
Altered Purinergic Signaling in Neurodevelopmental Disorders: Focus on P2 Receptors
 , , , and
, , , and
Abstract
:1. Introduction
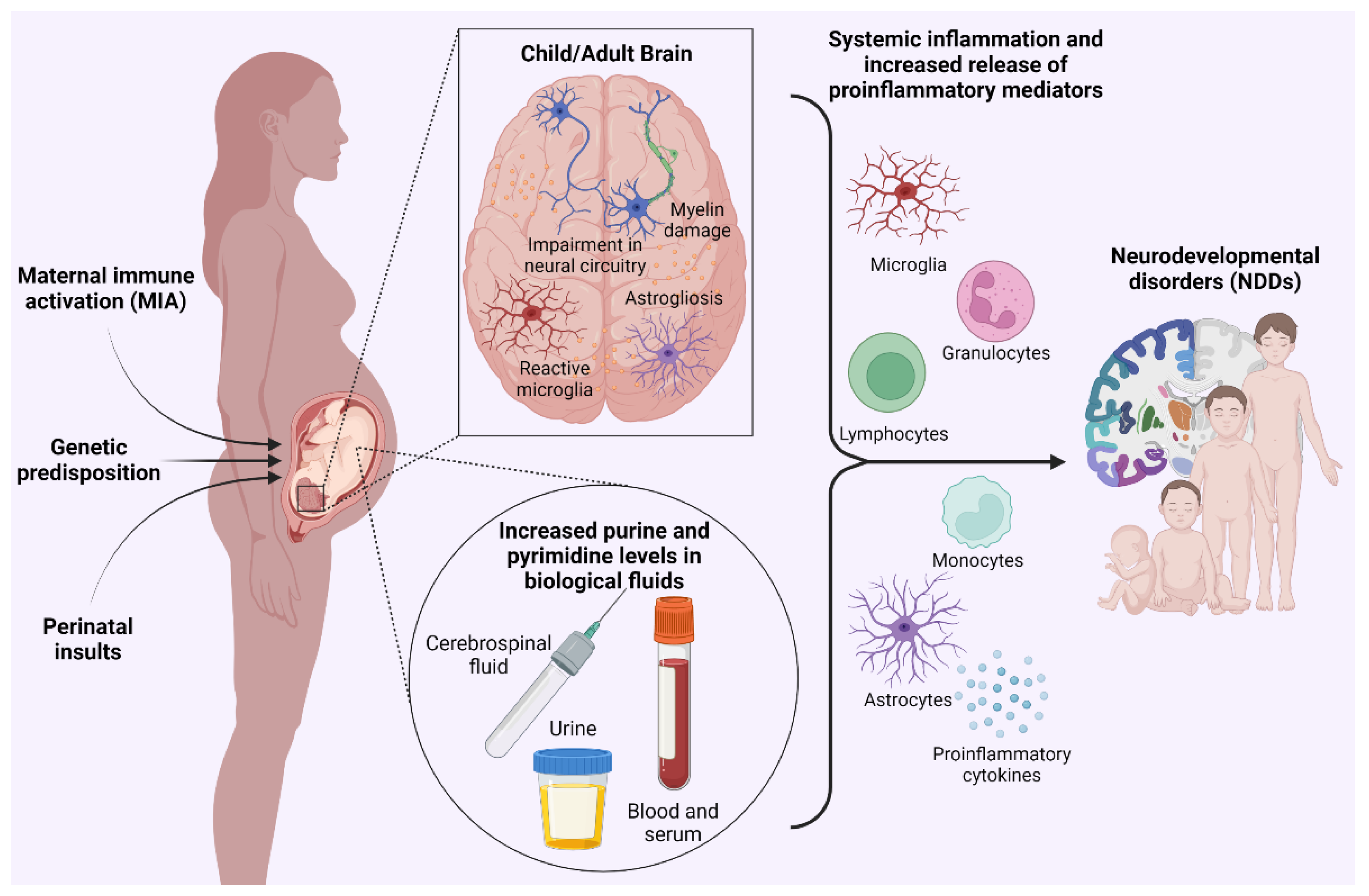
2. Clinical Evidence Linking Neurodevelopmental Disorders with Alterations of Purinergic Signaling and Purine Metabolism
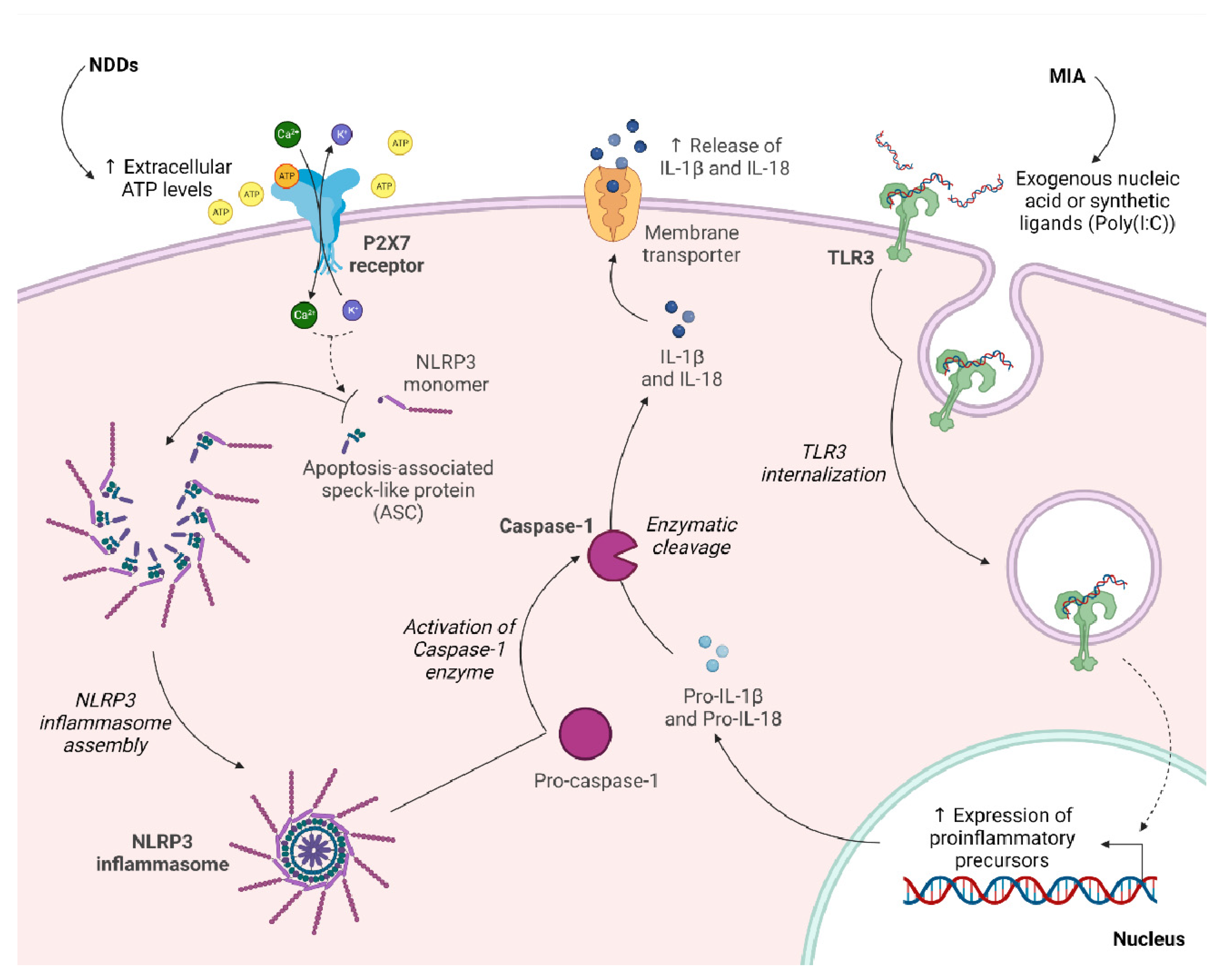
3. Therapeutic Potential of P2X7 Receptor in Maternal Immune Activation (MIA)
Clinical Study
4. Involvement of Purinergic Transmission in Perinatal White Matter Injuries
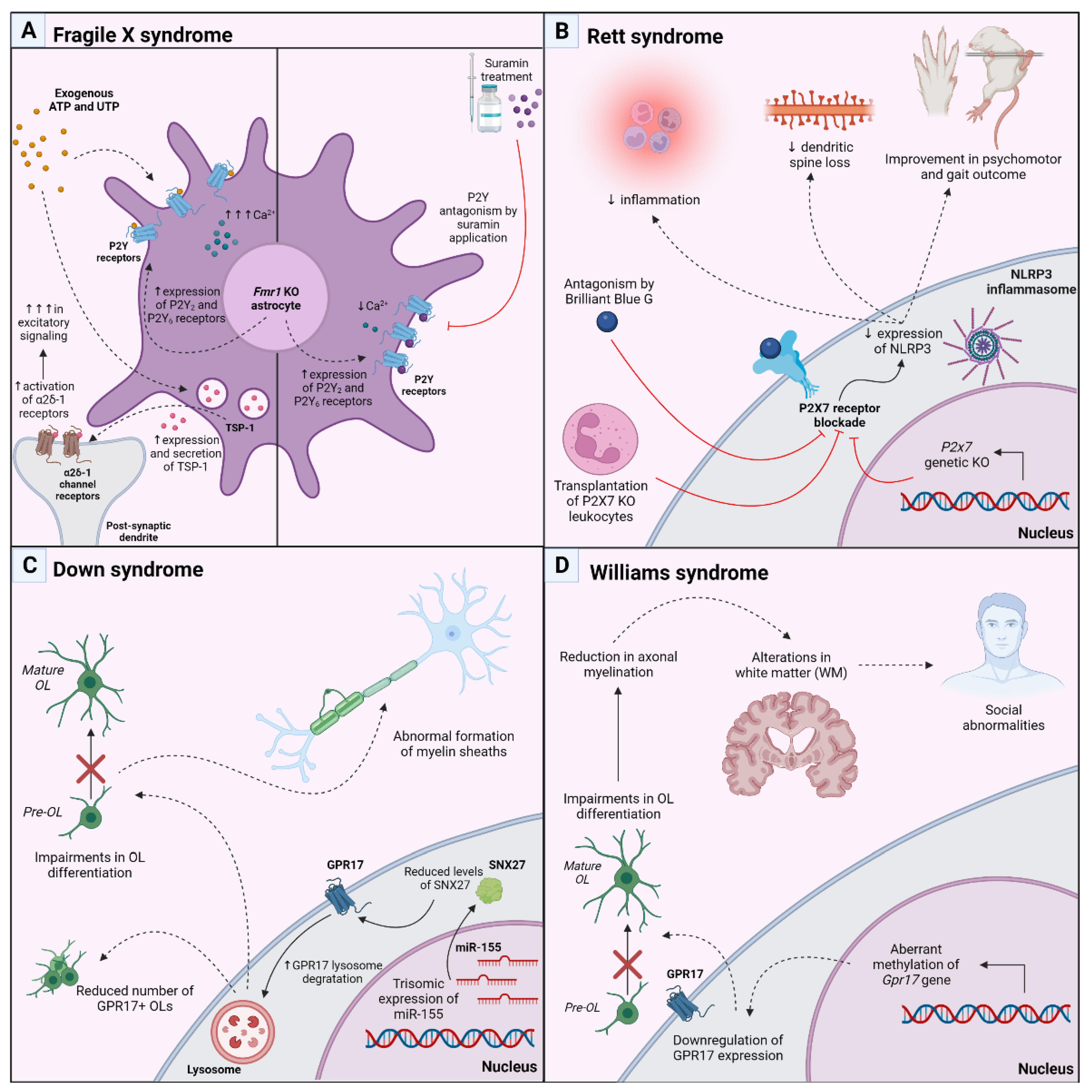
5. Therapeutic Approach Targeting P2 and P2Y-like Receptors in Genetic Models of Postnatal Neurodevelopmental Disorders
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental Disorders. Lancet Psychiatry 2017, 4, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends. Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Shook, L.L.; Sullivan, E.L.; Lo, J.O.; Perlis, R.H.; Edlow, A.G. COVID-19 in Pregnancy: Implications for Fetal Brain Development. Trends. Mol. Med. 2022, 28, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Nardelli, J. Cellular and Molecular Introduction to Brain Development. Neurobiol. Dis. 2016, 92, 3–17. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. Adenosine as a Neuromodulator and as a Homeostatic Regulator in the Nervous System: Different Roles, Different Sources and Different Receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Ceruti, S. Roles of P2 Receptors in Glial Cells: Focus on Astrocytes. Purinergic. Signal. 2006, 2, 595–604. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P.; Ceruti, S. Pathophysiological Role of Purines and Pyrimidines in Neurodevelopment: Unveiling New Pharmacological Approaches to Congenital Brain Diseases. Front. Pharmacol. 2017, 8, 941. [Google Scholar] [CrossRef]
- Boison, D.; Singer, P.; Shen, H.Y.; Feldon, J.; Yee, B.K. Adenosine Hypothesis of Schizophrenia—Opportunities for Pharmacotherapy. Neuropharmacology 2012, 62, 1527–1543. [Google Scholar] [CrossRef]
- Heine, C.; Sygnecka, K.; Franke, H. Purines in Neurite Growth and Astroglia Activation. Neuropharmacology 2016, 104, 255–271. [Google Scholar] [CrossRef]
- Alçada-Morais, S.; Gonçalves, N.; Moreno-Juan, V.; Andres, B.; Ferreira, S.; Marques, J.M.; Magalhães, J.; Rocha, J.M.M.; Xu, X.; Partidário, M.; et al. Adenosine A2A Receptors Contribute to the Radial Migration of Cortical Projection Neurons through the Regulation of Neuronal Polarization and Axon Formation. Cereb. Cortex. 2021, 31, 5652–5663. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Métin, C.; Fazeli, W.; Machado, N.J.; Darmopil, S.; Launay, P.S.; Ghestem, A.; Nesa, M.P.; Bassot, E.; Szabó, E.; et al. Adenosine Receptor Antagonists Including Caffeine Alter Fetal Brain Development in Mice. Sci. Transl. Med. 2013, 5, 197ra104. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Castro, F.; Zappettini, S.; Pressey, J.C.; Silva, C.G.; Russeau, M.; Gervasi, N.; Figueiredo, M.; Montmasson, C.; Renner, M.; Canas, P.M.; et al. Convergence of Adenosine and GABA Signaling for Synapse Stabilization during Development. Science 2021, 374, eabk2055. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Beleza, R.O.; Gonçalves, F.Q.; Valbuena, S.; Alçada-Morais, S.; Gonçalves, N.; Magalhães, J.; Rocha, J.M.M.; Ferreira, S.; Figueira, A.S.G.; et al. Adenosine A2A Receptors Control Synaptic Remodeling in the Adult Brain. Sci. Rep. 2022, 12, 14690. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Pedata, F.; Pugliese, A.M. New Insight into the Role of Adenosine in Demyelination, Stroke and Neuropathic Pain. Front. Pharmacol. 2021, 11, 2403. [Google Scholar] [CrossRef]
- Oliveira, Á.; Illes, P.; Ulrich, H. Purinergic Receptors in Embryonic and Adult Neurogenesis. Neuropharmacology 2016, 104, 272–281. [Google Scholar] [CrossRef]
- Ulrich, H.; Abbracchio, M.P.; Burnstock, G. Extrinsic Purinergic Regulation of Neural Stem/Progenitor Cells: Implications for CNS Development and Repair. Stem. Cell. Rev. 2012, 8, 755–767. [Google Scholar] [CrossRef]
- Cserép, C.; Schwarcz, A.D.; Pósfai, B.; László, Z.I.; Kellermayer, A.; Környei, Z.; Kisfali, M.; Nyerges, M.; Lele, Z.; Katona, I.; et al. Microglial Control of Neuronal Development via Somatic Purinergic Junctions. Cell. Rep. 2022, 40, 111369. [Google Scholar] [CrossRef]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the Damaged Brain: Molecular and Cellular Insights into Their Reactive Response and Healing Potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P. CNS Remyelination as a Novel Reparative Approach to Neurodegenerative Diseases: The Roles of Purinergic Signaling and the P2Y-like Receptor GPR17. Neuropharmacology 2016, 104, 82–93. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens, B. ATP: An Extracellular Signaling Molecule between Neurons and Glia. Trends. Neurosci. 2000, 23, 625–633. [Google Scholar] [CrossRef]
- Suyama, S.; Sunabori, T.; Kanki, H.; Sawamoto, K.; Gachet, C.; Koizumi, S.; Okano, H. Purinergic Signaling Promotes Proliferation of Adult Mouse Subventricular Zone Cells. J. Neurosci. 2012, 32, 9238–9247. [Google Scholar] [CrossRef]
- Boccazzi, M.; Rolando, C.; Abbracchio, M.P.; Buffo, A.; Ceruti, S. Purines Regulate Adult Brain Subventricular Zone Cell Functions: Contribution of Reactive Astrocytes. Glia 2014, 62, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Boda, E.; Nato, G.; Buffo, A. Emerging Pharmacological Approaches to Promote Neurogenesis from Endogenous Glial Cells. Biochem. Pharmacol. 2017, 141, 23–41. [Google Scholar] [CrossRef]
- del Puerto, A.; Díaz-Hernández, J.I.; Tapia, M.; Gomez-Villafuertes, R.; Benitez, M.J.; Zhang, J.; Miras-Portugal, M.T.; Wandosell, F.; Díaz-Hernández, M.; Garrido, J.J. Adenylate Cyclase 5 Coordinates the Action of ADP, P2Y1, P2Y13 and ATP-Gated P2X7 Receptors on Axonal Elongation. J. Cell. Sci. 2012, 125, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Serrano, A.; Engel, T.; De Diego-García, L.; Olivos-Oré, L.A.; Arribas-Blázquez, M.; Martínez-Frailes, C.; Pérez-Díaz, C.; Luis Millán, J.; Artalejo, A.R.; Miras-Portugal, M.T.; et al. Neurodevelopmental Alterations and Seizures Developed by Mouse Model of Infantile Hypophosphatasia Are Associated with Purinergic Signalling Deregulation. Hum. Mol. Genet. 2016, 25, 4143–4156. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.J.; Marques, J.M.; Cunha, R.A. Purinergic Signalling and Brain Development. Semin. Cell. Dev. Biol. 2019, 95, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Xie, P.; Liu, S.; Luan, G.; Li, T. Epilepsy and Autism Spectrum Disorder (ASD): The Underlying Mechanisms and Therapy Targets Related to Adenosine. Curr. Neuropharmacol. 2023, 21, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; Yee, B.K. The Adenosine Hypothesis of Schizophrenia into Its Third Decade: From Neurochemical Imbalance to Early Life Etiological Risks. Front. Cell. Neurosci. 2023, 17, 73. [Google Scholar] [CrossRef]
- Pasquini, S.; Contri, C.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K.; Vincenzi, F. Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 1219. [Google Scholar] [CrossRef]
- Pareek, V.; Pedley, A.M.; Benkovic, S.J. Human de Novo Purine Biosynthesis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lesch, M.; Nyhan, W.L. A Familial Disorder of Uric Acid Metabolism and Central Nervous System Function. Am. J. Med. 1964, 36, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.C. Lesch–Nyhan Syndrome and Its Variants. Curr. Opin. Psychiatry 2018, 31, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L.; James, J.A.; Teberg, A.J.; Sweetman, L.; Nelson, L.G. A New Disorder of Purine Metabolism with Behavioral Manifestations. J. Pediatr. 1969, 74, 20–27. [Google Scholar] [CrossRef]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary Metabolomics of Young Italian Autistic Children Supports Abnormal Tryptophan and Purine Metabolism. Mol. Autism. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Bitar, T.; Mavel, S.; Emond, P.; Nadal-Desbarats, L.; Lefèvre, A.; Mattar, H.; Soufia, M.; Blasco, H.; Vourc’h, P.; Hleihel, W.; et al. Identification of Metabolic Pathway Disturbances Using Multimodal Metabolomics in Autistic Disorders in a Middle Eastern Population. J. Pharm. Biomed. Anal. 2018, 152, 57–65. [Google Scholar] [CrossRef]
- Kurochkin, I.; Khrameeva, E.; Tkachev, A.; Stepanova, V.; Vanyushkina, A.; Stekolshchikova, E.; Li, Q.; Zubkov, D.; Shichkova, P.; Halene, T.; et al. Metabolome Signature of Autism in the Human Prefrontal Cortex. Commun. Biol. 2019, 2, 234. [Google Scholar] [CrossRef]
- Liang, Y.; Ke, X.; Xiao, Z.; Zhang, Y.; Chen, Y.; Li, Y.; Wang, Z.; Lin, L.; Yao, P.; Lu, J. Untargeted Metabolomic Profiling Using UHPLC-QTOF/MS Reveals Metabolic Alterations Associated with Autism. Biomed. Res. Int. 2020, 2020, 6105608. [Google Scholar] [CrossRef]
- Mussap, M.; Siracusano, M.; Noto, A.; Fattuoni, C.; Riccioni, A.; Rajula, H.S.R.; Fanos, V.; Curatolo, P.; Barberini, L.; Mazzone, L. The Urine Metabolome of Young Autistic Children Correlates with Their Clinical Profile Severity. Metabolites 2020, 10, 476. [Google Scholar] [CrossRef]
- Dai, S.; Lin, J.; Hou, Y.; Luo, X.; Shen, Y.; Ou, J. Purine Signaling Pathway Dysfunction in Autism Spectrum Disorders: Evidence from Multiple Omics Data. Front. Mol. Neurosci. 2023, 16, 1089871. [Google Scholar] [CrossRef]
- Torres, R.J.; Prior, C.; Garcia, M.G.; Puig, J.G. A Review of the Implication of Hypoxanthine Excess in the Physiopathology of Lesch–Nyhan Disease. Nucleosides Nucleotides Nucleic Acids 2016, 35, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Kristóf, Z.; Baranyi, M.; Tod, P.; Mut-Arbona, P.; Demeter, K.; Bitter, I.; Sperlágh, B. Elevated Serum Purine Levels in Schizophrenia: A Reverse Translational Study to Identify Novel Inflammatory Biomarkers. Int. J. Neuropsychopharmacol. 2022, 25, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Alnafisah, R.; Lundh, A.; Asah, S.M.; Hoeflinger, J.; Wolfinger, A.; Hamoud, A.-r.; McCullumsmith, R.E.; O’Donovan, S.M. Altered Purinergic Receptor Expression in the Frontal Cortex in Schizophrenia. Schizophrenia 2022, 8, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Ceruti, S.; Bramanti, P.; Abbracchio, M.P. Purinergic Signalling in Inflammation of the Central Nervous System. Trends. Neurosci. 2009, 32, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.J.; Tomé, A.R.; Cunha, R.A. ATP as a Multi-Target Danger Signal in the Brain. Front. Neurosci. 2015, 9, 148. [Google Scholar] [CrossRef]
- Naviaux, J.C.; Schuchbauer, M.A.; Li, K.; Wang, L.; Risbrough, V.B.; Powell, S.B.; Naviaux, R.K. Reversal of Autism-like Behaviors and Metabolism in Adult Mice with Single-Dose Antipurinergic Therapy. Transl. Psychiatry 2014, 4, e400. [Google Scholar] [CrossRef]
- Minakova, E.; Warner, B.B. Maternal Immune Activation, Central Nervous System Development and Behavioral Phenotypes. Birth. Defects. Res. 2018, 110, 1539–1550. [Google Scholar] [CrossRef]
- Selten, J.-P.; Morgan, V.A. Prenatal Exposure to Influenza and Major Affective Disorder. Bipolar. Disord. 2010, 12, 753–754. [Google Scholar] [CrossRef]
- Selten, J.P.; Termorshuizen, F. The Serological Evidence for Maternal Influenza as Risk Factor for Psychosis in Offspring Is Insufficient: Critical Review and Meta-Analysis. Schizophr. Res. 2017, 183, 2–9. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P.; Pharma Research, R.; Development, E.; et al. Maternal Immune Activation and Abnormal Brain Development across CNS Disorders. Nat. Publ. Group 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Meyer, U. Prenatal Poly(I:C) Exposure and Other Developmental Immune Activation Models in Rodent Systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Voget, M.; Willi, R.; Winter, C.; Riva, M.A.; Mortensen, P.B.; Schedlowski, M.; et al. Stress in Puberty Unmasks Latent Neuropathological Consequences of Prenatal Immune Activation in Mice. Science 2013, 339, 1100–1102. [Google Scholar] [CrossRef] [PubMed]
- Bucknor, M.C.; Gururajan, A.; Dale, R.C.; Hofer, M.J. A Comprehensive Approach to Modeling Maternal Immune Activation in Rodents. Front. Neurosci. 2022, 16, 1071976. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.M.; Lorusso, J.M.; Potter, H.G.; Neill, J.C.; Glazier, J.D.; Hager, R. Maternal Immune Activation in Rodent Models: A Systematic Review of Neurodevelopmental Changes in Gene Expression and Epigenetic Modulation in the Offspring Brain. Neurosci. Biobehav. Rev. 2021, 129, 389–421. [Google Scholar] [CrossRef] [PubMed]
- Garay, P.A.; Hsiao, E.Y.; Patterson, P.H.; McAllister, A.K. Maternal Immune Activation Causes Age- and Region-Specific Changes in Brain Cytokines in Offspring throughout Development. Brain Behav. Immun. 2013, 31, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Colucci, F.; Boulenouar, S.; Kieckbusch, J.; Moffett, A. How Does Variability of Immune System Genes Affect Placentation? Placenta 2011, 32, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Bergdolt, L.; Dunaevsky, A. Brain Changes in a Maternal Immune Activation Model of Neurodevelopmental Brain Disorders. Prog. Neurobiol. 2019, 175, 1–19. [Google Scholar] [CrossRef]
- Solek, C.M.; Farooqi, N.; Verly, M.; Lim, T.K.; Ruthazer, E.S.; Doi, D. Maternal Immune Activation in Neurodevelopmental Disorders. Dev. Dyn. 2018, 247, 588–619. [Google Scholar] [CrossRef]
- Haddad, F.L.; Patel, S.V.; Schmid, S. Maternal Immune Activation by Poly I:C as a Preclinical Model for Neurodevelopmental Disorders: A Focus on Autism and Schizophrenia. Neurosci. Biobehav. Rev. 2020, 113, 546–567. [Google Scholar] [CrossRef]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The Poly(I:C)-Induced Maternal Immune Activation Model in Preclinical Neuropsychiatric Drug Discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Zolkipli, Z.; Wang, L.; Nakayama, T.; Naviaux, J.C.; Le, T.P.; Schuchbauer, M.A.; Rogac, M.; Tang, Q.; Dugan, L.L.; et al. Antipurinergic Therapy Corrects the Autism-like Features in the Poly(IC) Mouse Model. PLoS ONE 2013, 8, e57380. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Jones, D.N.C. Emerging Role of the P2X7-NLRP3-IL1β Pathway in Mood Disorders. Psychoneuroendocrinology 2018, 98, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Vizi, E.S.; Wirkner, K.; Illes, P. P2X7 Receptors in the Nervous System. Prog. Neurobiol. 2006, 78, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Coutinho-Silva, R. Purinergic Signaling, DAMPs, and Inflammation. Am. J. Physiol.-Cell Physiol. 2020, 318, C832–C835. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.; Petiz, L.L.; Andrejew, R.; Turrini, N.; Silva, J.B.; Sack, U.; Ulrich, H. Role of P2X7 Receptors in Immune Responses during Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 662935. [Google Scholar] [CrossRef]
- Horváth, G.; Otrokocsi, L.; Beko, K.; Baranyi, M.; Kittel, Á.; Fritz-Ruenes, P.A.; Sperlágh, B. P2X7 Receptors Drive Poly(I:C) Induced Autism-like Behavior in Mice. J. Neurosci. 2019, 39, 2542–2561. [Google Scholar] [CrossRef] [PubMed]
- Szabó, D.; Tod, P.; Gölöncsér, F.; Román, V.; Lendvai, B.; Otrokocsi, L.; Sperlágh, B. Maternal P2X7 Receptor Inhibition Prevents Autism-like Phenotype in Male Mouse Offspring through the NLRP3-IL-1β Pathway. Brain Behav. Immun. 2022, 101, 318–332. [Google Scholar] [CrossRef]
- Mut-Arbona, P.; Huang, L.; Baranyi, M.; Tod, P.; Iring, A.; Calzaferri, F.; de Los Ríos, C.; Sperlágh, B. Dual Role of the P2X7 Receptor in Dendritic Outgrowth during Physiological and Pathological Brain Development. J. Neurosci. 2023, 43, 1125–1142. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Curtis, B.; Li, K.; Naviaux, J.C.; Bright, A.T.; Reiner, G.E.; Westerfield, M.; Goh, S.; Alaynick, W.A.; Wang, L.; et al. Low-Dose Suramin in Autism Spectrum Disorder: A Small, Phase I/II, Randomized Clinical Trial. Ann. Clin. Transl. Neurol. 2017, 4, 491–505. [Google Scholar] [CrossRef]
- Passera, S.; Boccazzi, M.; Bokobza, C.; Faivre, V.; Mosca, F.; Van Steenwinckel, J.; Fumagalli, M.; Gressens, P.; Fleiss, B. Therapeutic Potential of Stem Cells for Preterm Infant Brain Damage: Can We Move from the Heterogeneity of Preclinical and Clinical Studies to Established Therapeutics? Biochem. Pharmacol. 2021, 186, 114461. [Google Scholar] [CrossRef]
- Chawanpaiboon, S.; Vogel, J.P.; Moller, A.-B.; Lumbiganon, P.; Petzold, M.; Hogan, D.; Landoulsi, S.; Jampathong, N.; Kongwattanakul, K.; Laopaiboon, M.; et al. Global, Regional, and National Estimates of Levels of Preterm Birth in 2014: A Systematic Review and Modelling Analysis. Lancet Glob. Health 2019, 7, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Vanes, L.D.; Murray, R.M.; Nosarti, C. Adult Outcome of Preterm Birth: Implications for Neurodevelopmental Theories of Psychosis. Schizophr. Res. 2022, 247, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; Bokobza, C.; Saliba, E.; Chalon, S.; Gressens, P. Alteration of the Oligodendrocyte Lineage Varies According to the Systemic Inflammatory Stimulus in Animal Models That Mimic the Encephalopathy of Prematurity. Front. Physiol. 2022, 13, 881674. [Google Scholar] [CrossRef] [PubMed]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The Orphan Receptor GPR17 Identified as a New Dual Uracil Nucleotides/Cysteinyl-Leukotrienes Receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef]
- Mao, F.-X.; Li, W.-J.; Chen, H.-J.; Qian, L.-H.; Buzby, J.S. Periventricular Leukomalacia Long-Term Prognosis May Be Improved by Treatment with UDP-Glucose, GDNF, and Memantine in Neonatal Rats. Brain Res. 2012, 1486, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Mao, F.X.; Chen, H.J.; Qian, L.H.; Buzby, J.S. Treatment with UDP-Glucose, GDNF, and Memantine Promotes SVZ and White Matter Self-Repair by Endogenous Glial Progenitor Cells in Neonatal Rats with Ischemic PVL. Neuroscience 2015, 284, 444–458. [Google Scholar] [CrossRef]
- He, L.; Yang, H.; Feng, J.; Wei, T.; Huang, Y.; Zhang, X.; Wang, Z. Knockdown of G Protein-Coupled Receptor-17 (GPR17) Facilitates the Regeneration and Repair of Myelin Sheath Post-Periventricular Leukomalacia (PVL). Bioengineered 2021, 12, 7314–7324. [Google Scholar] [CrossRef]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic Inflammation Disrupts the Developmental Program of White Matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The Immune-Inflammatory Response of Oligodendrocytes in a Murine Model of Preterm White Matter Injury: The Role of TLR3 Activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef]
- Fumagalli, M.; Daniele, S.; Lecca, D.; Lee, P.R.; Parravicini, C.; Fields, R.D.; Rosa, P.; Antonucci, F.; Verderio, C.; Trincavelli, M.L.; et al. Phenotypic Changes, Signaling Pathway, and Functional Correlates of GPR17-Expressing Neural Precursor Cells during Oligodendrocyte Differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [Google Scholar] [CrossRef]
- Fratangeli, A.; Parmigiani, E.; Fumagalli, M.; Lecca, D.; Benfante, R.; Passafaro, M.; Buffo, A.; Abbracchio, M.P.; Rosa, P. The Regulated Expression, Intracellular Trafficking, and Membrane Recycling of the P2Y-like Receptor GPR17 in Oli-Neu Oligodendroglial Cells. J. Biol. Chem. 2013, 288, 5241–5256. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.; Ritter, K.; Thiemke, K.; Gillard, M.; Kostenis, E.; Müller, C.E. Development of [3H]2-Carboxy-4,6-Dichloro-1 H-Indole-3-Propionic Acid ([3H]PSB-12150): A Useful Tool for Studying GPR17. ACS Med. Chem. Lett. 2014, 5, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Gelosa, P.; Bonfanti, E.; Castiglioni, L.; Delgado-Garcia, J.M.; Gruart, A.; Fontana, L.; Gotti, M.; Tremoli, E.; Lecca, D.; Fumagalli, M.; et al. Improvement of Fiber Connectivity and Functional Recovery after Stroke by Montelukast, an Available and Safe Anti-Asthmatic Drug. Pharmacol. Res. 2019, 142, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020, 21, 2395. [Google Scholar] [CrossRef]
- Marschallinger, J.; Schäffner, I.; Klein, B.; Gelfert, R.; Rivera, F.J.; Illes, S.; Grassner, L.; Janssen, M.; Rotheneichner, P.; Schmuckermair, C.; et al. Structural and Functional Rejuvenation of the Aged Brain by an Approved Anti-Asthmatic Drug. Nat. Commun. 2015, 6, 8466. [Google Scholar] [CrossRef]
- Rivera, A.D.; Pieropan, F.; Chacon-De-La-Rocha, I.; Lecca, D.; Abbracchio, M.P.; Azim, K.; Butt, A.M. Functional Genomic Analyses Highlight a Shift in Gpr17-Regulated Cellular Processes in Oligodendrocyte Progenitor Cells and Underlying Myelin Dysregulation in the Aged Mouse Cerebrum. Aging Cell 2021, 20, e13335. [Google Scholar] [CrossRef]
- Prashad, S.; Gopal, P.P. RNA-Binding Proteins in Neurological Development and Disease. RNA Biol. 2021, 18, 972–987. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of Expression of the FMR-1 Gene in Fragile X Syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling Fragile X Syndrome in the Fmr1 Knockout Mouse. Intractable Rare. Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef]
- Naviaux, J.C.; Wang, L.; Li, K.; Bright, A.T.; Alaynick, W.A.; Williams, K.R.; Powell, S.B.; Naviaux, R.K. Antipurinergic Therapy Corrects the Autism-like Features in the Fragile X (Fmr1 Knockout) Mouse Model. Mol. Autism. 2015, 6, 1. [Google Scholar] [CrossRef]
- Hodges, J.L.; Yu, X.; Gilmore, A.; Bennett, H.; Tjia, M.; Perna, J.F.; Chen, C.C.; Li, X.; Lu, J.; Zuo, Y. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol. Psychiatry 2017, 82, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Doering, L.C. Astrocytes Prevent Abnormal Neuronal Development in the Fragile X Mouse. J. Neurosci. 2010, 30, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.E.; Wong, C.R.; Scott, A.L. Astrocyte-Mediated Purinergic Signaling Is Upregulated in a Mouse Model of Fragile X Syndrome. Glia 2021, 69, 1816–1832. [Google Scholar] [CrossRef]
- Krasovska, V.; Doering, L.C. Regulation of IL-6 Secretion by Astrocytes via TLR4 in the Fragile X Mouse Model. Front. Mol. Neurosci. 2018, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Shovlin, S.; Moriarty, M.; Bardoni, B.; Tropea, D. Rett Syndrome and Fragile X Syndrome: Different Etiology with Common Molecular Dysfunctions. Front. Cell. Neurosci. 2021, 15, 474. [Google Scholar] [CrossRef] [PubMed]
- Garré, J.M.; Silva, H.M.; Lafaille, J.J.; Yang, G. P2X7 Receptor Inhibition Ameliorates Dendritic Spine Pathology and Social Behavioral Deficits in Rett Syndrome Mice. Nat. Commun. 2020, 11, 1784. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The Oligodendrocyte-Specific G Protein-Coupled Receptor GPR17 Is a Cell-Intrinsic Timer of Myelination. Nat. Neurosci. 2009, 12, 1398–1406. [Google Scholar] [CrossRef]
- Angelini, J.; Marangon, D.; Raffaele, S.; Lecca, D.; Abbracchio, M.P. The Distribution of Gpr17-Expressing Cells Correlates with White Matter Inflammation Status in Brain Tissues of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2021, 22, 4574. [Google Scholar] [CrossRef]
- Franke, H.; Parravicini, C.; Lecca, D.; Zanier, E.R.; Heine, C.; Bremicker, K.; Fumagalli, M.; Rosa, P.; Longhi, L.; Stocchetti, N.; et al. Changes of the GPR17 Receptor, a New Target for Neurorepair, in Neurons and Glial Cells in Patients with Traumatic Brain Injury. Purinergic. Signal. 2013, 9, 451–462. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kino, Y.; Yanaizu, M.; Tosaki, Y.; Sakai, K.; Ishida, T.; Saito, Y. Expression of GPR17, a Regulator of Oligodendrocyte Differentiation and Maturation, in Nasu-Hakola Disease Brains. Intractable Rare. Dis. Res. 2017, 6, 50–54. [Google Scholar] [CrossRef]
- Boda, E.; Viganò, F.; Rosa, P.; Fumagalli, M.; Labat-Gest, V.; Tempia, F.; Abbracchio, M.P.; Dimou, L.; Buffo, A. The GPR17 Receptor in NG2 Expressing Cells: Focus on in Vivo Cell Maturation and Participation in Acute Trauma and Chronic Damage. Glia 2011, 59, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, T.; Zhao, Y.; Zheng, Q.; Thompson, R.C.; Bu, G.; Zhang, Y.; Hong, W.; Xu, H. Sorting Nexin 27 Regulates Aβ Production through Modulating γ-Secretase Activity. Cell Rep. 2014, 9, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Y.; Zhang, X.; Badie, H.; Zhou, Y.; Mu, Y.; Loo, L.S.; Cai, L.; Thompson, R.C.; Yang, B.; et al. Loss of Sorting Nexin 27 Contributes to Excitatory Synaptic Dysfunction by Modulating Glutamate Receptor Recycling in Down’s Syndrome. Nat. Med. 2013, 19, 473–480. [Google Scholar] [CrossRef]
- Chandra, M.; Kendall, A.K.; Jackson, L.P. Toward Understanding the Molecular Role of SNX27/Retromer in Human Health and Disease. Front. Cell Dev. Biol. 2021, 9, 642378. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Ulivi, A.F.; Boccazzi, M.; Valenza, F.; Fratangeli, A.; Passafaro, M.; Lecca, D.; Stagni, F.; Giacomini, A.; Bartesaghi, R.; et al. SNX27, a Protein Involved in down Syndrome, Regulates GPR17 Trafficking and Oligodendrocyte Differentiation. Glia 2016, 64, 1437–1460. [Google Scholar] [CrossRef]
- Nir, A.; Barak, B. White Matter Alterations in Williams Syndrome Related to Behavioral and Motor Impairments. Glia 2021, 69, 5–19. [Google Scholar] [CrossRef]
- Barak, B.; Zhang, Z.; Liu, Y.; Nir, A.; Trangle, S.S.; Ennis, M.; Levandowski, K.M.; Wang, D.; Quast, K.; Boulting, G.L.; et al. Neuronal Deletion of Gtf2i, Associated with Williams Syndrome, Causes Behavioral and Myelin Alterations Rescuable by a Remyelinating Drug. Nat. Neurosci. 2019, 22, 700–708. [Google Scholar] [CrossRef]
- Trangle, S.S.; Rosenberg, T.; Parnas, H.; Levy, G.; Bar, E.; Marco, A.; Barak, B. In Individuals with Williams Syndrome, Dysregulation of Methylation in Non-Coding Regions of Neuronal and Oligodendrocyte DNA Is Associated with Pathology and Cortical Development. Mol. Psychiatry 2023, 28, 1112–1127. [Google Scholar] [CrossRef]
- Calleri, E.; Ceruti, S.; Cristalli, G.; Martini, C.; Temporini, C.; Parravicini, C.; Volpini, R.; Daniele, S.; Caccialanza, G.; Lecca, D.; et al. Frontal Affinity Chromatography-Mass Spectrometry Useful for Characterization of New Ligands for GPR17 Receptor. J. Med. Chem. 2010, 53, 3489–3501. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Receptor | Implicated Diseases | Therapeutic Targets or Receptor Engagement |
|---|---|---|
| P2Y2 | Autism spectrum disorders (ASDs) | |
| Fragile X syndrome (FXS) | Upregulated in cultured Fmr1 KO astrocytes [93] | |
| P2Y6 | Autism spectrum disorders (ASDs) | Upregulated in the blood of children with ASDs [40] |
| Fragile X syndrome (FXS) | Upregulated in cultured Fmr1 KO astrocytes [93] | |
| P2X4 | Schizophrenia (SCZ) | mRNA significantly increased in the dorsolateral prefrontal cortex of schizophrenic subjects [43] |
| P2X5 | Schizophrenia (SCZ) | mRNA significantly increased in the dorsolateral prefrontal cortex of schizophrenic subjects [43] |
| P2X7 | Autism spectrum disorders (ASDs) |
|
| Schizophrenia (SCZ) | ||
| Rett syndrome (RTT) |
| |
| GPR17 | Periventricular Leukomalacia (PVL) | |
| Down syndrome |
| |
| Williams syndrome (WS) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccazzi, M.; Raffaele, S.; Zanettin, T.; Abbracchio, M.P.; Fumagalli, M. Altered Purinergic Signaling in Neurodevelopmental Disorders: Focus on P2 Receptors. Biomolecules 2023, 13, 856. https://doi.org/10.3390/biom13050856
Boccazzi M, Raffaele S, Zanettin T, Abbracchio MP, Fumagalli M. Altered Purinergic Signaling in Neurodevelopmental Disorders: Focus on P2 Receptors. Biomolecules. 2023; 13(5):856. https://doi.org/10.3390/biom13050856
Chicago/Turabian StyleBoccazzi, Marta, Stefano Raffaele, Thomas Zanettin, Maria P. Abbracchio, and Marta Fumagalli. 2023. "Altered Purinergic Signaling in Neurodevelopmental Disorders: Focus on P2 Receptors" Biomolecules 13, no. 5: 856. https://doi.org/10.3390/biom13050856