
β-Cyclocitral, a Master Regulator of Multiple Stress-Responsive Genes in Solanum lycopersicum L. Plants


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
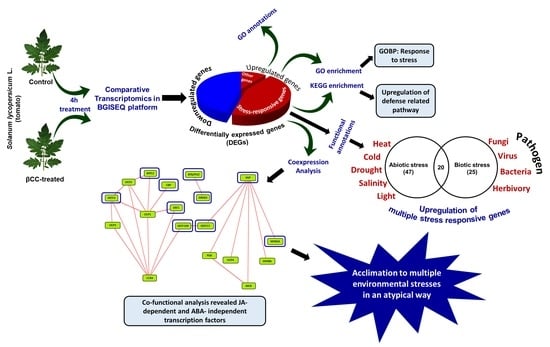
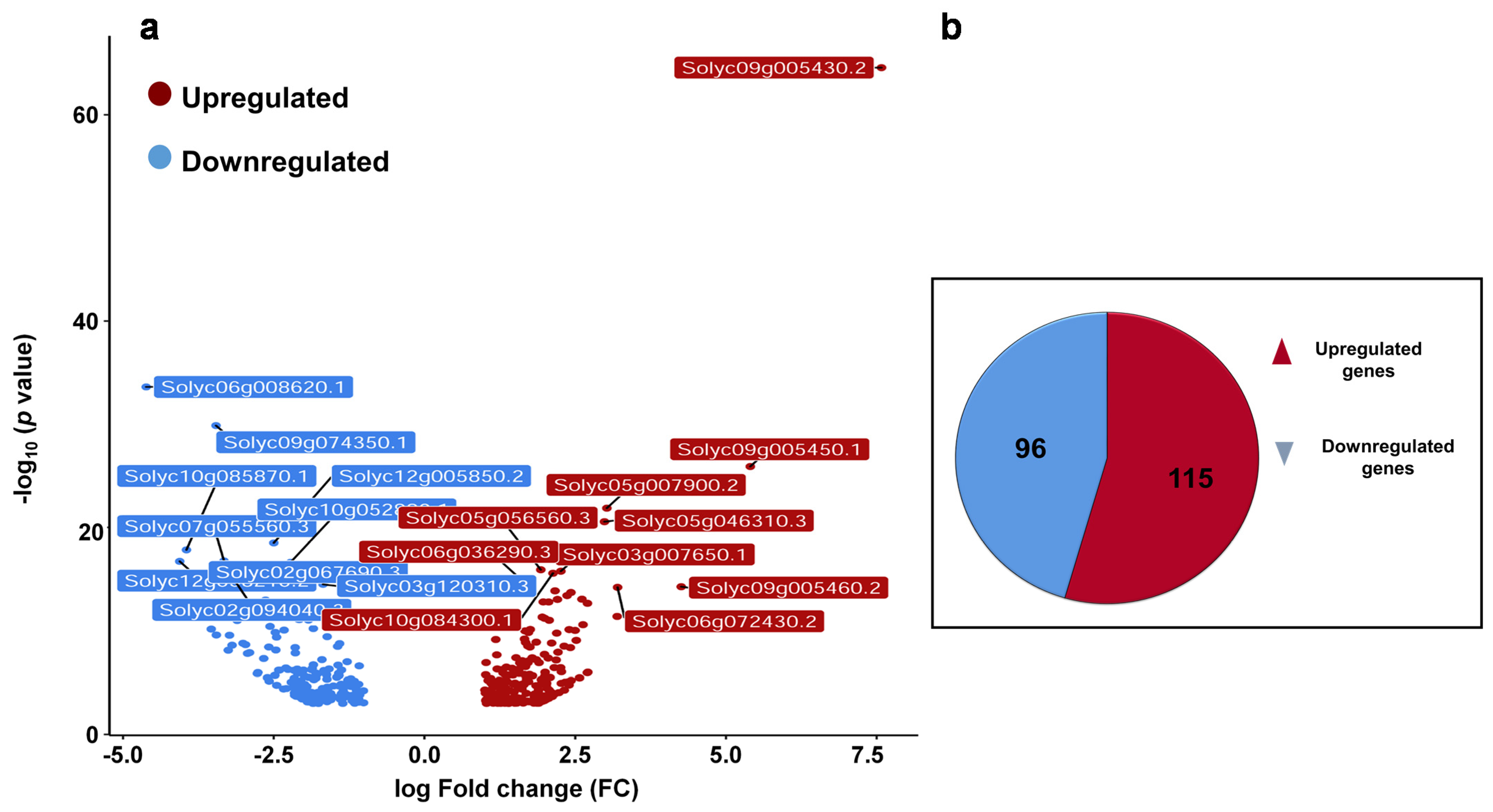
2.1. Effect of Exogenous βCC Treatment on Transcriptomic Profile of Tomato Plant
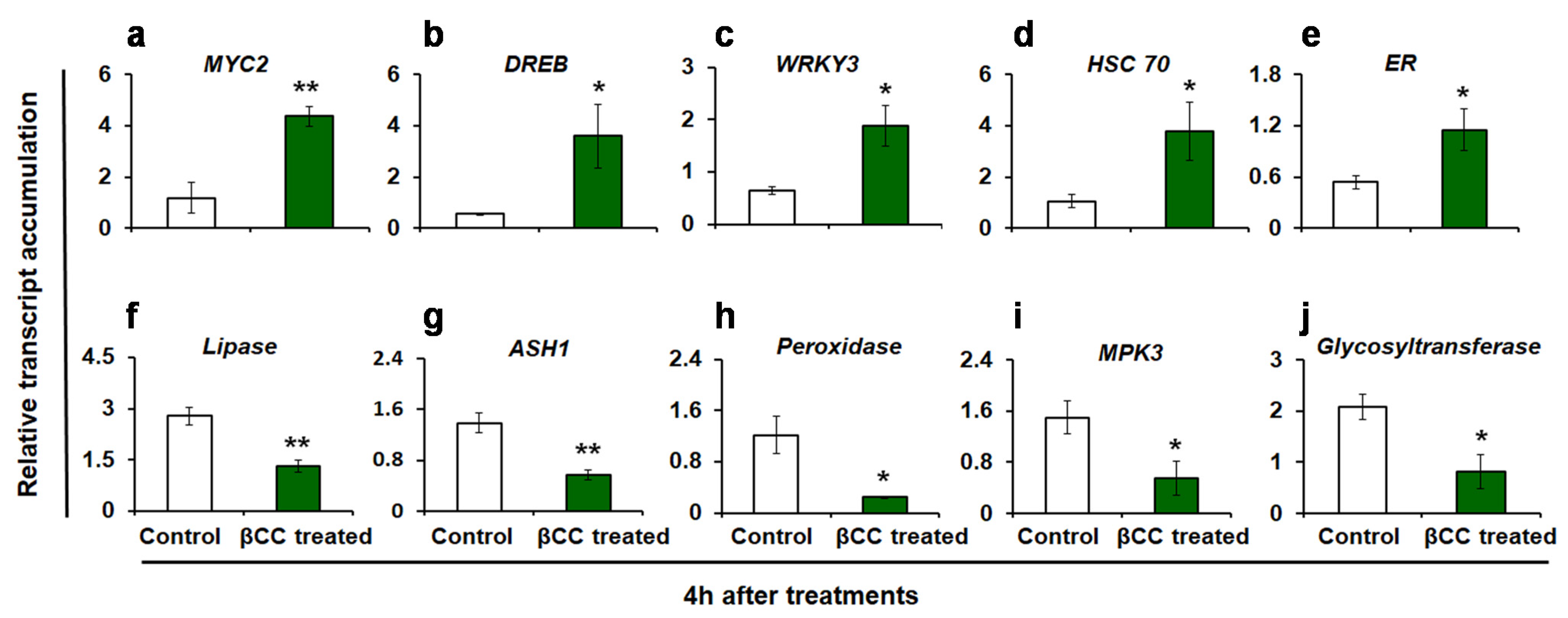
2.2. Validation of Differentially Expressed Transcripts by Quantitative Real-Time PCR
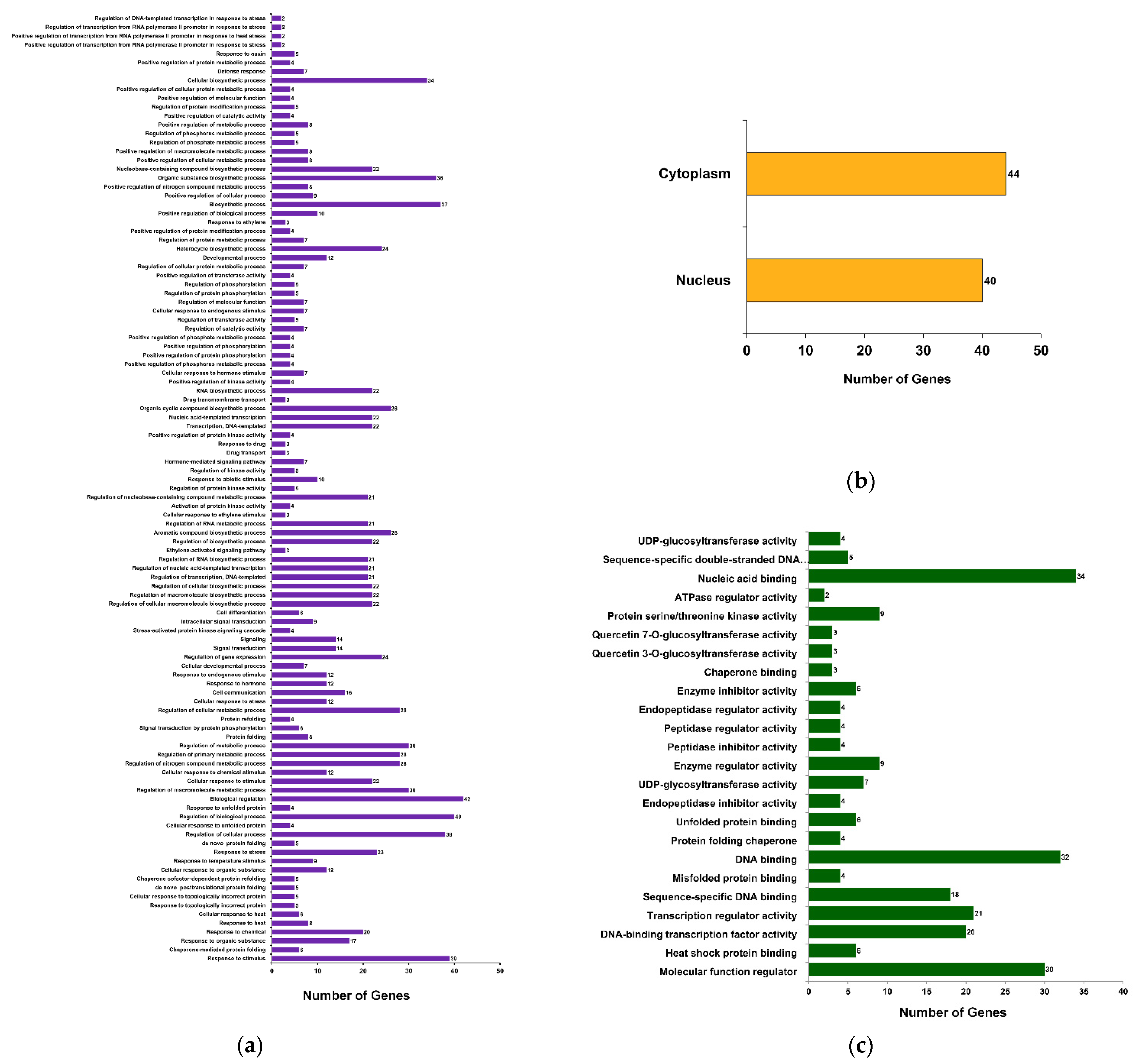
2.3. βCC-Induced Differentially Expressed Genes (DEGs) Are Mainly Coding the Proteins That Are Regulated by External Stimuli
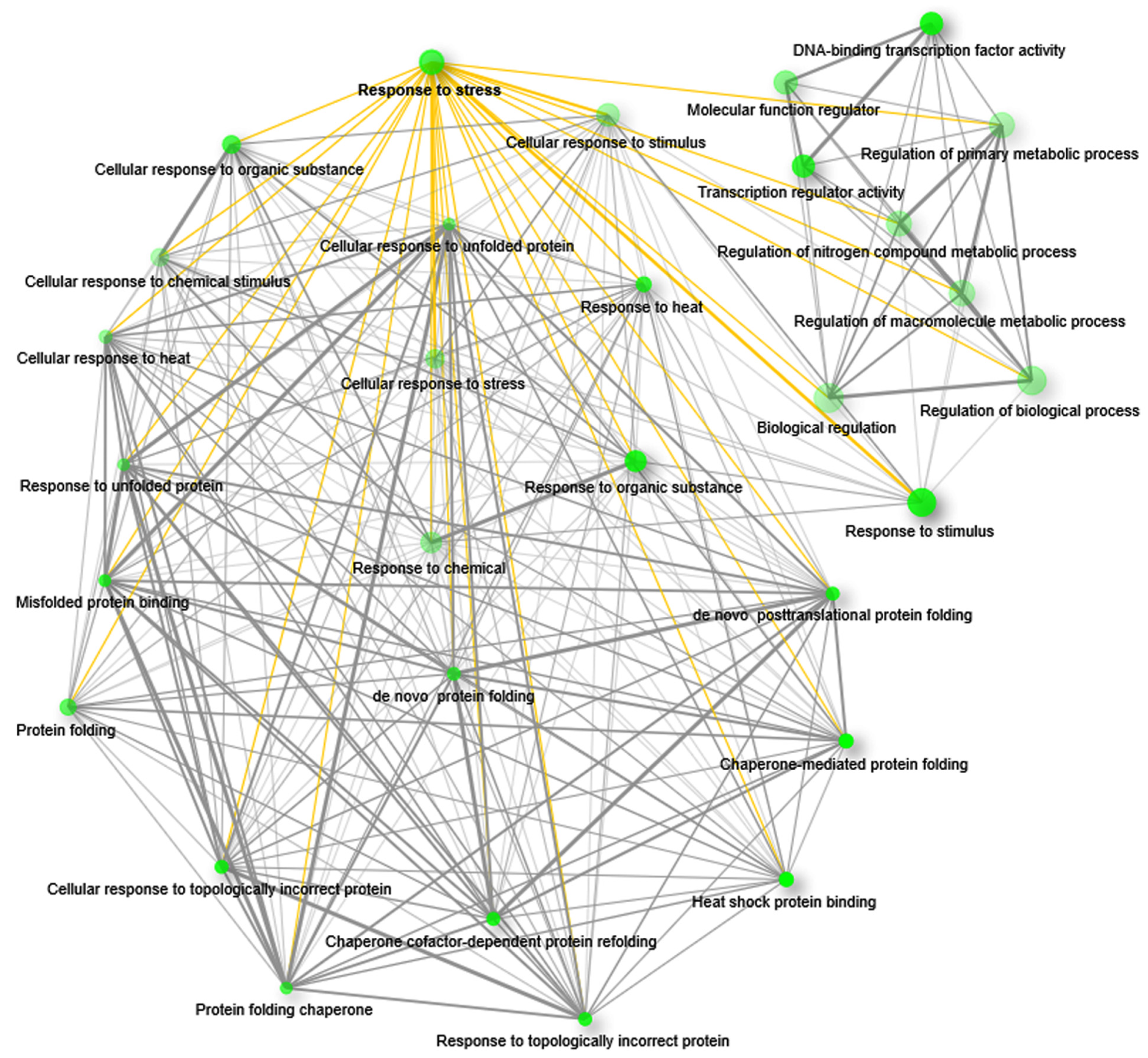
2.4. Upregulated Genes Are Significantly Enriched with Stress-Responsive Functions
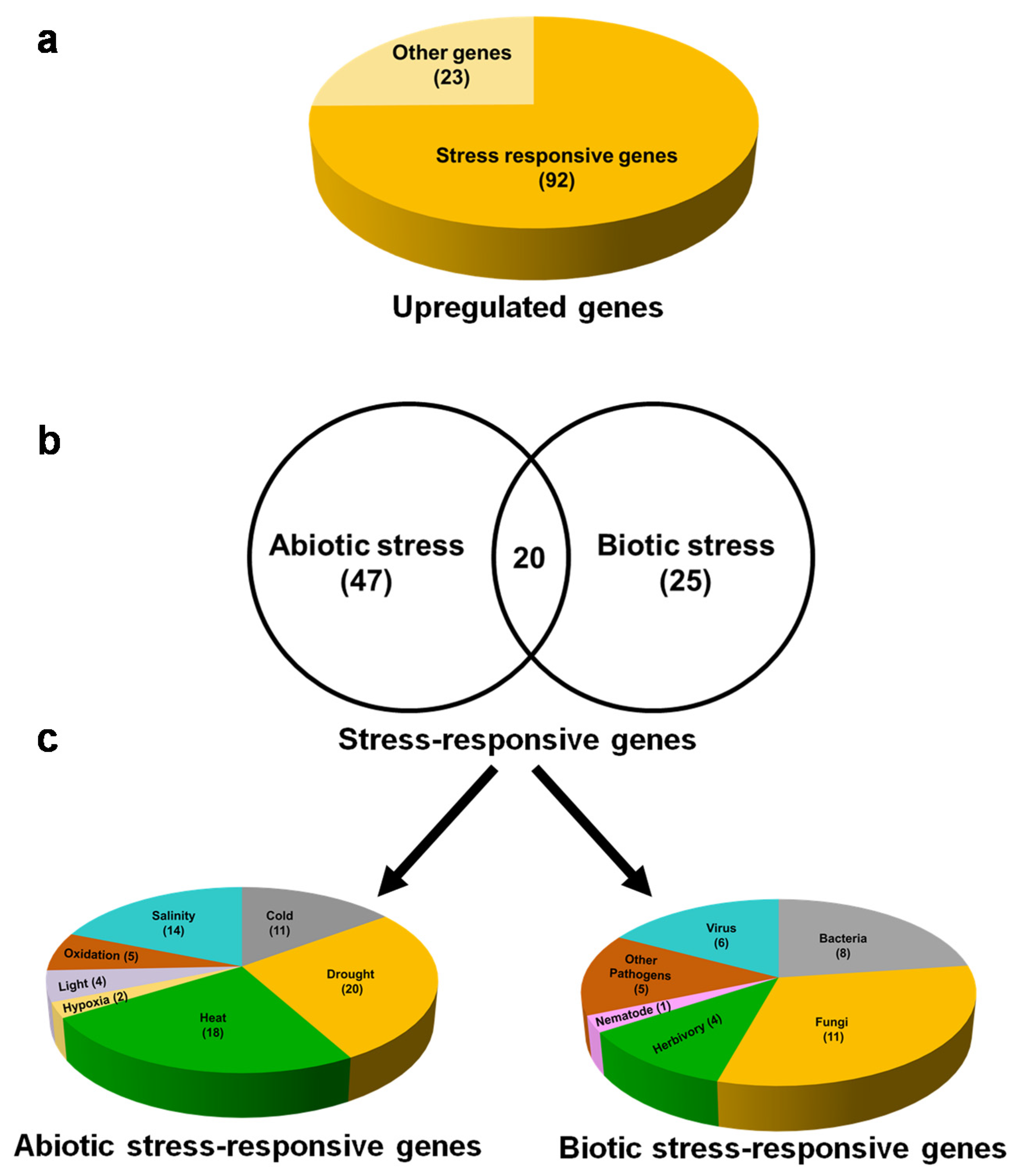
2.5. Exogenous βCC Treatment Specifically Upregulates Genes That Are Involved in Acclimation to the Plethora of Environmental Stresses
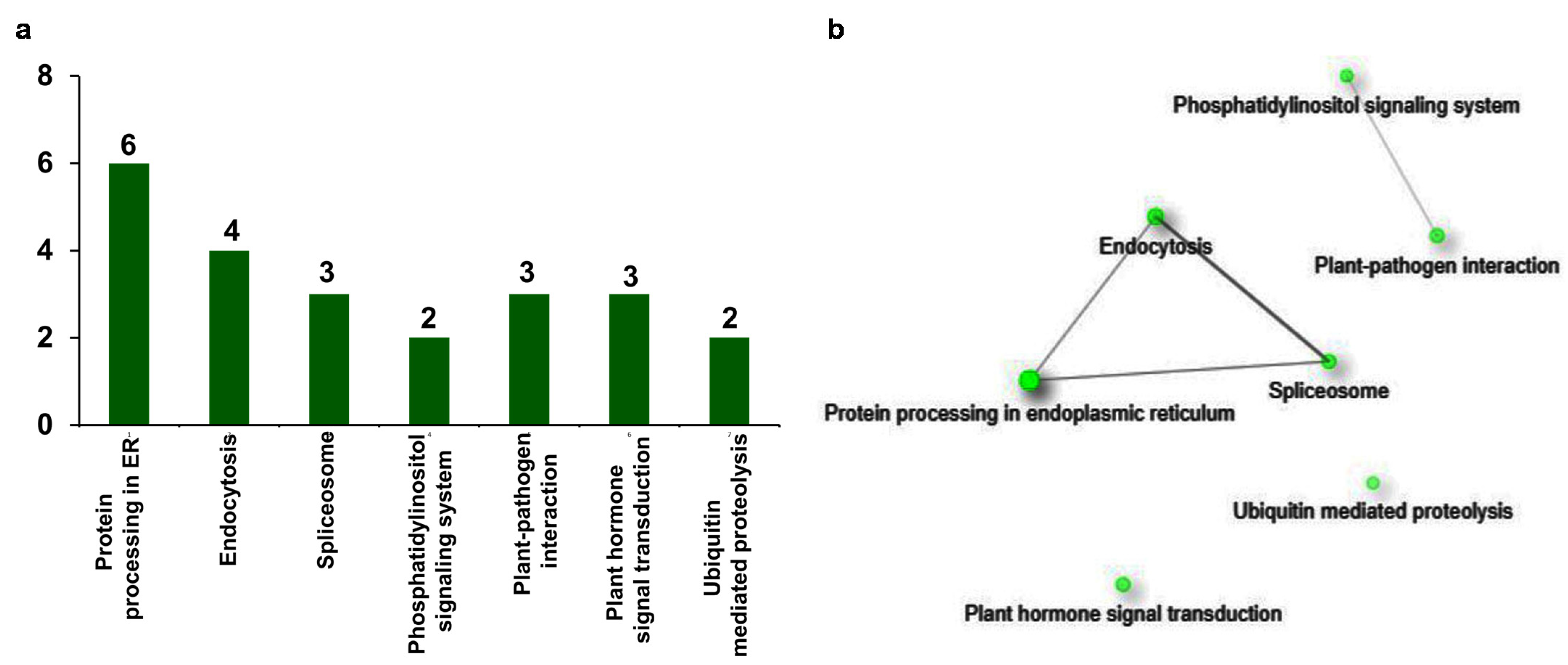
2.6. Upregulated DEGs Are Specifically Involved in Pathways for Defense Response
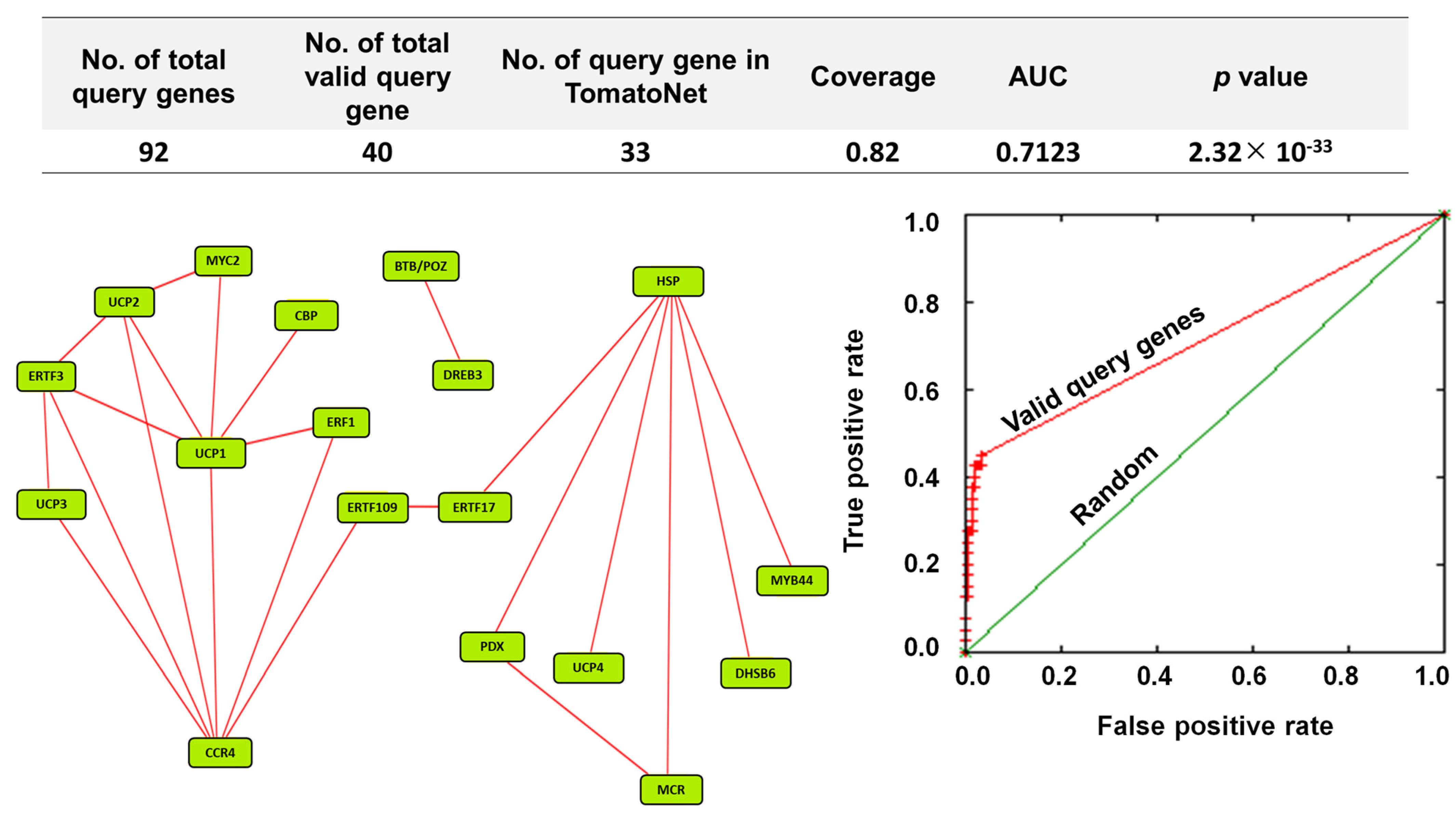
2.7. βCC Treatment Co-Expressed the Transcription Factors and Other Protein-Encoding Genes That Provide Acclimation to Multiple Stresses
3. Discussion
4. Materials and Methods
4.1. Plant Material, Growth Conditions, and βCC Treatment
4.2. RNA Extraction, cDNA Library Preparation, and RNA Sequencing
4.3. Quality Check and Analysis of RNA Sequence Data
4.4. Quantitative Real-Time PCR (RT-PCR)
4.5. Analysis of GO and Its Enrichment, Functional Annotations of Differentially Expressed Genes (DEGs), and Enrichment of KEGG
4.6. Generation of DEG Network
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thakur, B.; Singh, R.; Nelson, P. Quality attributes of processed tomato products: A review. Food Rev. Int. 1996, 12, 375–401. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, H.; Wang, X.; Zhou, X.; Zhang, K.; Chen, X.; Liu, J.; Han, J.; Wang, A. The roles of different types of trichomes in tomato resistance to cold, drought, whiteflies, and botrytis. Agronomy 2020, 10, 411. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, S.; Manoharan, R.; Mitra, S. Exogenous β-cyclocitral treatment primes tomato plants against drought by inducing tolerance traits, independent of abscisic acid. Plant Biol. 2021, 23, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Zurbriggen, M.D.; Carrillo, N.; Hajirezaei, M.-R. ROS signaling in the hypersensitive response: When, where and what for? Plant Signal. Behav. 2010, 5, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; He, Z.; Chen, N.; Tang, Z.; Wang, Q.; Cai, Y. The roles of environmental factors in regulation of oxidative stress in plant. Biomed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, S.; Mizokami, Y.; Legeret, B.; Havaux, M. The apocarotenoid β-cyclocitric acid elicits drought tolerance in plants. Iscience 2019, 19, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Sedlářová, M.; Kale, R.S.; Pospíšil, P. Lipoxygenase in singlet oxygen generation as a response to wounding: In vivo imaging in Arabidopsis thaliana. Sci. Rep. 2017, 7, 1–10. [Google Scholar]
- Havaux, M. β-Cyclocitral and derivatives: Emerging molecular signals serving multiple biological functions. Plant Physiol. Biochem. 2020, 155, 35–41. [Google Scholar] [CrossRef]
- Ramel, F.; Birtic, S.; Ginies, C.; Soubigou-Taconnat, L.; Triantaphylidès, C.; Havaux, M. Carotenoid oxidation products are stress signals that mediate gene responses to singlet oxygen in plants. Proc. Natl. Acad. Sci. USA 2012, 109, 5535–5540. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Estrada-Tejedor, R.; Volke, D.C.; Phillips, M.A.; Gershenzon, J.; Wright, L.P. Negative regulation of plastidial isoprenoid pathway by herbivore-induced β-cyclocitral in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2021, 118, e2008747118. [Google Scholar] [CrossRef]
- Taniguchi, S.; Hosokawa-Shinonaga, Y.; Tamaoki, D.; Yamada, S.; Akimitsu, K.; Gomi, K. Jasmonate induction of the monoterpene linalool confers resistance to rice bacterial blight and its biosynthesis is regulated by JAZ protein in rice. Plant Cell Environ. 2014, 37, 451–461. [Google Scholar] [CrossRef]
- Cohen, D.; Bogeat-Triboulot, M.-B.; Tisserant, E.; Balzergue, S.; Martin-Magniette, M.-L.; Lelandais, G.; Ningre, N.; Renou, J.-P.; Tamby, J.-P.; Le Thiec, D. Comparative transcriptomics of drought responses in Populus: A meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genom. 2010, 11, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejat, N.; Ramalingam, A.; Mantri, N. Advances in transcriptomics of plants. Plant Genet. Mol. Biol. 2018, 164, 161–185. [Google Scholar]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, X.; Tang, X.-M.; Xiao, L.; Sun, J.-L.; Yan, X.-F.; Li, D.; Deng, H.-Y.; Ma, X.-R. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, B.S.; Shim, J.E.; Hwang, S.; Yang, S.; Kim, E.; Iyer-Pascuzzi, A.S.; Lee, I. TomatoNet: A genome-wide co-functional network for unveiling complex traits of tomato, a model crop for fleshy fruits. Mol. Plant 2017, 10, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.C.; Mi, J.; Alagoz, Y.; AlMoreno, J.C.; Mi, J.; Alagoz, Y. Plant apocarotenoids: From retrograde signaling to interspecific communication. Plant J. 2021, 105, 351. [Google Scholar] [CrossRef]
- Hou, X.; Rivers, J.; León, P.; McQuinn, R.P.; Pogson, B.J. Synthesis and function of apocarotenoid signals in plants. Trends Plant Sci. 2016, 21, 792–803. [Google Scholar] [CrossRef]
- Knight, H.; Knight, M.R. Abiotic stress signalling pathways: Specificity and cross-talk. Trends Plant Sci. 2001, 6, 262–267. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Abe, H.; Kasuga, M.; Yamaguchi-Shinozaki, K.; Carninci, P.; Hayashizaki, Y.; Shinozaki, K. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell 2001, 13, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.; Budworth, P.; Han, B.; Brown, D.; Chang, H.-S.; Zou, G.; Wang, X. Toward elucidating the global gene expression patternsof developing Arabidopsis: Parallel analysis of 8 300 genesby a high-density oligonucleotide probe array. Plant Physiol. Biochem. 2001, 39, 221–242. [Google Scholar] [CrossRef]
- Nakajima, Y.; Suzuki, S. Environmental stresses induce misfolded protein aggregation in plant cells in a microtubule-dependent manner. Int. J. Mol. Sci. 2013, 14, 7771–7783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Srivastava, R.; Howell, S.H. Endoplasmic reticulum (ER) stress response and its physiological roles in plants. Int. J. Mol. Sci. 2013, 14, 8188–8212. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Deng, Y.; Howell, S.H. Stress sensing in plants by an ER stress sensor/transducer, bZIP28. Front. Plant Sci. 2014, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miernyk, J.A. Protein folding in the plant cell. Plant Physiol. 1999, 121, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Kragler, F.; Xoconostle-Cázares, B.; Lucas, W.J. A subclass of plant heat shock cognate 70 chaperones carries a motif that facilitates trafficking through plasmodesmata. Proc. Natl. Acad. Sci. USA 2002, 99, 16342–16347. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.C.; Sharma, A.; Mishra, M.; Mishra, R.K.; Chowdhuri, D.K. Heat shock proteins in toxicology: How close and how far? Life Sci. 2010, 86, 377–384. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255. [Google Scholar] [CrossRef] [Green Version]
- Sung, D.Y.; Kaplan, F.; Guy, C.L. Plant Hsp70 molecular chaperones: Protein structure, gene family, expression and function. Physiol. Plant. 2001, 113, 443–451. [Google Scholar] [CrossRef]
- Lee, J.H.; Schöffl, F. An Hsp70 antisense gene affects the expression of HSP70/HSC70, the regulation of HSF, and the acquisition of thermotolerance in transgenic Arabidopsis thaliana. Mol. Gen. Genet. MGG 1996, 252, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Alvim, F.C.; Carolino, S.M.; Cascardo, J.C.; Nunes, C.C.; Martinez, C.A.; Otoni, W.C.; Fontes, E.P. Enhanced accumulation of BiP in transgenic plants confers tolerance to water stress. Plant Physiol. 2001, 126, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambill, B.D.; Voos, W.; Kang, P.J.; Miao, B.; Langer, T.; Craig, E.A.; Pfanner, N. A dual role for mitochondrial heat shock protein 70 in membrane translocation of preproteins. J. Cell Biol. 1993, 123, 109–117. [Google Scholar] [CrossRef]
- Kourtz, L.; Ko, K. The early stage of chloroplast protein import involves Com70. J. Biol. Chem. 2001, 272, 2808–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, P.-H.; Li, H.-M. Arabidopsis stromal 70-kD heat shock proteins are essential for plant development and important for thermotolerance of germinating seeds. Plant Physiol. 2008, 146, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, H.; Saitoh, H.; Ito, A.; Fujisawa, S.; Kamoun, S.; Katou, S.; Yoshioka, H.; Terauchi, R. Cytosolic HSP90 and HSP70 are essential components of INF1-mediated hypersensitive response and non-host resistance to Pseudomonas cichorii in Nicotiana benthamiana. Mol. Plant Pathol. 2003, 4, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Hwang, B.K. Pepper heat shock protein 70a interacts with the type III effector AvrBsT and triggers plant cell death and immunity. Plant Physiol. 2015, 167, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hamada, S.; Fujiwara, M.; Zhu, T.; Thao, N.P.; Wong, H.L.; Krishna, P.; Ueda, T.; Kaku, H.; Shibuya, N. The Hop/Sti1-Hsp90 chaperone complex facilitates the maturation and transport of a PAMP receptor in rice innate immunity. Cell Host Microbe 2010, 7, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.; Pelloux, J.; Brownlee, C.; Harper, J.F. Calcium at the crossroads of signaling. Plant Cell 2002, 14, S401–S417. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.F.; Harmon, A. Plants, symbiosis and parasites: A calcium signalling connection. Nat. Rev. Mol. Cell Biol. 2005, 6, 555–566. [Google Scholar] [CrossRef] [PubMed]
- La Verde, V.; Dominici, P.; Astegno, A. Towards understanding plant calcium signaling through calmodulin-like proteins: A biochemical and structural perspective. Int. J. Mol. Sci. 2018, 19, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poovaiah, B.; Du, L.; Wang, H.; Yang, T. Recent advances in calcium/calmodulin-mediated signaling with an emphasis on plant-microbe interactions. Plant Physiol. 2013, 163, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-activated protein kinase cascades in plant hormone signaling. Front. Plant Sci. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Sinha, A.K.; Jaggi, M.; Raghuram, B.; Tuteja, N. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal. Behav. 2011, 6, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Staswick, P.E.; Tiryaki, I. The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell 2004, 16, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Allmann, S.; Wu, J.; Baldwin, I.T. Comparisons of LIPOXYGENASE3-and JASMONATE-RESISTANT4/6-silenced plants reveal that jasmonic acid and jasmonic acid-amino acid conjugates play different roles in herbivore resistance of Nicotiana attenuata. Plant Physiol. 2008, 146, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; An, F.; Feng, Y.; Li, P.; Xue, L.; Mu, A.; Jiang, Z.; Kim, J.-M.; To, T.K.; Li, W. Derepression of ethylene-stabilized transcription factors (EIN3/EIL1) mediates jasmonate and ethylene signaling synergy in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 12539–12544. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z. Molecular basis for jasmonate and ethylene signal interactions in Arabidopsis. J. Exp. Bot. 2014, 65, 5743–5748. [Google Scholar] [CrossRef]
- Solano, R.; Stepanova, A.; Chao, Q.; Ecker, J.R. Nuclear events in ethylene signaling: A transcriptional cascade mediated by ETHYLENE-INSENSITIVE3 and ETHYLENE-RESPONSE-FACTOR1. Genes Dev. 1998, 12, 3703–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boter, M.; Ruíz-Rivero, O.; Abdeen, A.; Prat, S. Conserved MYC transcription factors play a key role in jasmonate signaling both in tomato and Arabidopsis. Genes Dev. 2004, 18, 1577–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Kobayashi, T.; Seo, S. α-Ionone, an apocarotenoid, induces plant resistance to Western Flower Thrips, Frankliniella occidentalis, independently of jasmonic acid. Molecules 2020, 25, 17. [Google Scholar] [CrossRef] [Green Version]
- Luziatelli, F.; Gatti, L.; Ficca, A.G.; Medori, G.; Silvestri, C.; Melini, F.; Muleo, R.; Ruzzi, M. Metabolites Secreted by a Plant-Growth-Promoting Pantoea agglomerans Strain Improved Rooting of Pyrus communis L. cv Dar Gazi Cuttings. Front. Microbiol. 2020, 11, 2412. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Hagen, G.; Guilfoyle, T.J. Aux/IAA proteins contain a potent transcriptional repression domain. Plant Cell 2004, 16, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [CrossRef]
- Nagels Durand, A.; Pauwels, L.; Goossens, A. The ubiquitin system and jasmonate signaling. Plants 2016, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Santner, A.; Pozo, J.C.D.; Murray, J.A.; Estelle, M. Degradation of the cyclinlin system kinase inhibitor KRP1 is regulated by two different ubiquitin E3 ligases. Plant J. 2008, 53, 705–716. [Google Scholar] [CrossRef]
- Lu, H. Dissection of salicylic acid-mediated defense signaling networks. Plant Signal. Behav. 2009, 4, 713–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.; Zhou, J.; Zeng, L.; Xing, D. β-cyclocitral upregulates salicylic acid signalling to enhance excess light acclimation in Arabidopsis. J. Exp. Bot. 2015, 66, 4719–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangerl, A.R.; Hamilton, J.G.; Miller, T.J.; Crofts, A.R.; Oxborough, K.; Berenbaum, M.R.; de Lucia, E.H. Impact of folivory on photosynthesis is greater than the sum of its holes. Proc. Natl. Acad. Sci. USA 2002, 99, 1088–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herms, D.A.; Mattson, W.J. The Dilemma of Plants—To Grow or Defend. Q. Rev. Biol. 1992, 67, 283–335. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Wang, M.-H. Expression of dehydration responsive element-binding protein-3 (DREB3) under different abiotic stresses in tomato. BMB Rep. 2009, 42, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Weber, H.; Hellmann, H. Arabidopsis thaliana BTB/POZpsis thaliana H. Hract with members of the ERF/AP2 transcription factor family. FEBS J. 2009, 276, 6624–6635. [Google Scholar] [CrossRef]
- Larionov, A.; Krause, A.; Miller, W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.; Ryan, P. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshpande, S.; Purkar, V.; Mitra, S. β-Cyclocitral, a Master Regulator of Multiple Stress-Responsive Genes in Solanum lycopersicum L. Plants. Plants 2021, 10, 2465. https://doi.org/10.3390/plants10112465
Deshpande S, Purkar V, Mitra S. β-Cyclocitral, a Master Regulator of Multiple Stress-Responsive Genes in Solanum lycopersicum L. Plants. Plants. 2021; 10(11):2465. https://doi.org/10.3390/plants10112465
Chicago/Turabian StyleDeshpande, Shreyas, Vishwabandhu Purkar, and Sirsha Mitra. 2021. "β-Cyclocitral, a Master Regulator of Multiple Stress-Responsive Genes in Solanum lycopersicum L. Plants" Plants 10, no. 11: 2465. https://doi.org/10.3390/plants10112465