmitz, J.F.; Buckeridge, M.S.; Alves-Ferreira, M. Short-term responses of soybean roots to individual and combinatorial effects of elevated [CO2] and water deficit. Plant Sci. 2019, 280, 283–296. [Google Scholar] [CrossRef]
- Waters, B.M.; Amundsen, K.; Graef, G. Gene Expression Profiling of Iron Deficiency Chlorosis Sensitive and Tolerant Soybean Indicates Key Roles for Phenylpropanoids under Alkalinity Stress. Front. Plant Sci. 2018, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Lanubile, A.; Muppirala, U.K.; Severin, A.J.; Marocco, A.; Munkvold, G.P. Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genom. 2015, 16, 1089. [Google Scholar] [CrossRef] [Green Version]
- Redding, N.W.; Agudelo, P.; Wells, C.E. Multiple Nodulation Genes Are Up-Regulated during Establishment of Reniform Nematode Feeding Sites in Soybean. Phytopathology 2018, 108, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Xun, H.; Yang, X.; He, H.; Wang, M.; Guo, P.; Wang, Y.; Pang, J.; Dong, Y.; Feng, X.; Wang, S.; et al. Over-expression of GmKR3, a TIR–NBS–LRR type R gene, confers resistance to multiple viruses in soybean. Plant Mol. Biol. 2019, 99, 95–111. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Grant, D.; Nelson, R.T.; Cannon, S.B.; Shoemaker, R.C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2010, 38, D843–D846. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33). 2011. [Google Scholar]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, F.; Binder, H. pcaExplorer: An R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinform. 2019, 20, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadley, W. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sites Known For Catalytic Activity | Allosteric Sites (Lysine Inhibition) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 68 | 69 | 130 | 131 | 155 | 160 | 183 | 185 | 204 | 206 | 207 | 222 | 261 | 73 | 77 | 80 | 81 | 102 | 103 | 104 | 108 | 111 | 112 | |
| CONSENSUS | T | T | Y | Y | Y | R | K | C | G | D | D | I | N | Q | W | H | I | G | S | N | E | H | A |
| Pp.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Pp.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Sm.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Pa.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Pa.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Sb.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Sb.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Zm.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Zm.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Oz.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Oz.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | H | * | * | * | * | * | * | * | * | * |
| Ac.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | V | * | H | * | * | * | * | * | * | * | * | * |
| Ac.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | V | * | H | * | * | * | * | * | * | * | * | * |
| Vv.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Pt.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Pt.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | V | * | * | * | * | * | * | * | * | * | * | * |
| Eg.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Eg.DHDPS-A2 | A | V | * | * | * | * | * | * | * | * | Y | M | T | H | * | * | * | * | R | * | * | Q | * |
| Eg.DHDPS-A3 | * | A | * | * | * | * | * | * | * | * | * | M | * | H | * | * | * | * | * | * | * | E | * |
| Eg.DHDPS-A4 | * | A | * | * | * | * | * | * | * | * | H | M | A | H | * | * | * | * | * | * | * | Q | * |
| AtDHDPS2 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| AtDHDPS1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Lj.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Mt.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Mt.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Ps.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | V | * | * | * | * | * | * | * | * | * | * | * |
| Pv.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Vu.DHDPS-A | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Gm.DHDPS-A1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Gm.DHDPS-A2 | * | * | * | * | * | * | * | * | * | * | * | V | * | * | * | * | * | * | * | * | * | * | * |
| Lj.DHDPS-B1 | S | * | * | * | * | * | * | G | * | * | K | Q | T | * | * | Q | * | * | * | * | * | * | * |
| Lj.DHDPS-B2 | * | * | * | * | * | * | * | * | A | * | E | M | T | * | L | K | * | * | * | * | Q | S | I |
| Lj.DHDPS-B3 | * | * | * | * | * | * | * | Y | S | E | K | M | T | * | V | K | V | * | * | * | Q | S | I |
| Lj.DHDPS-B4 | * | * | * | * | * | * | * | * | A | Q | R | M | I | * | L | K | * | * | * | * | Q | S | I |
| Mt.DHDPS-B1 | * | * | * | * | * | * | * | * | * | * | E | H | A | Y | * | Q | * | * | * | * | * | T | * |
| Mt.DHDPS-B2 | S | * | * | * | * | * | * | * | - | * | I | Q | S | * | * | Q | * | * | * | * | * | N | * |
| Mt.DHDPS-B3 | S | * | * | * | * | * | * | * | - | * | I | Q | S | * | * | Q | * | * | * | * | * | N | * |
| Mt.DHDPS-B4 | * | * | * | * | * | * | * | * | A | Q | K | V | I | H | L | K | V | * | * | * | L | S | L |
| Ps.DHDPS-B1 | * | * | * | * | * | * | * | * | * | * | E | H | T | Y | * | Q | * | * | * | * | * | T | * |
| Ps.DHDPS-B2 | S | * | * | * | * | * | * | * | - | * | G | Q | S | * | * | Q | * | * | * | * | * | E | * |
| Ps.DHDPS-B3 | * | * | * | * | * | * | * | * | A | * | E | M | C | * | I | K | V | * | * | * | Q | T | I |
| Pv.DHDPS-B | S | * | * | * | * | * | * | * | * | * | K | Q | V | * | * | Q | * | * | * | * | * | K | * |
| Vu.DHDPS-B | S | * | * | * | * | * | * | * | * | * | K | Q | G | * | * | Q | * | * | * | * | * | K | * |
| Gm.DHDPS-B | * | * | * | * | * | * | * | * | * | * | K | Q | V | * | * | Q | * | * | * | * | * | K | * |

| Total Reads | Unique Reads (%) | Correlation | |||
|---|---|---|---|---|---|
| Sample | Severin et al. | This Paper | Severin et al. | This Paper | (Pearson’s r) |
| young leaf | 6,618,852 | 6,621,825 | 66% | 72% | 0.991 *** |
| flower | 5,829,223 | 5,800,039 | 60% | 67% | 0.962 *** |
| one cm pod | 6,181,917 | 6,149,218 | 58% | 64% | 0.977 *** |
| pod shell 10DAF | 6,464,386 | 6,426,175 | 58% | 63% | 0.992 *** |
| pod shell 14DAF | 5,983,354 | 5,921,485 | 50% | 57% | 0.909 *** |
| seed 10DAF | 6,962,047 | 6,936,823 | 44% | 47% | 0.999 *** |
| seed 14DAF | 5,888,849 | 5,845,764 | 43% | 47% | 0.999 *** |
| seed 21DAF | 2,711,453 | 2,692,219 | 39% | 44% | 0.985 *** |
| seed 25DAF | 7,835,063 | 7,142,607 | 37% | 48% | 0.989 *** |
| seed 28DAF | 9,673,118 | 8,010,459 | 26% | 35% | 0.991 *** |
| seed 35DAF | 9,102,649 | 8,791,274 | 52% | 64% | 0.997 *** |
| seed 42DAF | 7,052,993 | 6,884,047 | 49% | 60% | 0.998 *** |
| root | 8,402,716 | 8,402,716 | 57% | 68% | 0.933 *** |
| nodule | 8,930,860 | 8,930,860 | 61% | 68% | 0.995 *** |
| SRA Study | Genotype, Experiment, Tissue | Enzyme | Log2Fold Change | Base Mean |
|---|---|---|---|---|
| SRP050050 | Wm82, Ethylene 12 h, Leaf Abscission Zone | Gm.DHDPS-A1 | 1.3 * | 27.9 |
| SRP155375 | Himok, P. soja 4 dpi, Root | Gm.DHDPS-A1 | −1 * | 166.6 |
| SRP050050 | Wm82, Ethylene 48 h, Leaf Petiole | Gm.DHDPS-A1 | −1.1 * | 23.5 |
| SRP045932 | Benning, Water Deficit 24 h, Shoot | Gm.DHDPS-A1 | −1.1 *** | 95.7 |
| SRP050050 | Wm82, Ethylene 72 h, Leaf Abscission Zone | Gm.DHDPS-A1 | −3.4 * | 5.1 |
| SRP050050 | Wm82, Ethylene 72 h, Leaf Petiole | Gm.DHDPS-A1 | −3.7 *** | 10.2 |
| SRP050050 | Wm82, Ethylene 48 h, Leaf Petiole | Gm.DHDPS-A2 | −1.8 *** | 30.7 |
| SRP045932 | Benning, Water Deficit 24 h, Shoot | Gm.DHDPS-A2 | −2.4 *** | 65.1 |
| SRP050050 | Wm82, Ethylene 48 h, Leaf Abscission Zone | Gm.DHDPS-A2 | −2.9 *** | 17.9 |
| SRP050050 | Wm82, Ethylene 72 h, Leaf Petiole | Gm.DHDPS-A2 | −3.1 *** | 15.8 |
| SRP050050 | Wm82, Ethylene 72 h, Leaf Abscission Zone | Gm.DHDPS-A2 | −3.6 *** | 15.3 |
| SRP050050 | Wm82, Ethylene 48 h, Leaf Petiole | Gm.DHDPS-B | 4.1 * | 1.8 |
| SRP050050 | Wm82, Ethylene 24 h, Leaf Petiole | Gm.DHDPS-B | 4.0 * | 1.6 |
| SRP076153 | Wm82, Flooding, Leaf | Gm.DHDPS-B | 2.7 * | 5.6 |
| SRP045932 | PI416937, Water Deficit 12 h, Shoot | Gm.DHDPS-B | 1.8 ** | 54.4 |
| SRP045932 | Benning, Water Deficit 12 h, Shoot | Gm.DHDPS-B | 1.7 * | 12.4 |
| SRP009826 | Be Sweet 292, Ozone, Leaf | Gm.DHDPS-B | 1.5 *** | 26.0 |
| SRP045932 | PI416937, Water Deficit 24 h, Shoot | Gm.DHDPS-B | 1.3 * | 30.5 |
| SRP135932 | Heidou, Nematode 15 dpi, Root | Gm.DHDPS-B | 1.2 *** | 316.0 |
| SRP041622 | Wm82, Salt 12 h, Root | Gm.DHDPS-B | 1.0 *** | 148.0 |
| SRP132150 | C08, Salt 2 h, Root | Gm.DHDPS-B | −1.7 *** | 350.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiekens, R.; de Koning, R.; Toili, M.E.M.; Angenon, G. The Hidden Potential of High-Throughput RNA-Seq Re-Analysis, a Case Study for DHDPS, Key Enzyme of the Aspartate-Derived Lysine Biosynthesis Pathway and Its Role in Abiotic and Biotic Stress Responses in Soybean. Plants 2022, 11, 1762. https://doi.org/10.3390/plants11131762
Kiekens R, de Koning R, Toili MEM, Angenon G. The Hidden Potential of High-Throughput RNA-Seq Re-Analysis, a Case Study for DHDPS, Key Enzyme of the Aspartate-Derived Lysine Biosynthesis Pathway and Its Role in Abiotic and Biotic Stress Responses in Soybean. Plants. 2022; 11(13):1762. https://doi.org/10.3390/plants11131762
Chicago/Turabian StyleKiekens, Raphaël, Ramon de Koning, Mary Esther Muyoka Toili, and Geert Angenon. 2022. "The Hidden Potential of High-Throughput RNA-Seq Re-Analysis, a Case Study for DHDPS, Key Enzyme of the Aspartate-Derived Lysine Biosynthesis Pathway and Its Role in Abiotic and Biotic Stress Responses in Soybean" Plants 11, no. 13: 1762. https://doi.org/10.3390/plants11131762