
Suberoylanilide Hydroxamic Acid (SAHA) Is a Driver Molecule of Neuroplasticity: Implication for Neurological Diseases

 ,
, 
Abstract
:1. Introduction
2. The Role of SAHA as a Neuroactive Compound
2.1. Neuronal Maturation and Plasticity
2.2. Activation of Autophagy
2.3. Microtubule Organization
2.4. Regulation of Alternative Splicing Switch
3. SAHA in Neurodevelopmental and Psychiatric Diseases: Current Evidence
3.1. Fragile X Syndrome
3.2. Autism Spectrum Disorders
3.3. Epilepsy
3.4. Depression
4. SAHA in Other Neurological Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- von Bernhardi, R.; Bernhardi, L.E.; Eugenín, J. What Is Neural Plasticity? Adv. Exp. Med. Biol. 2017, 1015, 1–15. [Google Scholar]
- Carafa, V.; Nebbioso, A.; Altucci, L. Histone deacetylase inhibitors: Recent insights from basic to clinical knowledge & patenting of anti-cancer actions. Recent Pat. Anticancer Drug Discov. 2011, 6, 131–145. [Google Scholar]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Guntner, A.S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 2020, 8, 78. [Google Scholar] [CrossRef]
- Gottesfeld, J.M.; Pandolfo, M. Development of histone deacetylase inhibitors as therapeutics for neurological disease. Future Neurol. 2009, 4, 775–784. [Google Scholar] [CrossRef]
- Athira, K.V.; Sadanandan, P.; Chakravarty, S. Repurposing Vorinostat for the Treatment of Disorders Affecting Brain. Neuromolecular Med. 2021, 23, 449–465. [Google Scholar] [CrossRef]
- Ciptasari, U.; van Bokhoven, H. The phenomenal epigenome in neurodevelopmental disorders. Hum. Mol. Genet. 2020, 29, R42–R50. [Google Scholar] [CrossRef]
- Mossink, B.; Negwer, M.; Schubert, D.; Nadif Kasri, N. The emerging role of chromatin remodelers in neurodevelopmental disorders: A developmental perspective. Cell Mol. Life Sci. 2021, 78, 2517–2563. [Google Scholar] [CrossRef]
- Goey, A.K.; Sissung, T.M.; Peer, C.J.; Figg, W.D. Pharmacogenomics and histone deacetylase inhibitors. Pharmacogenomics 2016, 17, 1807–1815. [Google Scholar] [CrossRef]
- Suresh, P.S.; Devaraj, V.C.; Srinivas, N.R.; Mullangi, R. Review of bioanalytical assays for the quantitation of various HDAC inhibitors such as vorinostat, belinostat, panobinostat, romidepsin and chidamine. Biomed. Chromatogr. 2017, 31, 3807. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shariat-Madar, Z.; Walker, L.A.; Tekwani, B.L. Mechanism for neurotropic action of vorinostat; a pan histone deacetylase inhibitor. Mol. Cell. Neurosci. 2016, 77, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Franci, G.; Casalino, L.; Petraglia, F.; Miceli, M.; Menafra, R.; Radic, B.; Tarallo, V.; Vitale, M.; Scarfò, M.; Pocsfalvi, G.; et al. The class I-specific HDAC inhibitor MS-275 modulates the differentiation potential of mouse embryonic stem cells. Biol. Open 2013, 22, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Poeta, L.; Padula, A.; Attianese, B.; Valentino, M.; Verrillo, L.; Filosa, S.; Shoubridge, C.; Barra, A.; Schwartz, C.E.; Christensen, J.; et al. Histone demethylase KDM5C is a SAHA-sensitive central hub at the crossroads of transcriptional axes involved in multiple neurodevelopmental disorders. Hum. Mol. Genet. 2019, 28, 4089–4102. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide hydroxamic acid; a histone deacetylase inhibitor; ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef]
- Benito, E.; Urbanke, H.; Ramachandran, B.; Barth, J.; Halder, R.; Awasthi, A.; Jain, G.; Capece, V.; Burkhardt, S.; Navarro-Sala, M.; et al. HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J. Clin. Investig. 2015, 125, 3572–3584. [Google Scholar] [CrossRef]
- Elvir, L.; Duclot, F.; Wang, Z.; Kabbaj, M. Epigenetic regulation of motivated behaviors by histone deacetylase inhibitors. Neurosci. Biobehav. Rev. 2019, 105, 305–317. [Google Scholar] [CrossRef]
- Lunke, S.; Maxwell, S.; Khurana, I.; Kaipananickal, H.; Okabe, J.; Al-Hasani, K.; El-Osta, A. Epigenetic evidence of an Ac/Dc axis by VPA and SAHA. Clin. Epigenet. 2021, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Kakoty, V.; Sarathlal, K.C.; Dubey, S.K.; Yang, C.H.; Marathe, S.A.; Taliyan, R. Epigenetic regulation and autophagy modulation debilitates insulin resistance associated Alzheimer’s disease condition in rats. Metab. Brain Dis. 2022, 37, 927–944. [Google Scholar] [CrossRef]
- Feng, X.L.; Deng, H.B.; Wang, Z.G.; Wu, Y.; Ke, J.J.; Feng, X.B. Suberoylanilide Hydroxamic Acid Triggers Autophagy by Influencing the mTOR Pathway in the Spinal Dorsal Horn in a Rat Neuropathic Pain Model. Neurochem. Res. 2019, 44, 450–464. [Google Scholar] [CrossRef]
- Fang, P.; Chen, C.; Zheng, F.; Jia, J.; Chen, T.; Zhu, J.; Chang, J.; Zhang, Z. NLRP3 inflammasome inhibition by histone acetylation ameliorates sevoflurane-induced cognitive impairment in aged mice by activating the autophagy pathway. Brain Res. Bull. 2021, 172, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Neault, N.; Ravel-Chapuis, A.; Baird, S.D.; Lunde, J.A.; Poirier, M.; Staykov, E.; Plaza-Diaz, J.; Medina, G.; Abadía-Molina, F.; Jasmin, B.J.; et al. Vorinostat Improves Myotonic Dystrophy Type 1 Splicing Abnormalities in DM1 Muscle Cell Lines and Skeletal Muscle from a DM1 Mouse Model. Int. J. Mol. Sci. 2023, 24, 3794. [Google Scholar] [CrossRef]
- Rahhal, R.; Seto, E. Emerging roles of histone modifications and HDACs in RNA splicing. Nucleic Acids Res. 2019, 47, 4911–4926. [Google Scholar] [CrossRef]
- Nakano, Y.; Kelly, M.C.; Rehman, A.U.; Boger, E.T.; Morell, R.J.; Kelley, M.W.; Friedman, T.B.; Bánfi, B. Defects in the Alternative Splicing-Dependent Regulation of REST Cause Deafness. Cell 2018, 174, 536–548. [Google Scholar] [CrossRef]
- Takada, N.; Nakamura, Y.; Ikeda, K.; Takaoka, N.; Hisaoka-Nakashima, K.; Sanoh, S.; Kotake, Y.; Nakata, Y.; Morioka, N. Treatment with Histone Deacetylase Inhibitor Attenuates Peripheral Inflammation-Induced Cognitive Dysfunction and Microglial Activation: The Effect of SAHA as a Peripheral HDAC Inhibitor. Neurochem. Res. 2021, 46, 2285–2296. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Koppel, I.; Timmusk, T. Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115. [Google Scholar] [CrossRef]
- Hossain, M.S.; Oomura, Y.; Katafuchi, T. Glucose Can Epigenetically Alter the Gene Expression of Neurotrophic Factors in the Murine Brain Cells. Mol. Neurobiol. 2018, 55, 3408–3425. [Google Scholar] [CrossRef]
- Rusconi, F.; Grillo, B.; Ponzoni, L.; Bassani, S.; Toffolo, E.; Paganini, L.; Mallei, A.; Braida, D.; Passafaro, M.; Popoli, M.; et al. LSD1 modulates stress-evoked transcription of immediate early genes and emotional behavior. Proc. Natl. Acad. Sci. USA 2016, 113, 3651–3656. [Google Scholar] [CrossRef]
- de la Fuente Revenga, M.; Ibi, D.; Saunders, J.M.; Cuddy, T.; Ijaz, M.K.; Toneatti, R.; Kurita, M.; Holloway, T.; Shen, L.; Seto, J.; et al. HDAC2-dependent Antipsychotic-like Effects of Chronic Treatment with the HDAC Inhibitor SAHA in Mice. Neuroscience 2018, 388, 102–117. [Google Scholar] [PubMed]
- Cavallo, F.; Troglio, F.; Fagà, G.; Fancelli, D.; Shyti, R.; Trattaro, S.; Zanella, M.; D’Agostino, G.; Hughes, J.M.; Cera, M.R.; et al. High-throughput screening identifies histone deacetylase inhibitors that modulate GTF2I expression in 7q11.23 microduplication autism spectrum disorder patient-derived cortical neurons. Mol. Autism 2020, 11, 88. [Google Scholar] [PubMed]
- Luo, K.; Wang, Z.; Zhuang, K.; Yuan, S.; Liu, F.; Liu, A. Suberoylanilide hydroxamic acid suppresses axonal damage and neurological dysfunction after subarachnoid hemorrhage via the HDAC1/HSP70/TDP-43 axis. Exp. Mol. Med. 2022, 54, 1423–1433. [Google Scholar] [PubMed]
- Colucci-D’Amato, L.; Perrone-Capano, C.; di Porzio, U. Chronic activation of ERK and neurodegenerative diseases. Bioessays 2003, 25, 1085–1095. [Google Scholar]
- Medina, J.H.; Viola, H. ERK1/2: A Key Cellular Component for the Formation, Retrieval, Reconsolidation and Persistence of Memory. Front. Mol. Neurosci. 2018, 11, 361. [Google Scholar] [PubMed]
- Fusco, F.R.; Anzilotti, S.; Giampà, C.; Dato, C.; Laurenti, D.; Leuti, A.; Colucci D’Amato, L.; Perrone, L.; Bernardi, G.; Melone, M.A. Changes in the expression of extracellular regulated kinase (ERK 1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol. Dis. 2012, 46, 225–233. [Google Scholar]
- Nizzari, M.; Barbieri, F.; Gentile, M.T.; Passarella, D.; Caorsi, C.; Diaspro, A.; Taglialatela, M.; Pagano, A.; Colucci-D’Amato, L.; Florio, T.; et al. Amyloid-β protein precursor regulates phosphorylation and cellular compartmentalization of microtubule associated protein tau. J. Alzheimer’s Dis. 2012, 29, 211–227. [Google Scholar] [CrossRef]
- Chen, S.H.; Wu, H.M.; Ossola, B.; Schendzielorz, N.; Wilson, B.C.; Chu, C.H.; Chen, S.L.; Wang, Q.; Zhang, D.; Qian, L.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 2012, 165, 494–505. [Google Scholar]
- Kurundkar, D.; Srivastava, R.K.; Chaudhary, S.C.; Ballestas, M.E.; Kopelovich, L.; Elmets, C.A.; Athar, M. Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol. Appl. Pharmacol. 2013, 266, 233–244. [Google Scholar]
- Swarup, V.; Hinz, F.I.; Rexach, J.E.; Noguchi, K.I.; Toyoshiba, H.; Oda, A.; Hirai, K.; Sarkar, A.; Seyfried, N.T.; Cheng, C.; et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat. Med. 2019, 25, 152–164. [Google Scholar]
- Lv, M.; Ma, Q. Autophagy in Neurodevelopmental Disorders. Adv. Exp. Med. Biol. 2020, 1207, 171–182. [Google Scholar]
- Thomas, S.D.; Jha, N.K.; Ojha, S.; Sadek, B. mTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, 28, 1889. [Google Scholar]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012, 11, 332–341. [Google Scholar]
- Moloney, P.B.; Cavalleri, G.L.; Delanty, N. Epilepsy in the mTORopathies: Opportunities for precision medicine. Brain Commun. 2021, 3, 4. [Google Scholar]
- Drongitis, D.; Caterino, M.; Verrillo, L.; Santonicola, P.; Costanzo, M.; Poeta, L.; Attianese, B.; Barra, A.; Terrone, G.; Lioi, M.B.; et al. Deregulation of microtubule organization and RNA metabolism in Arx models for lissencephaly and developmental epileptic encephalopathy. Hum. Mol. Genet. 2022, 31, 1884–1908. [Google Scholar]
- Gold, W.A.; Lacina, T.A.; Cantrill, L.C.; Christodoulou, J. MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med. 2015, 93, 63–72. [Google Scholar] [PubMed]
- Cappelletti, G.; Calogero, A.M.; Rolando, C. Microtubule acetylation: A reading key to neural physiology and degeneration. Neurosci. Lett. 2021, 755, 135900. [Google Scholar]
- Adalbert, R.; Kaieda, A.; Antoniou, C.; Loreto, A.; Yang, X.; Gilley, J.; Hoshino, T.; Uga, K.; Makhija, M.T.; Coleman, M.P. Novel HDAC6 Inhibitors Increase Tubulin Acetylation and Rescue Axonal Transport of Mitochondria in a Model of Charcot-Marie-Tooth Type 2F. ACS Chem. Neurosci. 2020, 11, 258–267. [Google Scholar]
- Su, C.H.; Dhananjaya, D.; Tarn, W.Y. Alternative Splicing in Neurogenesis and Brain Development. Front. Mol. Biosci. 2018, 12, 12. [Google Scholar]
- Joglekar, A.; Prjibelski, A.; Mahfouz, A.; Collier, P.; Lin, S.; Schlusche, A.K.; Marrocco, J.; Williams, S.R.; Haase, B.; Hayes, A.; et al. A spatially resolved brain region- and cell type-specific isoform atlas of the postnatal mouse brain. Nat. Commun. 2021, 12, 463. [Google Scholar] [PubMed]
- Ding, Q.; Wu, X.; Li, X.; Wang, H. Vorinostat Corrects Cognitive and Non-Cognitive Symptoms in a Mouse Model of Fragile X Syndrome. Int. J. Neuropsychopharmacol. 2022, 25, 147–159. [Google Scholar]
- Uchida, S.; Yamagata, H.; Seki, T.; Watanabe, Y. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018, 72, 212–227. [Google Scholar] [PubMed]
- Ibhazehiebo, K.; Gavrilovici, C.; de la Hoz, C.L.; Ma, S.C.; Rehak, R.; Kaushik, G.; Meza Santoscoy, P.L.; Scott, L.; Nath, N.; Kim, D.Y.; et al. A novel metabolism-based phenotypic drug discovery platform in zebrafish uncovers HDACs 1 and 3 as a potential combined anti-seizure drug target. Brain 2018, 141, 744–761. [Google Scholar] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [PubMed]
- Li, Y.; Stockton, M.E.; Eisinger, B.E.; Zhao, Y.; Miller, J.L.; Bhuiyan, I.; Gao, Y.; Wu, Z.; Peng, J.; Zhao, X. Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat. Commun. 2018, 9, 2494. [Google Scholar]
- Sarathlal, K.C.; Kakoty, V.; Krishna, K.V.; Dubey, S.K.; Chitkara, D.; Taliyan, R. Neuroprotective Efficacy of Co-Encapsulated Rosiglitazone and Vorinostat Nanoparticle on Streptozotocin Induced Mice Model of Alzheimer Disease. ACS Chem. Neurosci. 2021, 12, 1528–1541. [Google Scholar]
- Gao, Y.; Aljazi, M.B.; He, J. Neural Hyperactivity Is a Core Pathophysiological Change Induced by Deletion of a High Autism Risk Gene Ash1L in the Mouse Brain. Front. Behav. Neurosci. 2022, 16, 873466. [Google Scholar]
- Mielcarek, M.; Benn, C.L.; Franklin, S.A.; Smith, D.L.; Woodman, B.; Marks, P.A.; Bates, G.P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011, 11, e27746. [Google Scholar] [CrossRef]
- Fuchikami, M.; Yamamoto, S.; Morinobu, S.; Okada, S.; Yamawaki, Y.; Yamawaki, S. The potential use of histone deacetylase inhibitors in the treatment of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 320–324. [Google Scholar]
- Chen, W.Y.; Zhang, H.; Gatta, E.; Glover, E.J.; Pandey, S.C.; Lasek, A.W. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 2019, 78, 79–87. [Google Scholar]
- Ershadi, A.S.B.; Amini-Khoei, H.; Hosseini, M.J.; Dehpour, A.R. SAHA Improves Depressive Symptoms; Cognitive Impairment and Oxidative Stress: Rise of a New Antidepressant Class. Neurochem. Res. 2021, 46, 1252–1263. [Google Scholar] [CrossRef]
- Perron, N.R.; Nasarre, C.; Bandyopadhyay, M.; Beeson, C.C.; Rohrer, B. SAHA is neuroprotective in in vitro and in situ models of retinitis pigmentosa. Mol. Vis. 2021, 27, 151–160. [Google Scholar] [PubMed]
- Basu, T.; O’Riordan, K.J.; Schoenike, B.A.; Khan, N.N.; Wallace, E.P.; Rodriguez, G.; Maganti, R.K.; Roopra, A. Histone deacetylase inhibitors restore normal hippocampal synaptic plasticity and seizure threshold in a mouse model of Tuberous Sclerosis Complex. Sci. Rep. 2019, 9, 5266. [Google Scholar] [CrossRef]
- Hong, D.; Iakoucheva, L.M. Therapeutic strategies for autism: Targeting three levels of the central dogma of molecular biology. Transl. Psychiatry 2023, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent De Novo CNVs; including duplications of the 7q11.23 Williams syndrome region; are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef]
- Pober, B.R. Williams-Beuren syndrome. N. Engl. J. Med. 2010, 36, 239–252. [Google Scholar] [CrossRef]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact; Potential Mechanisms; and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Chiarello, D.; Gogou, M.; Silvennoinen, K.; Puvirajasinghe, C.; Jones, W.D.; Reif, P.; Klein, K.M.; Rosenow, F.; Weber, Y.; et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1044–1052. [Google Scholar] [CrossRef]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Paulhus, K.; Ammerman, L.; Glasscock, E. Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int. J. Mol. Sci. 2020, 21, 2802. [Google Scholar] [CrossRef]
- Nabbout, R.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D Amato, L.; d’Augères, G.B.; de Vries, P.J.; et al. Epilepsy in tuberous sclerosis complex: Findings from the TOSCA Study. Epilepsia Open 2018, 4, 73–84. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Primers 2016, 2, 16065. [Google Scholar] [CrossRef] [PubMed]
- Athira, K.V.; Madhana, R.M.; Js, I.C.; Lahkar, M.; Sinha, S.; Naidu, V.G.M. Antidepressant activity of vorinostat is associated with amelioration of oxidative stress and inflammation in a corticosterone-induced chronic stress model in mice. Behav. Brain Res. 2018, 344, 73–84. [Google Scholar]
- Misztak, P.; Sowa-Kućma, M.; Szewczyk, B.; Nowak, G. Vorinostat (SAHA) May Exert Its Antidepressant-Like Effects Through the Modulation of Oxidative Stress Pathways. Neurotox. Res. 2021, 39, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.M.; Fridell, R.A.; Boger, E.T.; San Agustin, T.B.; Madeo, A.C.; Griffith, A.J.; Friedman, T.B.; Morell, R.J. A locus for autosomal dominant progressive non-syndromic hearing loss; DFNA27; is on chromosome 4q12-13.1. Clin. Genet. 2008, 73, 367–372. [Google Scholar] [CrossRef]
- Baldelli, P.; Meldolesi, J. The Transcription Repressor REST in Adult Neurons: Physiology, Pathology, and Diseases. eNeuro 2015, 2, e0010-15.2015. [Google Scholar] [CrossRef]
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef]
- Svechnikova, I.; Almqvist, P.M.; Ekström, T.J. HDAC inhibitors effectively induce cell type-specific differentiation in human glioblastoma cell lines of different origin. Int. J. Oncol. 2008, 32, 821–827. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
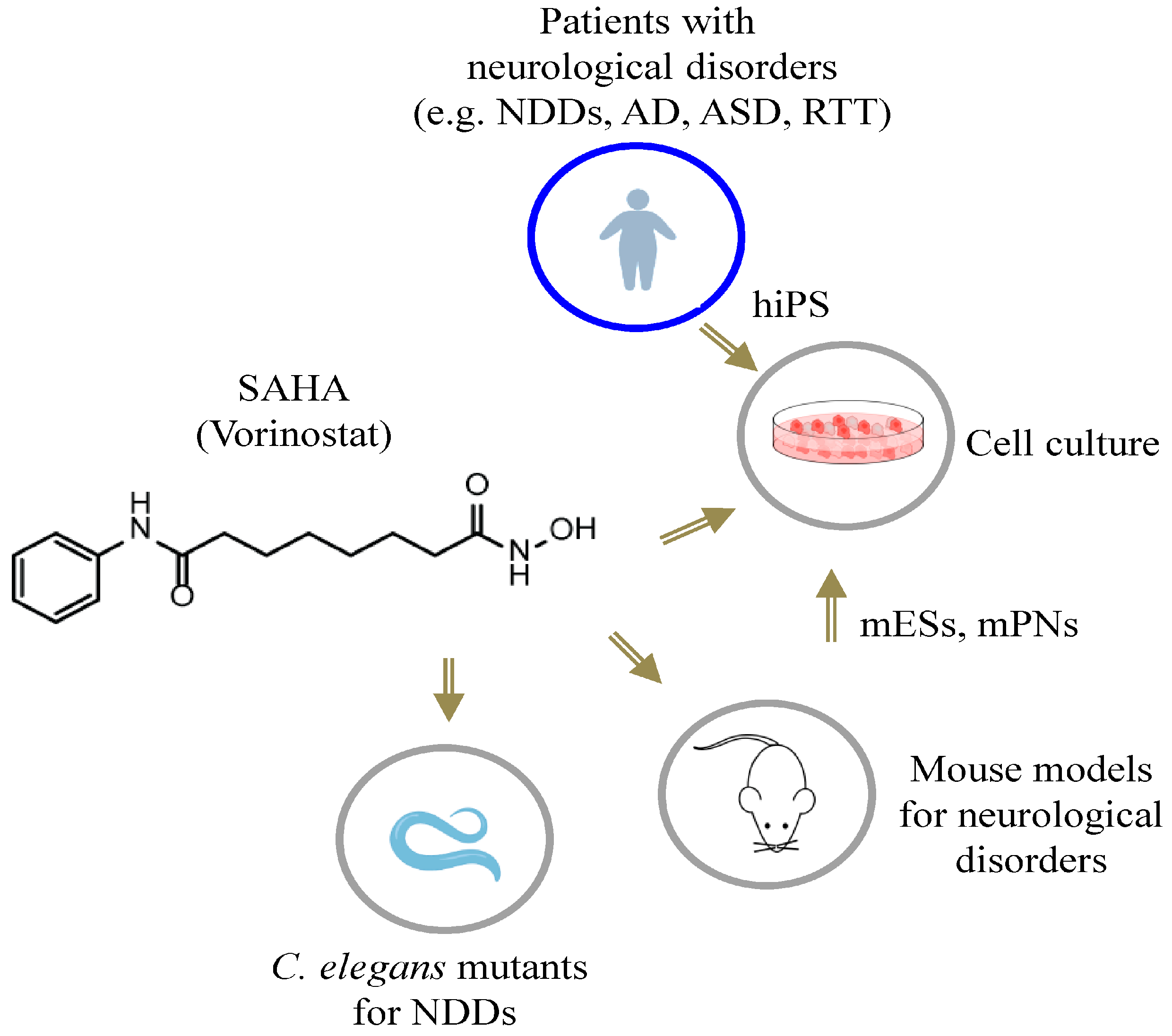{kind=link}
{kind=link}
| Mechanism | Molecular Pathway/Mechanism of Action | Ref. |
|---|---|---|
| Induction of neuronal differentiation | Restoring of KDM5C-H3K4me3 signaling; induction of Bdnf transcription | [14] |
| Regulation of autophagy | Activation of the ULK1 complex; suppression of mTOR; increase in the autophagy markers LC3 and P62 | [19,21,27] |
| Regulation of axonal transport of BDNF | Increase in α-tubulin acetylation, enhancing kinesin-1 motility along MTs | [22] |
| Increase in neurite outgrowth | ERK phosphorylation; increase in α-tubulin and H3 and H4 histones acetylation | [11] |
| Memory formation | Induction of Bdnf transcription | [28,29] |
| Neuronal morphogenesis and synaptic plasticity | Increase in the expression of plasticity-related genes | [30,31,32] |
| Inhibition of axonal damage | HSP70 acetylation | [33] |
| Human Disease | Disease Models | Molecular Pathway/Mechanism of Action | Ref. |
|---|---|---|---|
| Alzheimer’s disease (AD) [MIM: 104300] | AD mice | improvement of the oxidative stress response; increase in neurotrophic factor levels; improvement of cognitive functions (in combination with Rosiglitazone) | [56] |
| Autism spectrum disorder (ASD) [MIM: 209850] | 7Dup iPSC-derived cortical neurons | correction of the aberrant transcriptional level of GTF2I | [32] |
| Ash1L-Nes-cKO mice | amelioration of AS-like behaviors and ID phenotype | [57] | |
| Deafness autosomal dominant 27 (DFNA27) [MIM: 612431] | Rest-exon 4 deficient mice | increase in REST-target gene expression; rescue of inner and outer hair cells stereocilia formation; partial rescue of hearing defects | [25] |
| Episodic ataxia/myokymia syndrome [MIM: 176270] | Kcna1-null zebrafish Kcna1-KO mice | seizure frequency reduction | [53] |
| Fragile X syndrome (FXS) [MIM: 300604] | FMR1 KO mice | amelioration of memory, repetitive behavior, and social interaction deficits | [51] |
| Huntington’s disease (HD) [MIM: 143100] | HTT- R6/2 mice | improvement of motor impairments; decrease in insoluble aggregates in the cortex and brain stem; restoration of Bdnf transcript levels in the cortex | [15,58] |
| Major depressive disorder (MDD) [MIM: 608516] | chronic-stress mice model | correction of defective neurotrophin levels; rescue of depressive-like behavior | [52,59] |
| alcohol withdrawal stress rat model | rescue of H3K9 acetylation in hippocampus; rescue of depressive-like behavior | [60] | |
| maternal separation stress mice model | rescue of depressive-like behavior and counteraction of neuroinflammation | [61] | |
| Myotonic Dystrophy Type 1 (DM1) [MIM: 160900] | DM1 patient myoblast DM1- HSASR mice | inhibition of RNA foci formation and release of MBNL1 splicing factor; improvement of aberrant mRNA splicing | [23] |
| Retinitis Pigmentosa (RP) [MIM: 268000] | RP-rd1 mice | improvement of mitochondrial respiration in photoreceptor cells; increase in photoreceptor cell survival | [62] |
| Tuberous sclerosis (TSC) [MIM: 191100] | TSC2+/− mice | amelioration of synaptic plasticity and reduction of seizure threshold | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verrillo, L.; Di Palma, R.; de Bellis, A.; Drongitis, D.; Miano, M.G. Suberoylanilide Hydroxamic Acid (SAHA) Is a Driver Molecule of Neuroplasticity: Implication for Neurological Diseases. Biomolecules 2023, 13, 1301. https://doi.org/10.3390/biom13091301
Verrillo L, Di Palma R, de Bellis A, Drongitis D, Miano MG. Suberoylanilide Hydroxamic Acid (SAHA) Is a Driver Molecule of Neuroplasticity: Implication for Neurological Diseases. Biomolecules. 2023; 13(9):1301. https://doi.org/10.3390/biom13091301
Chicago/Turabian StyleVerrillo, Lucia, Rosita Di Palma, Alberto de Bellis, Denise Drongitis, and Maria Giuseppina Miano. 2023. "Suberoylanilide Hydroxamic Acid (SAHA) Is a Driver Molecule of Neuroplasticity: Implication for Neurological Diseases" Biomolecules 13, no. 9: 1301. https://doi.org/10.3390/biom13091301