
Cyclosporines Antagonize the Antiviral Activity of IFITMProteins by Redistributing Them toward the Golgi Apparatus


{kind=link}
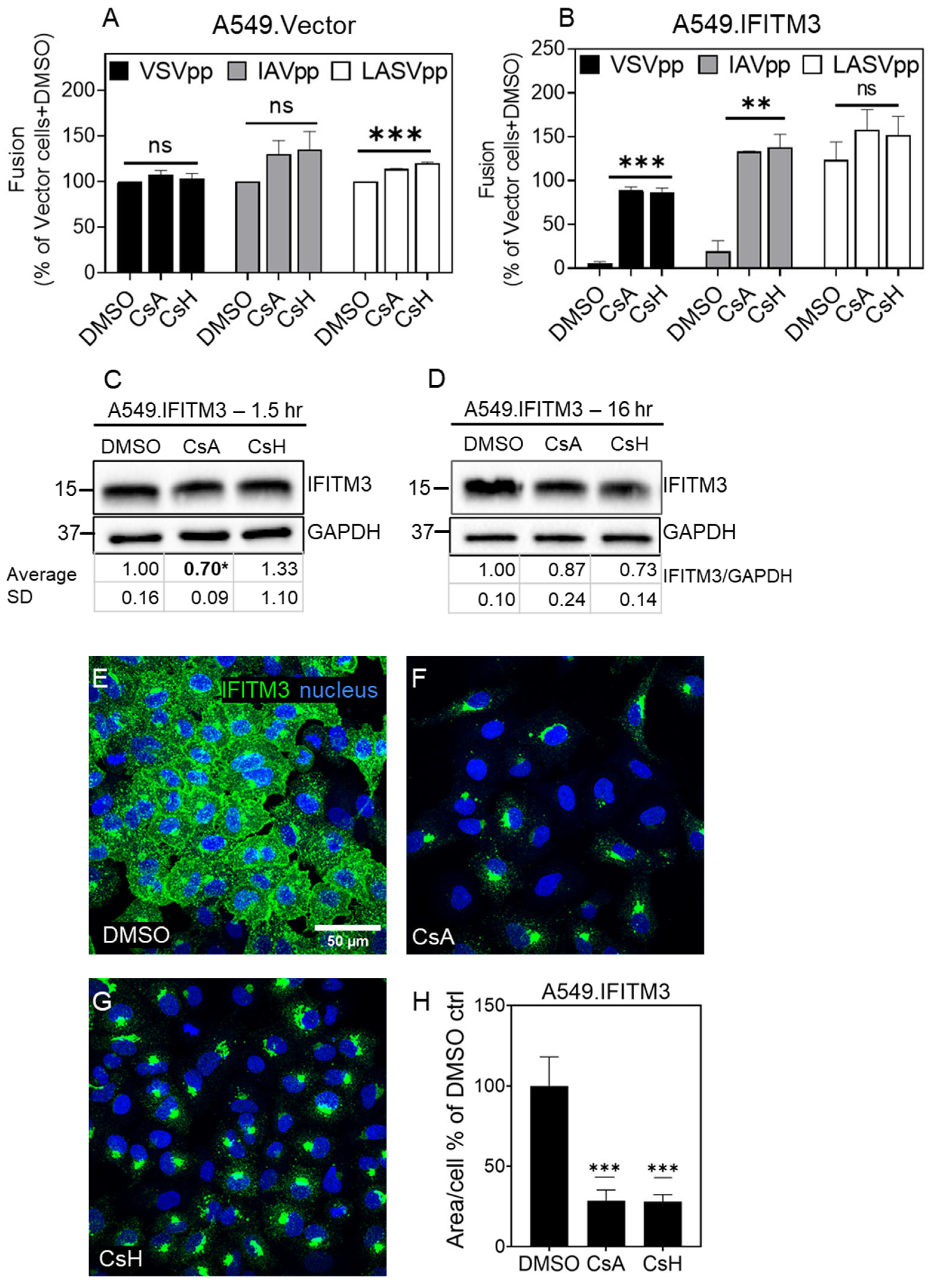{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Plasmids, and Reagents
2.2. Pseudovirus Production and Characterization
2.3. Virus–Cell Fusion Assay
2.4. LOPAC Library Screen
2.5. Western Blot Analysis
2.6. Immunostaining, Microscopy, and Image Analysis
2.7. Statistical Analysis
3. Results
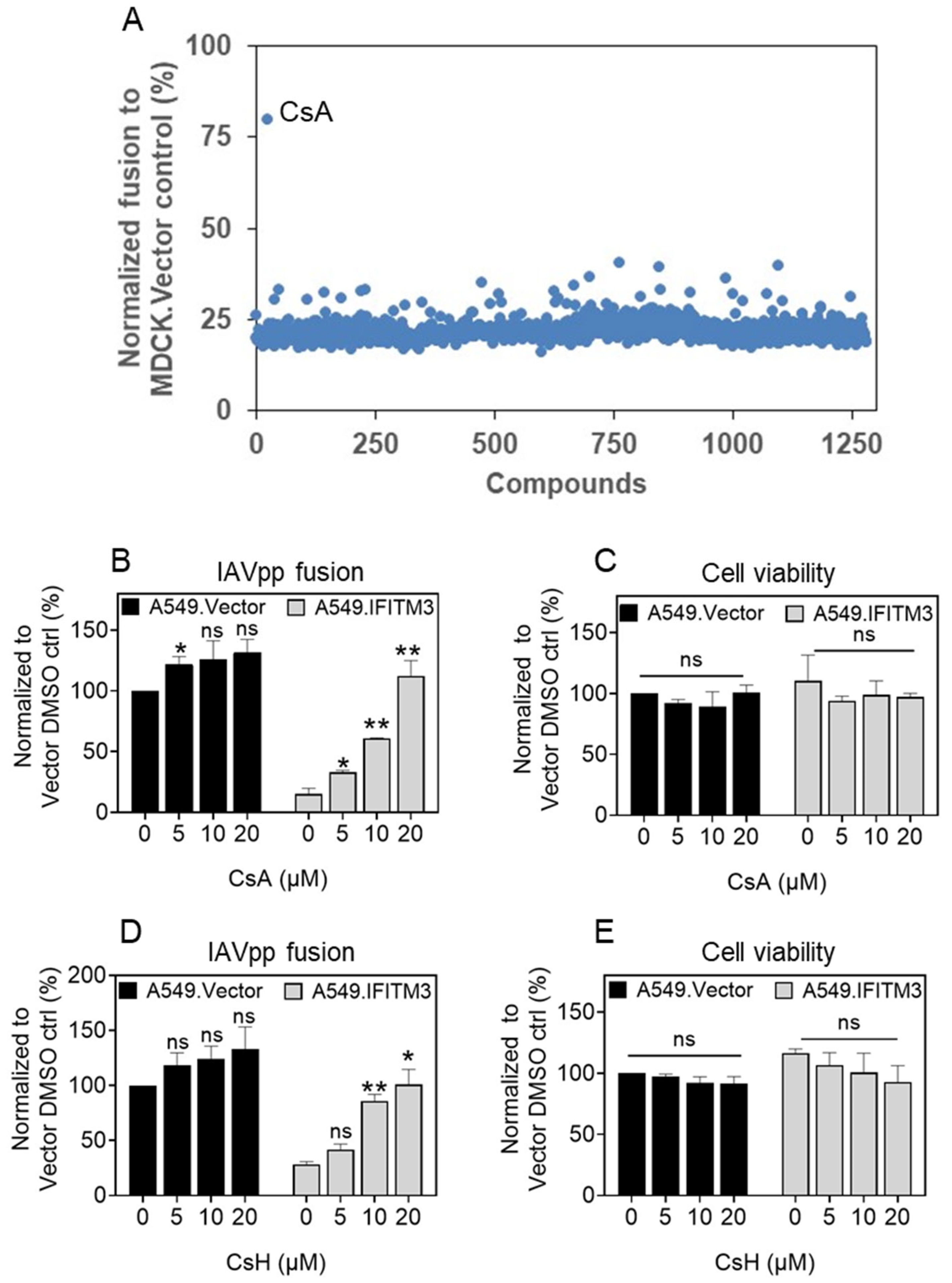
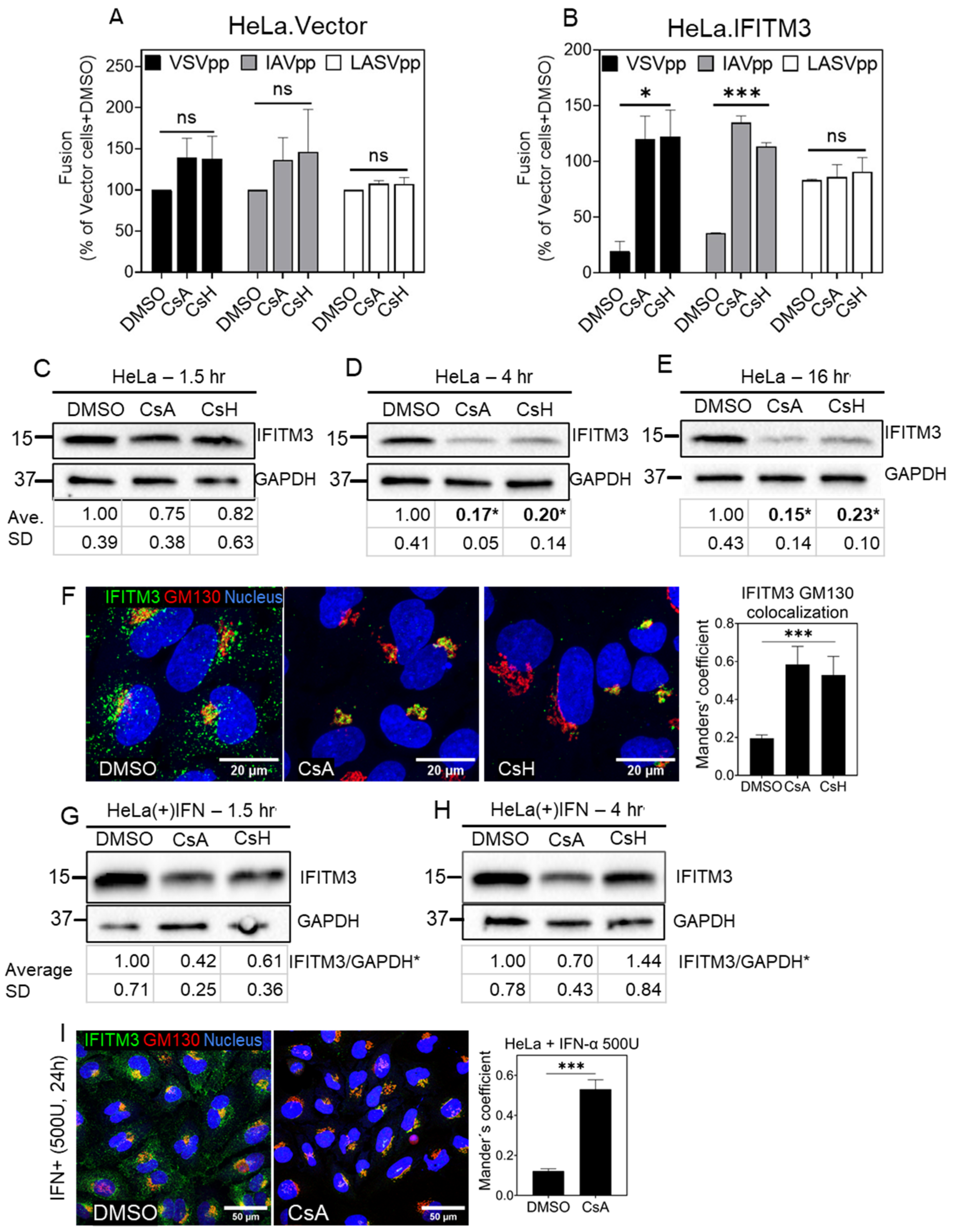
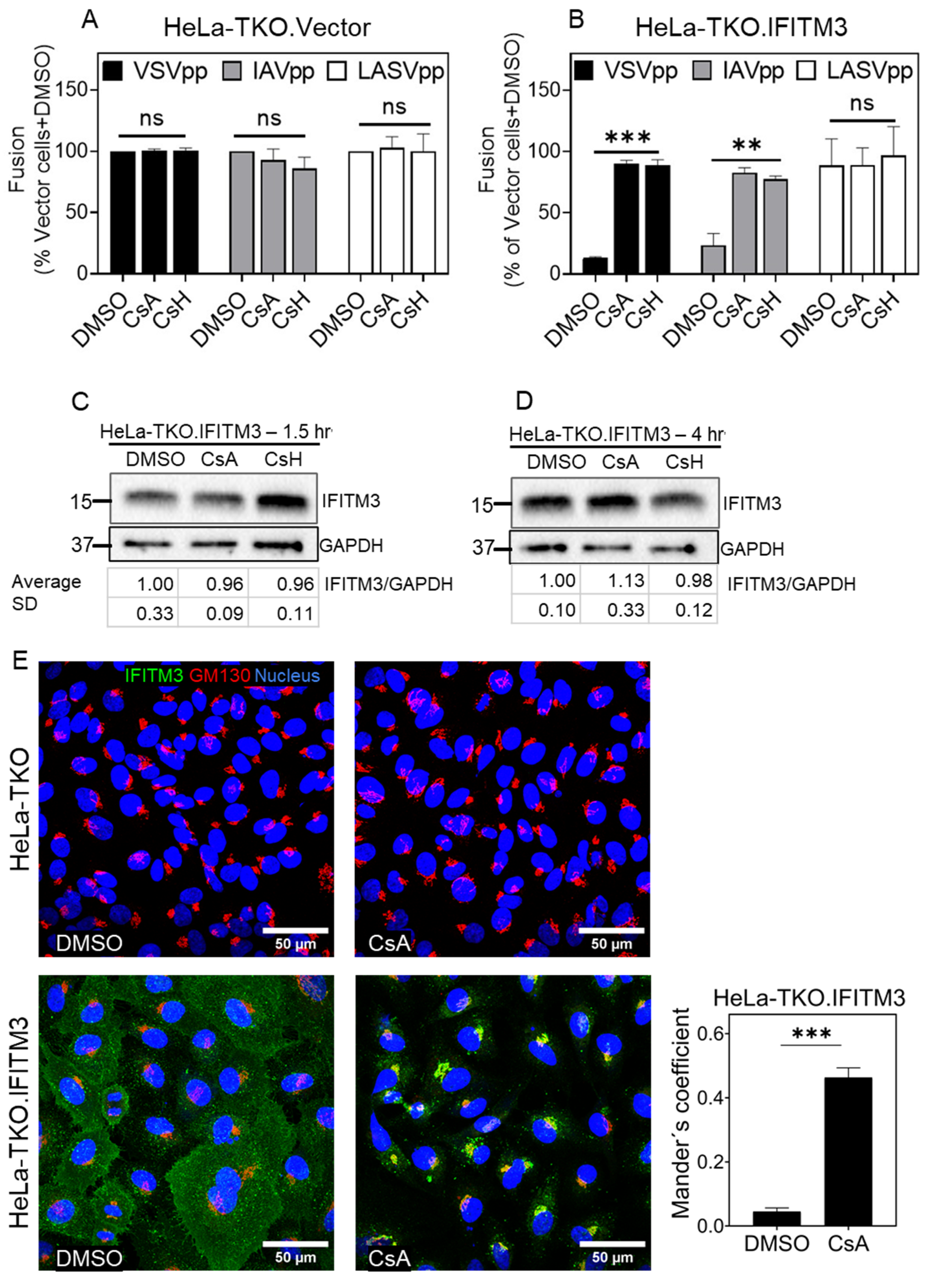
3.1. Cyclosporine a Antagonizes the Antiviral Activity of IFITM3
3.2. Cyclosporines can Promote Virus Infection without Inducing IFITM3 Degradation

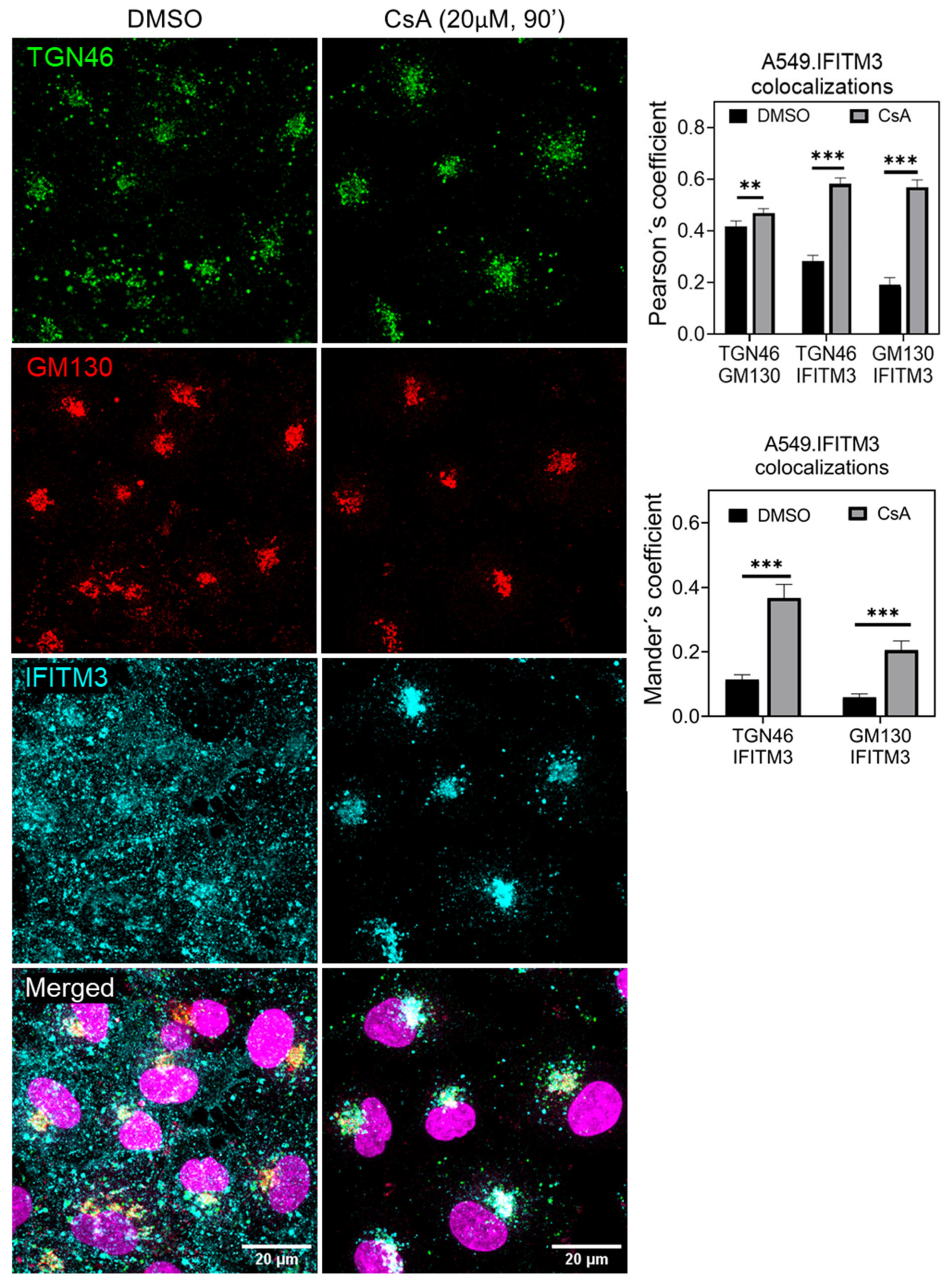
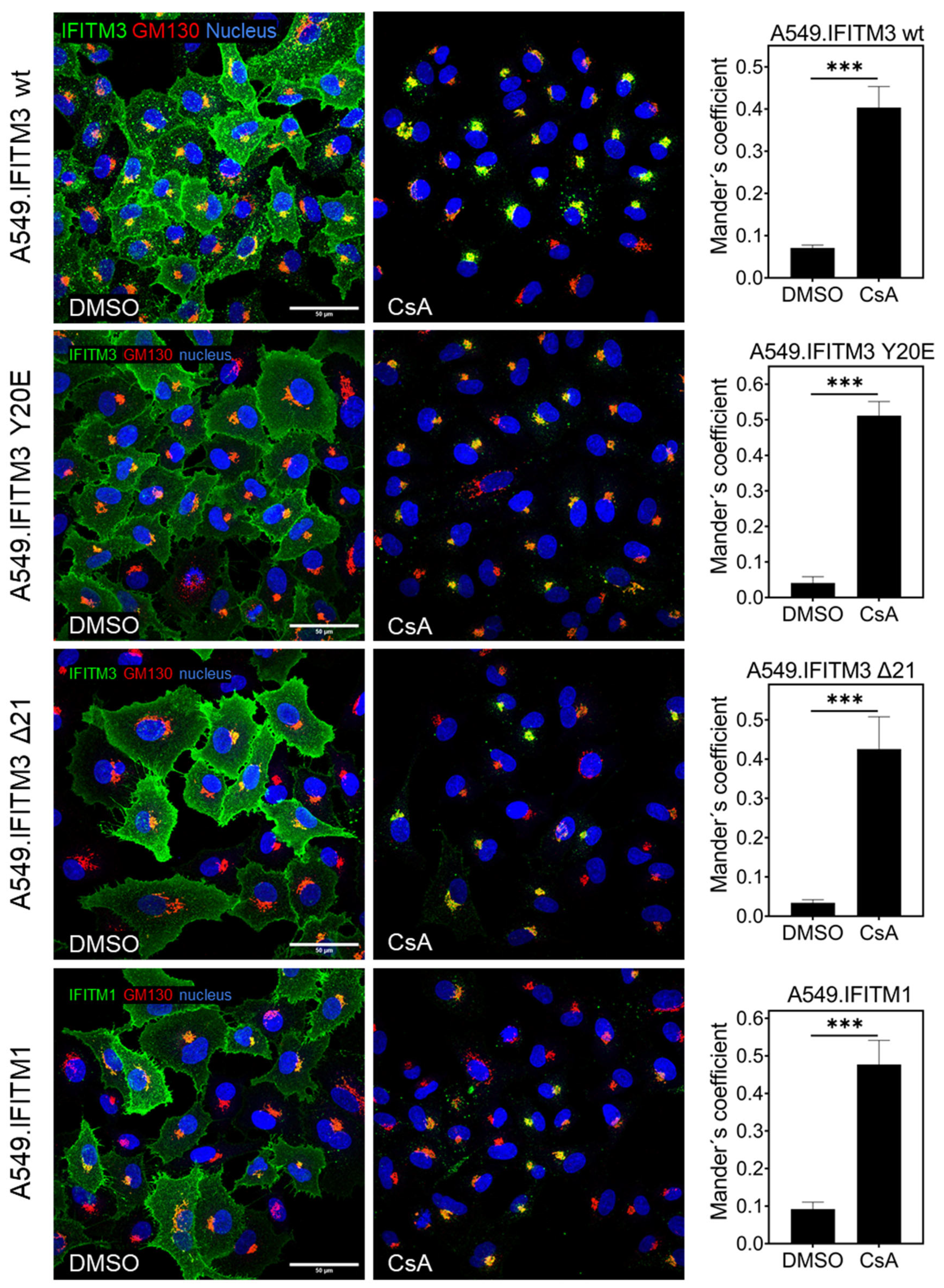
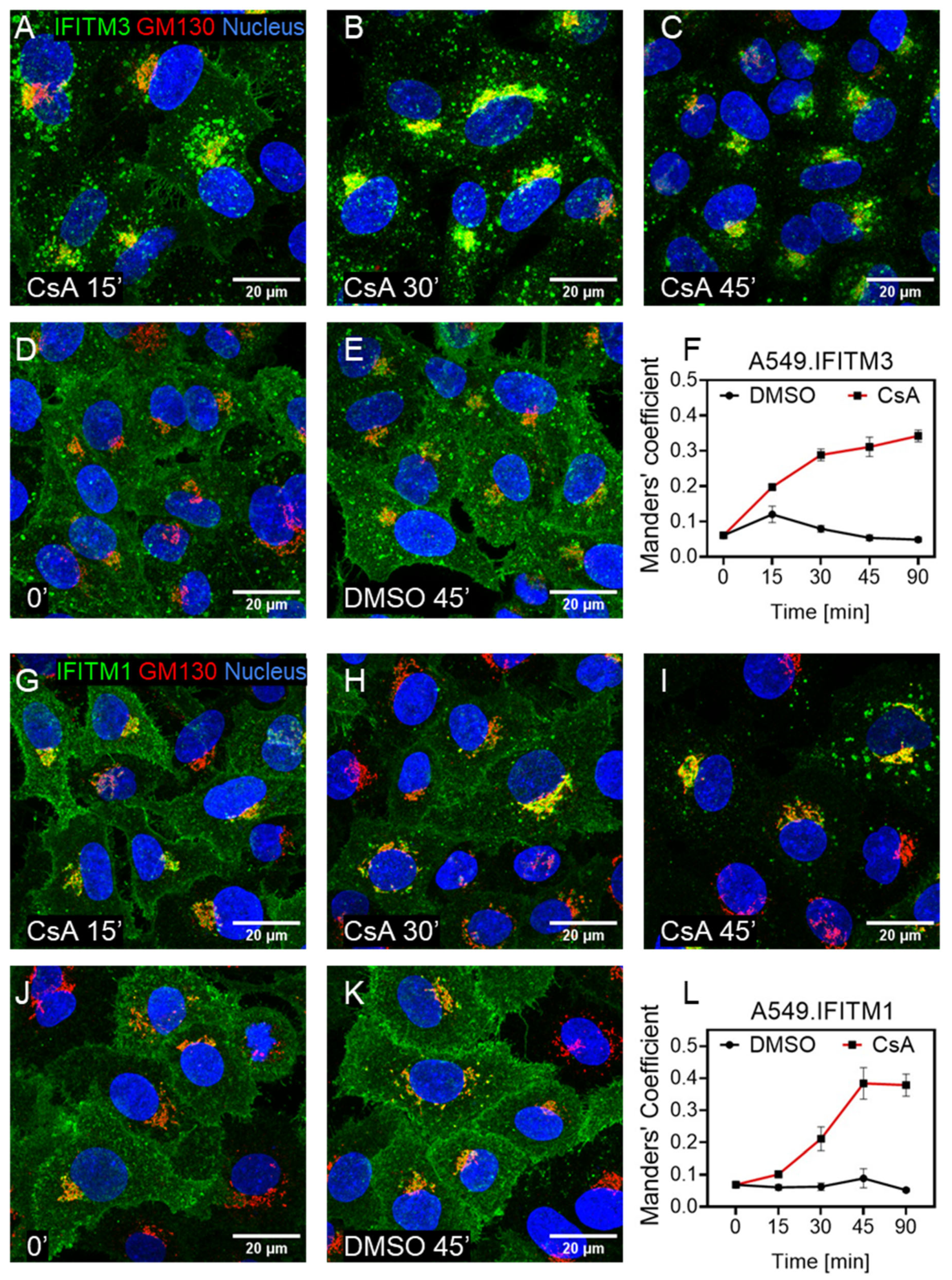
3.3. Cyclosporine Treatment Quickly Relocates IFITMs toward the Golgi Area
3.4. Prolonged CsA Exposure induces IFITM3 Degradation in Cells Expressing Low/Endogenous Levels of This Protein
3.5. Rapamycin Induces IFITM Redistribution and Rescues Viral Fusion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [Green Version]
- Marziali, F.; Cimarelli, A. Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs. Cells 2021, 10, 1171. [Google Scholar] [CrossRef]
- Yánez, D.C.; Ross, S.; Crompton, T. The IFITM protein family in adaptive immunity. Immunology 2019, 159, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Huang, I.-C.; Kam, C.; Farzan, M. Ifitm3 Limits the Severity of Acute Influenza in Mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef] [Green Version]
- Everitt, A.R.; Clare, S.; McDonald, J.U.; Kane, L.; Harcourt, K.; Ahras, M.; Lall, A.; Hale, C.; Rodgers, A.; Young, D.B.; et al. Defining the range of pathogens susceptible to Ifitm3 restriction using a knockout mouse model. PLoS ONE 2013, 8, e80723. [Google Scholar] [CrossRef] [Green Version]
- Everitt, A.R.; The GenISIS Investigators; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Zani, A.; Yount, J.S. Antiviral Protection by IFITM3 In Vivo. Curr. Clin. Microbiol. Rep. 2018, 5, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Sakhaee, F.; Sotoodehnejadnematalahi, F.; Zamani, M.S.; Ahmadi, I.; Anvari, E.; Fateh, A. Increased risk of COVID-19 mortality rate in IFITM3 rs6598045 G allele carriers infected by SARS-CoV-2 delta variant. Hum. Genom. 2022, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Jeong, M.-J.; Jeong, B.-H. Strong association of regulatory single nucleotide polymorphisms (SNPs) of the IFITM3 gene with influenza H1N1 2009 pandemic virus infection. Cell. Mol. Immunol. 2020, 17, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Schönfelder, K.; Breuckmann, K.; Elsner, C.; Dittmer, U.; Fistera, D.; Herbstreit, F.; Risse, J.; Schmidt, K.; Sutharsan, S.; Taube, C.; et al. The influence of IFITM3 polymorphisms on susceptibility to SARS-CoV-2 infection and severity of COVID-19. Cytokine 2021, 142, 155492. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, L.N.; Li, W.; Zi, H.R.; Guo, Y.; Yan, W.J.; Chen, X.B.; Wei, P.M. IFITM3 rs12252 T>C polymorphism is associated with the risk of severe influenza: A meta-analysis. Epidemiol. Infect. 2015, 143, 2975–2984. [Google Scholar] [CrossRef]
- Friedlová, N.; Kokáš, F.Z.; Hupp, T.R.; Vojtěšek, B.; Nekulová, M. IFITM protein regulation and functions: Far beyond the fight against viruses. Front. Immunol. 2022, 13, 1042368. [Google Scholar] [CrossRef]
- Lee, J. Does IFITM3 link inflammation to tumorigenesis? BMB Rep. 2022, 55, 602–608. [Google Scholar] [CrossRef]
- Rajapaksa, U.S.; Jin, C.; Dong, T. Malignancy and IFITM3: Friend or Foe? Front. Oncol. 2020, 10, 593245. [Google Scholar] [CrossRef]
- Buchrieser, J.; Degrelle, S.A.; Couderc, T.; Nevers, Q.; Disson, O.; Manet, C.; Donahue, D.A.; Porrot, F.; Hillion, K.-H.; Perthame, E.; et al. IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science 2019, 365, 176–180. [Google Scholar] [CrossRef]
- McMichael, T.M.; Zhang, L.; Chemudupati, M.; Hach, J.C.; Kenney, A.D.; Hang, H.C.; Yount, J.S. The palmitoyltransferase ZDHHC20 enhances interferon-induced transmembrane protein 3 (IFITM3) palmitoylation and antiviral activity. J. Biol. Chem. 2017, 292, 21517–21526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesarino, N.; McMichael, T.M.; Yount, J.S. E3 Ubiquitin Ligase NEDD4 Promotes Influenza Virus Infection by Decreasing Levels of the Antiviral Protein IFITM3. PLoS Pathog. 2015, 11, e1005095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yount, J.; Karssemeijer, R.A.; Hang, H.C. S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J. Biol. Chem. 2012, 287, 19631–19641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesarino, N.M.; McMichael, T.M.; Hach, J.C.; Yount, J.S. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J. Biol. Chem. 2014, 289, 11986–11992. [Google Scholar] [CrossRef] [Green Version]
- Chesarino, N.M.; McMichael, T.M.; Yount, J.S. Regulation of the trafficking and antiviral activity of IFITM3 by post-translational modifications. Futur. Microbiol. 2014, 9, 1151–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Z.; Han, Q.; Nie, J.; Cao, X.; Chen, Z.; Yin, S.; Gao, Y.; Lin, F.; Zhou, X.; Xu, K.; et al. Negative regulation of interferon-induced transmembrane protein 3 by SET7-mediated lysine monomethylation. J. Biol. Chem. 2013, 288, 35093–35103. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Kondur, H.R.; Huang, I.-C.; Farzan, M. Interferon-induced transmembrane protein 3 is a type II transmembrane protein. J. Biol. Chem. 2013, 288, 32184–32193. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef]
- Weston, S.; Czieso, S.; White, I.J.; Smith, S.E.; Kellam, P.; Marsh, M. A membrane topology model for human interferon inducible transmembrane protein 1. PLoS ONE 2014, 9, e104341. [Google Scholar] [CrossRef]
- Ling, S.; Zhang, C.; Wang, W.; Cai, X.; Yu, L.; Wu, F.; Zhang, L.; Tian, C. Combined approaches of EPR and NMR illustrate only one transmembrane helix in the human IFITM3. Sci. Rep. 2016, 6, 24029. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Jia, R.; Li, M.; Zheng, Y.-M.; Miao, C.; Yao, Y.; Ji, H.-L.; Geng, Y.; Qiao, W.; Albritton, L.M.; et al. A sorting signal suppresses IFITM1 restriction of viral entry. J. Biol. Chem. 2015, 290, 4248–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzo, C.P.; Nchioua, R.; Volcic, M.; Koepke, L.; Krüger, J.; Schütz, D.; Heller, S.; Stürzel, C.M.; Kmiec, D.; Conzelmann, C.; et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM 3 requires an amphipathic helix for antiviral activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Helenius, A. Endocytosis of Viruses and Bacteria. Cold Spring Harb. Perspect. Biol. 2014, 6, a016972. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [Green Version]
- Barrow, E.; Nicola, A.V.; Liu, J. Multiscale perspectives of virus entry via endocytosis. Virol. J. 2013, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.-T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2019, 9, 3228. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Du, S.; Xu, W.; Li, T.; Wu, S.; Jin, N.; Li, C. Current Progress on Host Antiviral Factor IFITMs. Front. Immunol. 2020, 11, 543444. [Google Scholar] [CrossRef]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [Green Version]
- Weidner, J.M.; Jiang, D.; Pan, X.-B.; Chang, J.; Block, T.M.; Guo, J.-T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Busse, D.C.; Binter, S.; Weston, S.; Soria, C.D.; Laksono, B.M.; Clare, S.; Van Nieuwkoop, S.; Hoogen, B.G.V.D.; Clement, M.; et al. Interferon-Induced Transmembrane Protein 1 Restricts Replication of Viruses That Enter Cells via the Plasma Membrane. J. Virol. 2019, 93, e02003-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Pan, Q.; Liu, S.-L.; Liang, C. HIV-1 mutates to evade IFITM1 restriction. Virology 2014, 454–455, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wang, Y.; Li, L.; Qu, X.; Liu, Q.; Li, T.; Wu, S.; Liao, M.; Jin, N.; Du, S.; et al. Transmembrane domain of IFITM3 is responsible for its interaction with influenza virus HA2 subunit. Virol. Sin. 2022, 37, 664–675. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Steinkühler, J.; Marin, M.; Li, X.; Lu, W.; Dimova, R.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Diverse Enveloped Viruses by Altering Mechanical Properties of Cell Membranes. ACS Nano 2021, 15, 8155–8170. [Google Scholar] [CrossRef]
- Kühnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.E.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late Endosomal/Lysosomal Cholesterol Accumulation Is a Host Cell-Protective Mechanism Inhibiting Endosomal Escape of Influenza A Virus. MBio 2018, 9, e01345-18. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [Green Version]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; Golani, G.; Lolicato, F.; Lahr, C.; Beyer, D.; Herrmann, A.; Wachsmuth-Melm, M.; Reddmann, N.; Brecht, R.; Hosseinzadeh, M.; et al. IFITM3 blocks influenza virus entry by sorting lipids and stabilizing hemifusion. Cell Host Microbe 2023, 31, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Gaillard, J.B.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef] [Green Version]
- Ahi, Y.S.; Yimer, D.; Shi, G.; Majdoul, S.; Rahman, K.; Rein, A.; Compton, A.A. IFITM3 Reduces Retroviral Envelope Abundance and Function and Is Counteracted by glycoGag. MBio 2020, 11, e03088-19. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-Y.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM proteins inhibit HIV-1 protein synthesis. Sci. Rep. 2018, 8, 14551. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Chiramel, A.I.; Li, T.; Lai, K.K.; Kenney, A.D.; Zani, A.; Eddy, A.C.; Majdoul, S.; Zhang, L.; Dempsey, T.; et al. Rapalogs downmodulate intrinsic immunity and promote cell entry of SARS-CoV-2. J. Clin. Investig. 2022, 132, e160766. [Google Scholar] [CrossRef]
- Shi, G.; Ozog, S.; Torbett, B.E.; Compton, A.A. mTOR inhibitors lower an intrinsic barrier to virus infection mediated by IFITM3. Proc. Natl. Acad. Sci. USA 2018, 115, E10069–E10078. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, C.; Thorne, L.G.; Unali, G.; Schiroli, G.; Giordano, A.M.S.; Piras, F.; Cuccovillo, I.; Petit, S.J.; Ahsan, F.; Noursadeghi, M.; et al. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing in Human Hematopoietic Stem Cells. Cell Stem Cell 2018, 23, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of Mature HIV-1 Particles Leads to Complete Release of a Gag-GFP-Based Content Marker and Raises the Intraviral pH. PLoS ONE 2013, 8, e71002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jiménez, V.; Scholte, F.; García-Sastre, A.; Rottier, P.J.M.; de Haan, C.A.M. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosset, F.-L.; Marianneau, P.; Verney, G.; Gallais, F.; Tordo, N.; Pecheur, E.; ter Meulen, J.; Deubel, V.; Bartosch, B. Characterization of Lassa virus cell entry and neutralization with Lassa virus pseudoparticles. J. Virol. 2009, 83, 3228–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammonds, J.E.; Beeman, N.; Ding, L.; Takushi, S.; Francis, A.C.; Wang, J.-J.; Melikyan, G.B.; Spearman, P. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog. 2017, 13, e1006181. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, M.; Du, Y.; Giroud, C.; Kim, J.H.; Qui, M.; Fu, H.; Melikyan, G.B. High-Throughput HIV–Cell Fusion Assay for Discovery of Virus Entry Inhibitors. ASSAY Drug Dev. Technol. 2015, 13, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224 Pt 3, 213–232. [Google Scholar] [CrossRef]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.-M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [Green Version]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Kummer, S.; Avinoam, O.; Kräusslich, H.-G. IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses 2019, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.; Coomer, C.A.; Majdoul, S.; Ding, S.Y.; Padilla-Parra, S.; Compton, A.A.; Dynamics, H.; Program, R.; Center for Cancer Research; National Cancer Institute; et al. Homology-guided identification of a conserved motif linking the antiviral functions of IFITM3 to its oligomeric state. eLife 2020, 9, e58537. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The Antiviral Effector IFITM3 Disrupts Intracellular Cholesterol Homeostasis to Block Viral Entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-Y.; Chin, C.R.; Everitt, A.R.; Clare, S.; Perreira, J.M.; Savidis, G.; Aker, A.M.; John, S.P.; Sarlah, D.; Carreira, E.M.; et al. Amphotericin B Increases Influenza A Virus Infection by Preventing IFITM3-Mediated Restriction. Cell Rep. 2013, 5, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Spence, J.S.; Das, T.; Yuan, X.; Chen, C.; Zhang, Y.; Li, Y.; Sun, Y.; Chandran, K.; Hang, H.C.; et al. Site-Specific Photo-Crosslinking Proteomics Reveal Regulation of IFITM3 Trafficking and Turnover by VCP/p97 ATPase. Cell Chem. Biol. 2020, 27, 571–585. [Google Scholar] [CrossRef]
- Nakamura, N.; Rabouille, C.; Watson, R.; Nilsson, T.; Hui, N.; Slusarewicz, P.; Kreis, T.E.; Warren, G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995, 131 Pt 2, 1715–1726. [Google Scholar] [CrossRef] [Green Version]
- Prescott, A.R.; Lucocq, J.; James, J.; Lister, J.M.; Ponnambalam, S. Distinct compartmentalization of TGN46 and beta 1,4-galactosyltransferase in HeLa cells. Eur. J. Cell Biol. 1997, 72, 238–246. [Google Scholar]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Song, Y.; Marziali, F.; Uzbekov, R.; Nguyen, X.-N.; Journo, C.; Roingeard, P.; Cimarelli, A. A novel domain within the CIL regulates egress of IFITM3 from the Golgi and reveals a regulatory role of IFITM3 on the secretory pathway. Life Sci. Alliance 2022, 5, e202101174. [Google Scholar] [CrossRef] [PubMed]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.H.; Everitt, A.R.; Vora, M.; et al. The CD225 Domain of IFITM3 Is Required for both IFITM Protein Association and Inhibition of Influenza A Virus and Dengue Virus Replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Gallon, M.; Cullen, P.J. Retromer and sorting nexins in endosomal sorting. Biochem. Soc. Trans. 2015, 43, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Multiple routes of protein transport from endosomes to the trans Golgi network. FEBS Lett. 2009, 583, 3811–3816. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Xiao, Z.; Jiang, W.; Chen, G.; Wang, Z. Overexpression of IFITM3 predicts poor prognosis in stage IIA esophageal squamous cell carcinoma after Ivor Lewis esophagectomy. Thorac. Cancer 2017, 8, 592–599. [Google Scholar] [CrossRef]
- Lee, J.; Robinson, M.E.; Ma, N.; Artadji, D.; Ahmed, M.A.; Xiao, G.; Sadras, T.; Deb, G.; Winchester, J.; Cosgun, K.N.; et al. IFITM3 functions as a PIP3 scaffold to amplify PI3K signalling in B cells. Nature 2020, 588, 491–497. [Google Scholar] [CrossRef]
- Perrin, P.; Janssen, L.; Janssen, H.; Broek, B.V.D.; Voortman, L.M.; van Elsland, D.; Berlin, I.; Neefjes, J. Retrofusion of intralumenal MVB membranes parallels viral infection and coexists with exosome release. Curr. Biol. 2021, 31, 3884–3893. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prikryl, D.; Marin, M.; Desai, T.M.; Du, Y.; Fu, H.; Melikyan, G.B. Cyclosporines Antagonize the Antiviral Activity of IFITMProteins by Redistributing Them toward the Golgi Apparatus. Biomolecules 2023, 13, 937. https://doi.org/10.3390/biom13060937
Prikryl D, Marin M, Desai TM, Du Y, Fu H, Melikyan GB. Cyclosporines Antagonize the Antiviral Activity of IFITMProteins by Redistributing Them toward the Golgi Apparatus. Biomolecules. 2023; 13(6):937. https://doi.org/10.3390/biom13060937
Chicago/Turabian StylePrikryl, David, Mariana Marin, Tanay M. Desai, Yuhong Du, Haian Fu, and Gregory B. Melikyan. 2023. "Cyclosporines Antagonize the Antiviral Activity of IFITMProteins by Redistributing Them toward the Golgi Apparatus" Biomolecules 13, no. 6: 937. https://doi.org/10.3390/biom13060937