

Hyperactivity in Mice Induced by Opioid Agonists with Partial Intrinsic Efficacy and Biased Agonism Administered Alone and in Combination with Morphine

Abstract
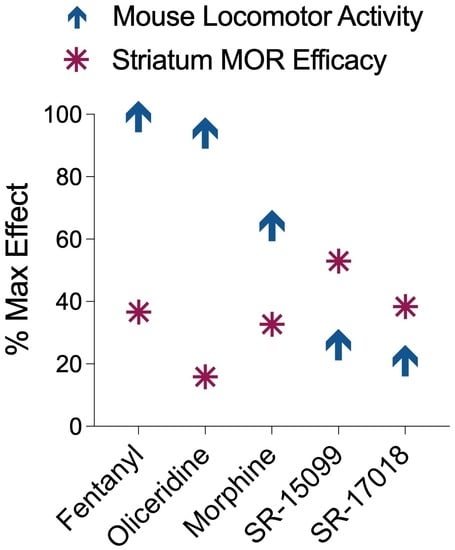
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Care and Use
2.2. Compounds
2.3. Drug Solutions Preparation
2.4. 35S-GTPγS Binding Assay in Mouse Striata
2.5. Locomotor Activity
2.6. Opioid–Opioid Interactions in the Acute Thermal Antinociceptive Responses
2.7. Software and Statistical Analysis
3. Results
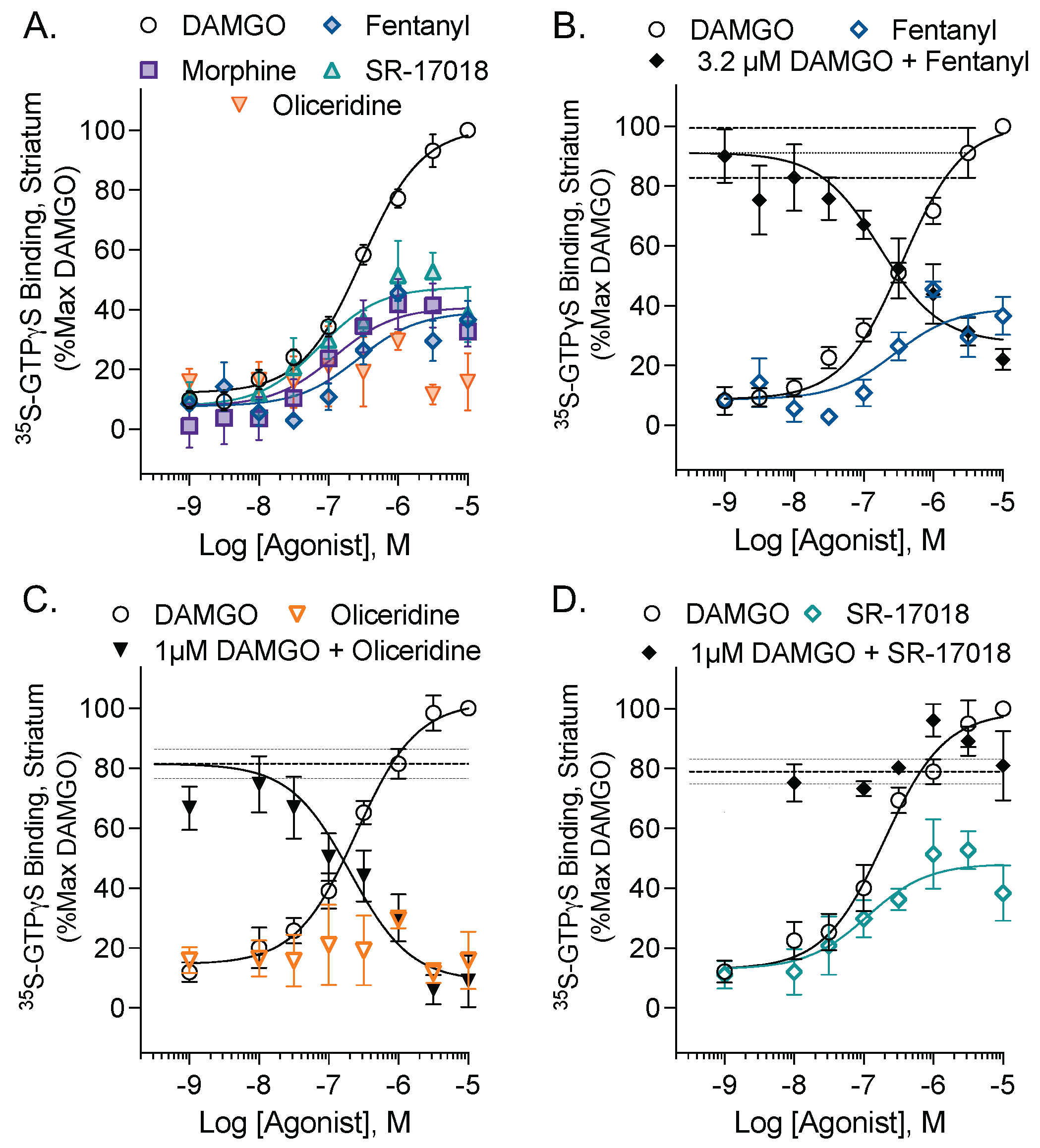
3.1. Evaluation of the Intrinsic Efficacy and Competitive Nature of Partial Agonists in Mouse Striatal Membranes
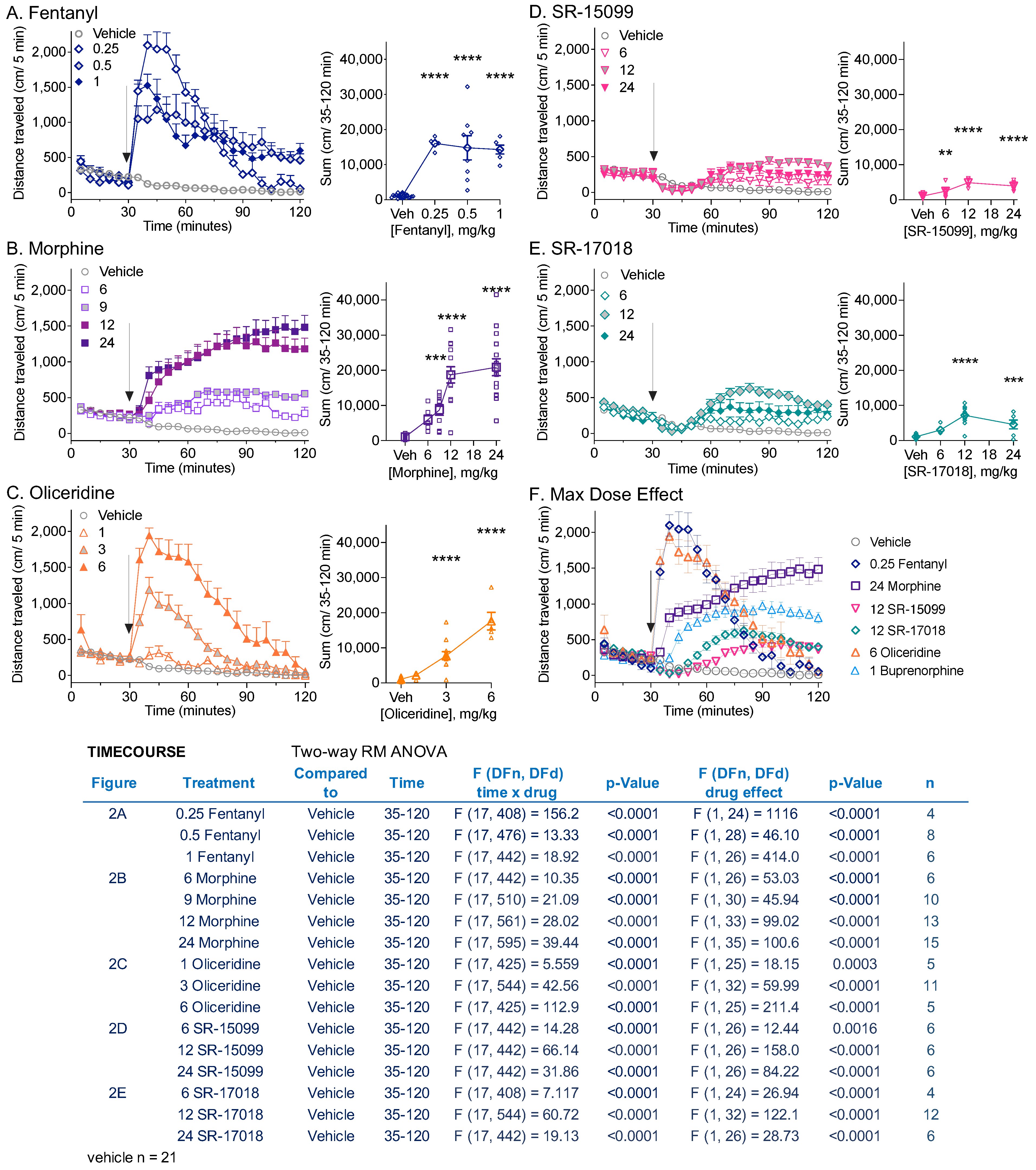
3.2. Comparison of Locomotor Activity by Different Opioid Agonists
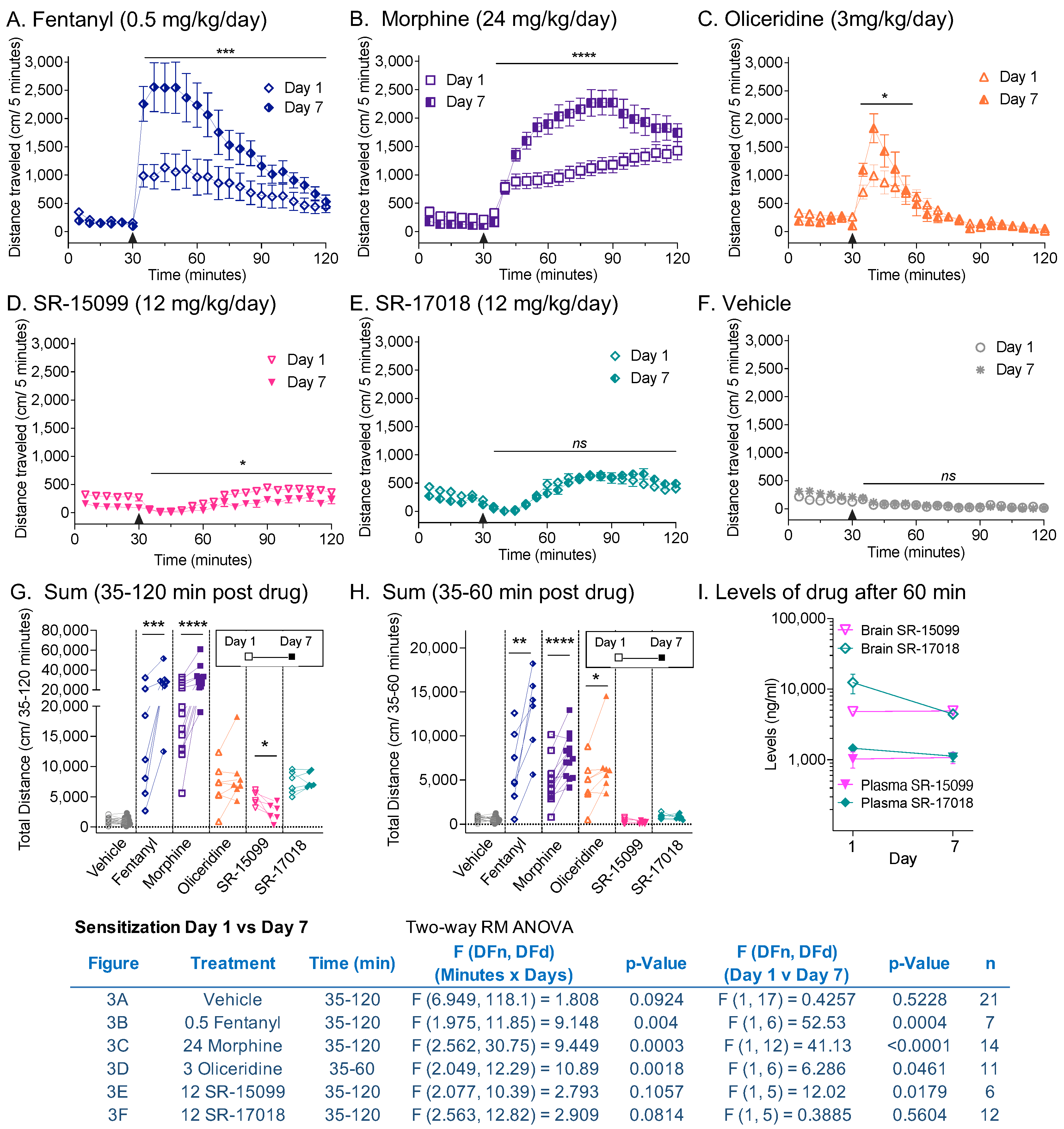
3.3. Sensitization following Chronic Opioid Exposure
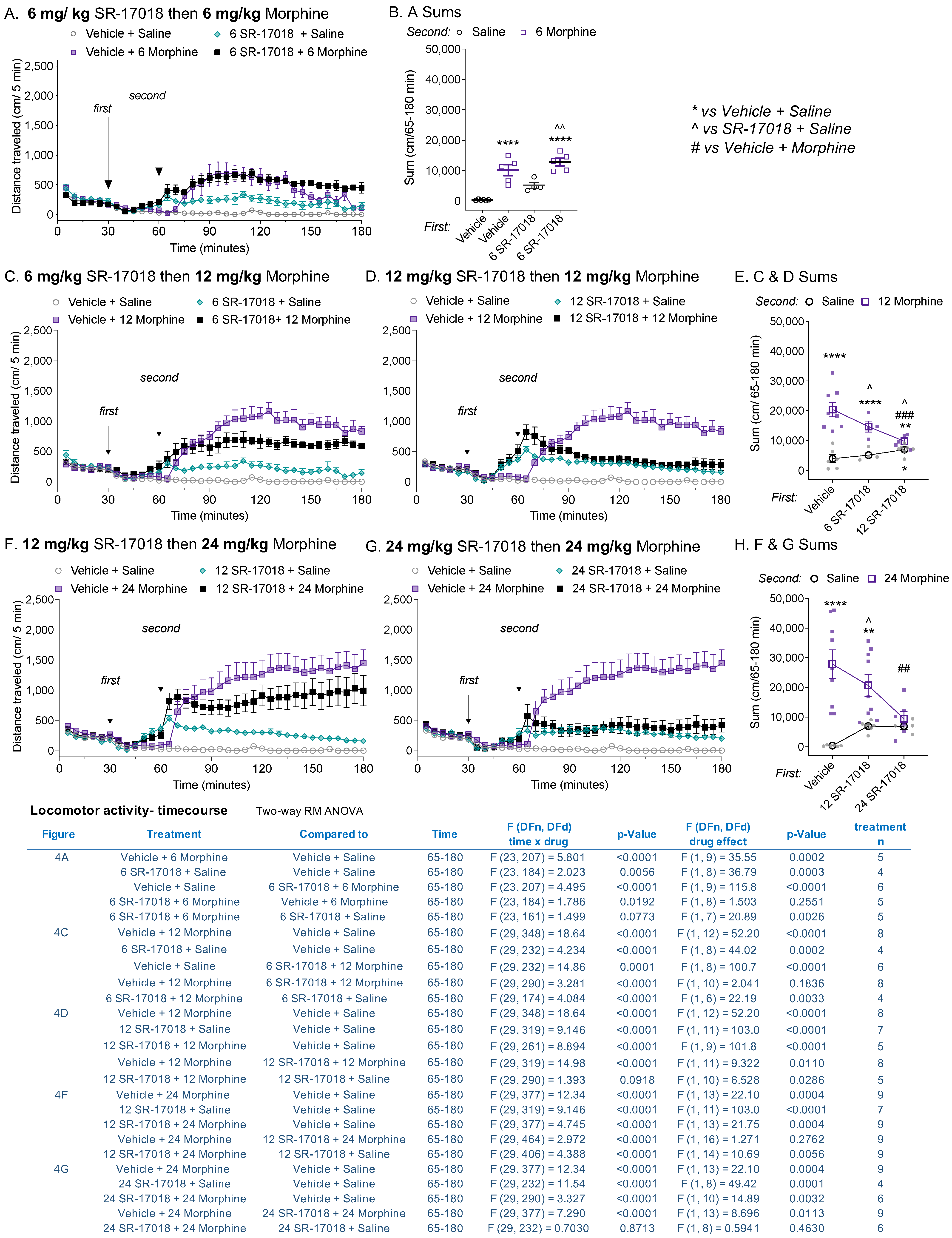
3.4. SR-17018 Attenuates Morphine-Induced Locomotor Activity
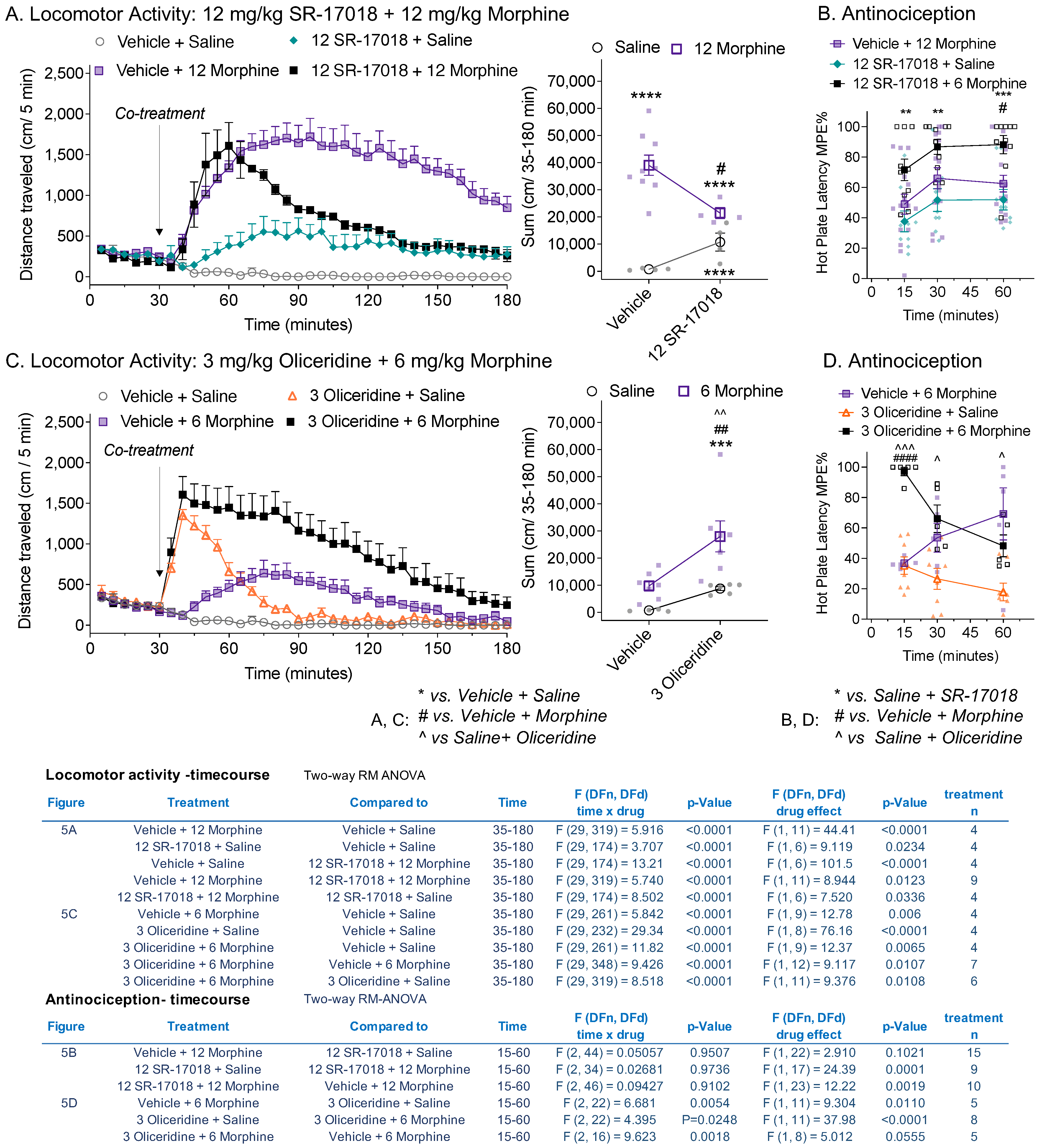
3.5. Co-Treatment of Partial Agonists Differentially Effects Morphine-Induced Hyperactivity and Antinociception
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rethy, C.R.; Smith, C.B.; Villareal, J.E. Effects of Narcotic Analgesics Upon the Locomotor Activity and Brain Cathecolamine Content of the Mouse. J. Pharmacol. Exp. Ther. 1970, 176, 472–479. [Google Scholar]
- Bailey, A.; Metaxas, A.; Al-Hasani, R.; Keyworth, H.L.; Forster, D.M.; Kitchen, I. Mouse strain differences in locomotor, sensitisation and rewarding effect of heroin; Association with alterations in MOP-r activation and dopamine transporter binding. Eur. J. Neurosci. 2010, 31, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Varshneya, N.B.; Walentiny, D.M.; Moisa, L.T.; Walker, T.D.; Akinfiresoye, L.R.; Beardsley, P.M. Opioid-like antinociceptive and locomotor effects of emerging fentanyl-related substances. Neuropharmacology 2019, 151, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Hall, F.S.; Li, X.F.; Goeb, M.; Roff, S.; Hoggatt, H.; Sora, I.; Uhl, G.R. Congenic C57BL/6 mu opiate receptor (MOR) knockout mice: Baseline and opiate effects. Genes Brain Behav. 2003, 2, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Sora, I.; Elmer, G.; Funada, M.; Pieper, J.; Li, X.F.; Hall, F.S.; Uhl, G.R. Mu opiate receptor gene dose effects on different morphine actions: Evidence for differential in vivo mu receptor reserve. Neuropsychopharmacology 2001, 25, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.-T. Enhanced Morphine Analgesia in Mice Lacking Beta-Arrestin 2. Science 1999, 286, 2495–2499. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef] [Green Version]
- Mittal, N.; Tan, M.; Egbuta, O.; Desai, N.; Crawford, C.; Xie, C.W.; Evans, C.; Walwyn, W. Evidence that behavioral phenotypes of morphine in beta-arr2−/− mice are due to the unmasking of JNK signaling. Neuropsychopharmacology 2012, 37, 1953–1962. [Google Scholar] [CrossRef] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e1113. [Google Scholar] [CrossRef] [Green Version]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef]
- Cornelissen, J.C.; Blough, B.E.; Bohn, L.M.; Negus, S.S.; Banks, M.L. Some effects of putative G-protein biased mu-opioid receptor agonists in male rhesus monkeys. Behav. Pharmacol. 2021, 32, 453. [Google Scholar] [CrossRef]
- Grim, T.W.; Schmid, C.L.; Stahl, E.L.; Pantouli, F.; Ho, J.H.; Acevedo-Canabal, A.; Kennedy, N.M.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. A G protein signaling-biased agonist at the mu-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 2019, 45, 416–425. [Google Scholar] [CrossRef]
- Pantouli, F.; Grim, T.W.; Schmid, C.L.; Acevedo-Canabal, A.; Kennedy, N.M.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Comparison of morphine, oxycodone and the biased MOR agonist SR-17018 for tolerance and efficacy in mouse models of pain. Neuropharmacology 2021, 185, 108439. [Google Scholar] [CrossRef]
- Stahl, E.L.; Schmid, C.L.; Acevedo-Canabal, A.; Read, C.; Grim, T.W.; Kennedy, N.M.; Bannister, T.D.; Bohn, L.M. G protein signaling-biased mu opioid receptor agonists that produce sustained G protein activation are noncompetitive agonists. Proc. Natl. Acad. Sci. USA 2021, 118, e2102178118. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Stahl, E.L.; Lovell, K.M.; Frankowski, K.J.; Prisinzano, T.E.; Aube, J.; Bohn, L.M. Characterization of kappa opioid receptor mediated, dynorphin-stimulated [35S]GTPgammaS binding in mouse striatum for the evaluation of selective KOR ligands in an endogenous setting. Neuropharmacology 2015, 99, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Takemura, Y.; Arima, T.; Iwase, Y.; Narita, M.; Miyano, K.; Hamada, Y.; Suda, Y.; Matsuzawa, A.; Sugita, K.; et al. Further investigation of the rapid-onset and short-duration action of the G protein-biased mu-ligand oliceridine. Biochem. Biophys. Res. Commun. 2021, 534, 988–994. [Google Scholar] [CrossRef]
- Mori, T.; Ito, S.; Narita, M.; Suzuki, T.; Sawaguchi, T. Combined effects of psychostimulants and morphine on locomotor activity in mice. J. Pharmacol. Sci. 2004, 96, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, K.A.; Smith, M.L.; Guaderrama, M.M. Powerful behavioral interactions between methamphetamine and morphine. Pharmacol. Biochem. Behav. 2011, 99, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selley, D.E.; Sim, L.J.; Xiao, R.; Liu, Q.; Childers, S.R. μ-Opioid Receptor-Stimulated Guanosine-5′-O-(γ-thio)-triphosphate Binding in Rat Thalamus and Cultured Cell Lines: Signal Transduction Mechanisms Underlying Agonist Efficacy. Mol. Pharmacol. 1997, 51, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selley, D.E.; Liu, Q.; Childers, S.R. Signal transduction correlates of mu opioid agonist intrinsic efficacy: Receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther. 1998, 285, 496–505. [Google Scholar] [PubMed]
- Lester, P.A.; Traynor, J.R. Comparison of the in vitro efficacy of mu, delta, kappa and ORL1 receptor agonists and non-selective opioid agonists in dog brain membranes. Brain. Res. 2006, 1073–1074, 290–296. [Google Scholar] [CrossRef]
- Varshneya, N.B.; Hassanien, S.H.; Holt, M.C.; Stevens, D.L.; Layle, N.K.; Bassman, J.R.; Iula, D.M.; Beardsley, P.M. Respiratory depressant effects of fentanyl analogs are opioid receptor-mediated. Biochem. Pharmacol. 2022, 195, 114805. [Google Scholar] [CrossRef]
- Santos, E.J.; Banks, M.L.; Negus, S.S. Role of Efficacy as a Determinant of Locomotor Activation by Mu Opioid Receptor Ligands in Female and Male Mice. J. Pharmacol. Exp. Ther. 2022, 382, 44–53. [Google Scholar] [CrossRef]
- Varshneya, N.B.; Walentiny, D.M.; Stevens, D.L.; Walker, T.D.; Akinfiresoye, L.R.; Beardsley, P.M. Structurally diverse fentanyl analogs yield differential locomotor activities in mice. Pharmacol. Biochem. Behav. 2023, 222, 173496. [Google Scholar] [CrossRef]
- Sim-Selley, L.J.; Scoggins, K.L.; Cassidy, M.P.; Smith, L.A.; Dewey, W.L.; Smith, F.L.; Selley, D.E. Region-dependent attenuation of mu opioid receptor-mediated G-protein activation in mouse CNS as a function of morphine tolerance. Br. J. Pharmacol. 2007, 151, 1324–1333. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.; Miranda, H.F.; Puig, M.M. Analysis of the opioid-opioid combinations according to the nociceptive stimulus in mice. Pharmacol. Res. 2010, 61, 511–518. [Google Scholar] [CrossRef]
- Varshneya, N.B.; Thakrar, A.P.; Hobelmann, J.G.; Dunn, K.E.; Huhn, A.S. Evidence of Buprenorphine-precipitated Withdrawal in Persons Who Use Fentanyl. J. Addict. Med. 2022, 16, e265–e268. [Google Scholar] [CrossRef]
- Lutfy, K.; Eitan, S.; Bryant, C.D.; Yang, Y.C.; Saliminejad, N.; Walwyn, W.; Kieffer, B.L.; Takeshima, H.; Carroll, F.I.; Maidment, N.T.; et al. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J. Neurosci. 2003, 23, 10331–10337. [Google Scholar] [CrossRef] [Green Version]
- Khroyan, T.V.; Wu, J.; Polgar, W.E.; Cami-Kobeci, G.; Fotaki, N.; Husbands, S.M.; Toll, L. BU08073 a buprenorphine analogue with partial agonist activity at mu-receptors in vitro but long-lasting opioid antagonist activity in vivo in mice. Br. J. Pharmacol. 2014, 172, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Canabal, A.; Pantouli, F.; Ravichandran, A.; Rullo, L.; Bohn, L.M. 3.21—Pharmacological Diversity in Opioid Analgesics: Lessons From Clinically Useful Drugs. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Oxford, UK, 2022; pp. 478–493. [Google Scholar] [CrossRef]
- Torralva, R.; Janowsky, A. Noradrenergic Mechanisms in Fentanyl-Mediated Rapid Death Explain Failure of Naloxone in the Opioid Crisis. J. Pharmacol. Exp. Ther. 2019, 371, 453–475. [Google Scholar] [CrossRef]
- Hill, R.; Santhakumar, R.; Dewey, W.; Kelly, E.; Henderson, G. Fentanyl depression of respiration: Comparison with heroin and morphine. Br. J. Pharmacol. 2020, 177, 254–266. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular recognition of morphine and fentanyl by the human mu-opioid receptor. Cell 2022, 185, 4361–4375.e4319. [Google Scholar] [CrossRef]
- Lalley, P.M. Opioidergic and dopaminergic modulation of respiration. Respir. Physiol. Neurobiol. 2008, 164, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Kudla, L.; Bugno, R.; Podlewska, S.; Szumiec, L.; Wiktorowska, L.; Bojarski, A.J.; Przewlocki, R. Comparison of an Addictive Potential of μ-Opioid Receptor Agonists with G Protein Bias: Behavioral and Molecular Modeling Studies. Pharmaceutics 2022, 14, 55. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acevedo-Canabal, A.; Grim, T.W.; Schmid, C.L.; McFague, N.; Stahl, E.L.; Kennedy, N.M.; Bannister, T.D.; Bohn, L.M. Hyperactivity in Mice Induced by Opioid Agonists with Partial Intrinsic Efficacy and Biased Agonism Administered Alone and in Combination with Morphine. Biomolecules 2023, 13, 935. https://doi.org/10.3390/biom13060935
Acevedo-Canabal A, Grim TW, Schmid CL, McFague N, Stahl EL, Kennedy NM, Bannister TD, Bohn LM. Hyperactivity in Mice Induced by Opioid Agonists with Partial Intrinsic Efficacy and Biased Agonism Administered Alone and in Combination with Morphine. Biomolecules. 2023; 13(6):935. https://doi.org/10.3390/biom13060935
Chicago/Turabian StyleAcevedo-Canabal, Agnes, Travis W. Grim, Cullen L. Schmid, Nina McFague, Edward L. Stahl, Nicole M. Kennedy, Thomas D. Bannister, and Laura M. Bohn. 2023. "Hyperactivity in Mice Induced by Opioid Agonists with Partial Intrinsic Efficacy and Biased Agonism Administered Alone and in Combination with Morphine" Biomolecules 13, no. 6: 935. https://doi.org/10.3390/biom13060935