
Pharmacological Characterization of P626, a Novel Dual Adenosine A2A/A2B Receptor Antagonist, on Synaptic Plasticity and during an Ischemic-like Insult in CA1 Rat Hippocampus

 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
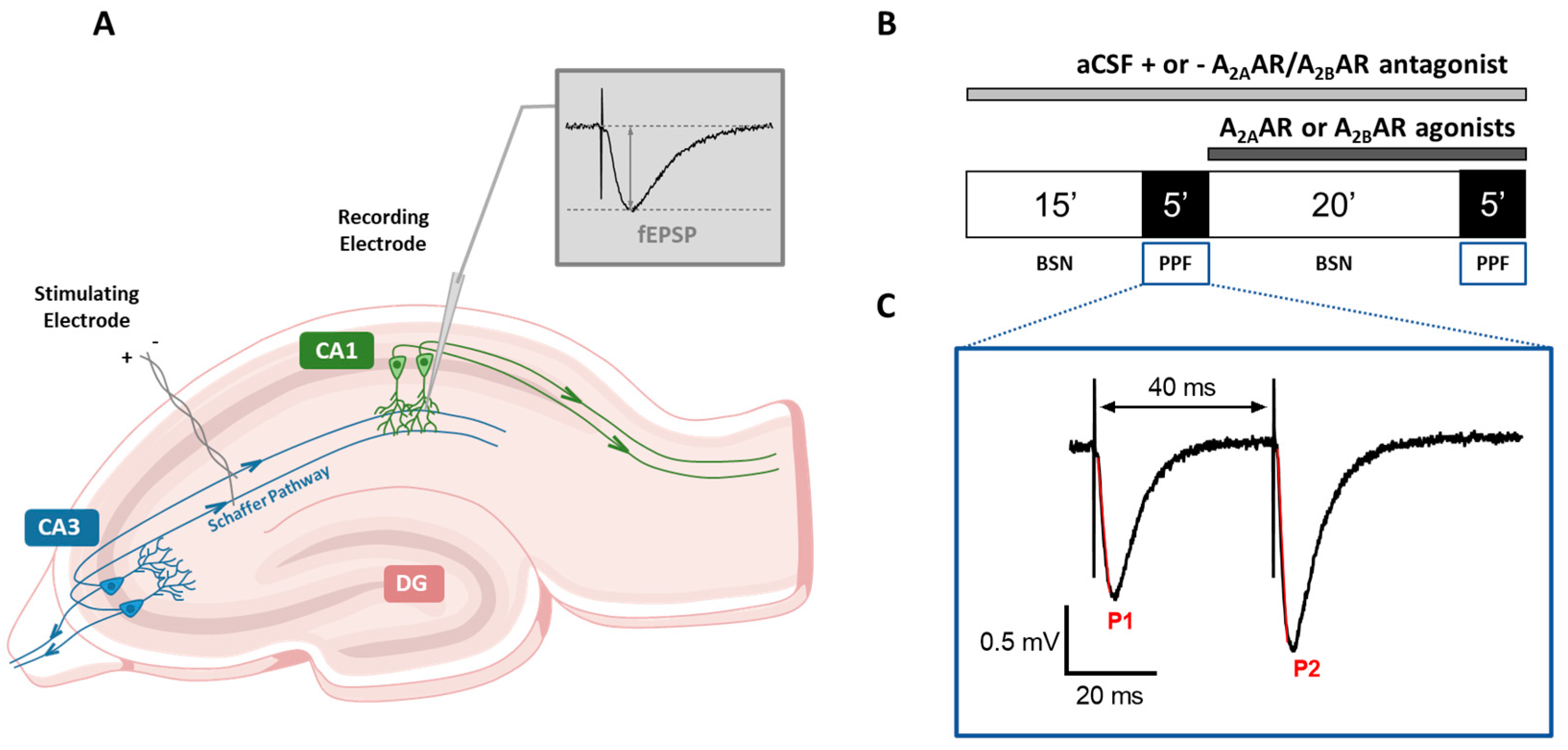
2.1. Preparation of Acute Hippocampal Slices
2.2. Extracellular Recordings
2.3. Paired-Pulse Facilitation
2.4. Oxygen-Glucose Deprivation
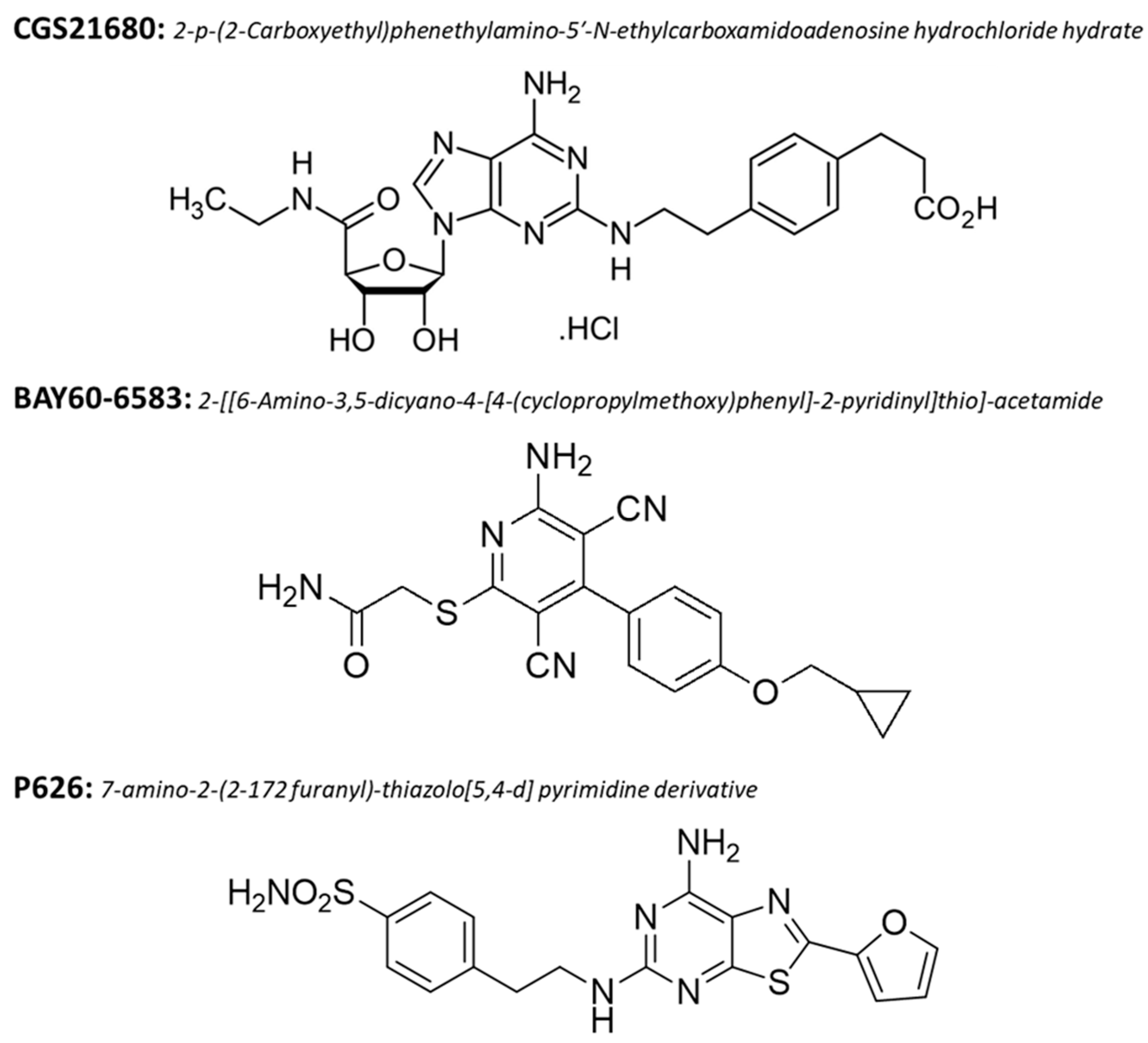
2.5. Drugs
2.6. Statistical Analysis
3. Results
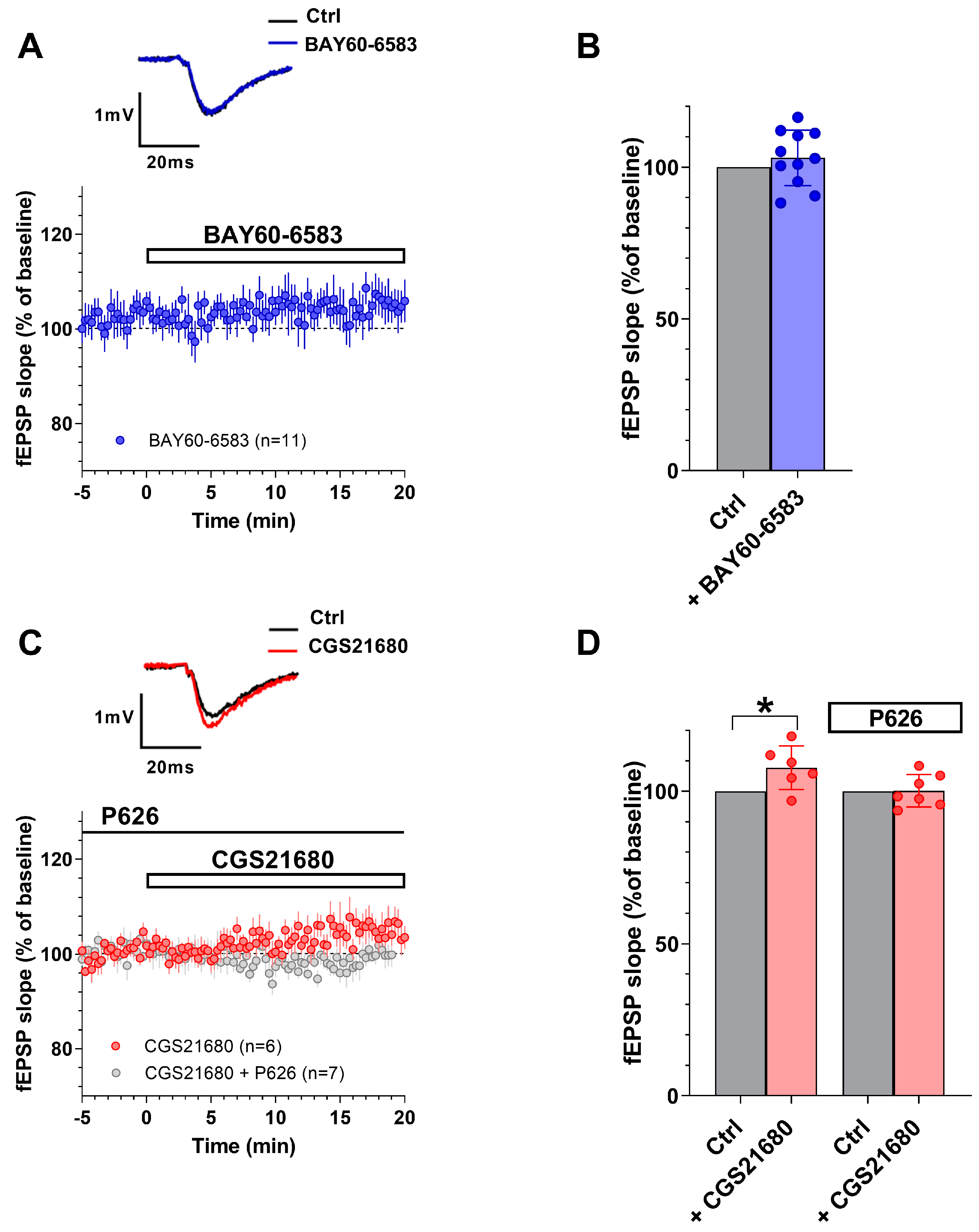
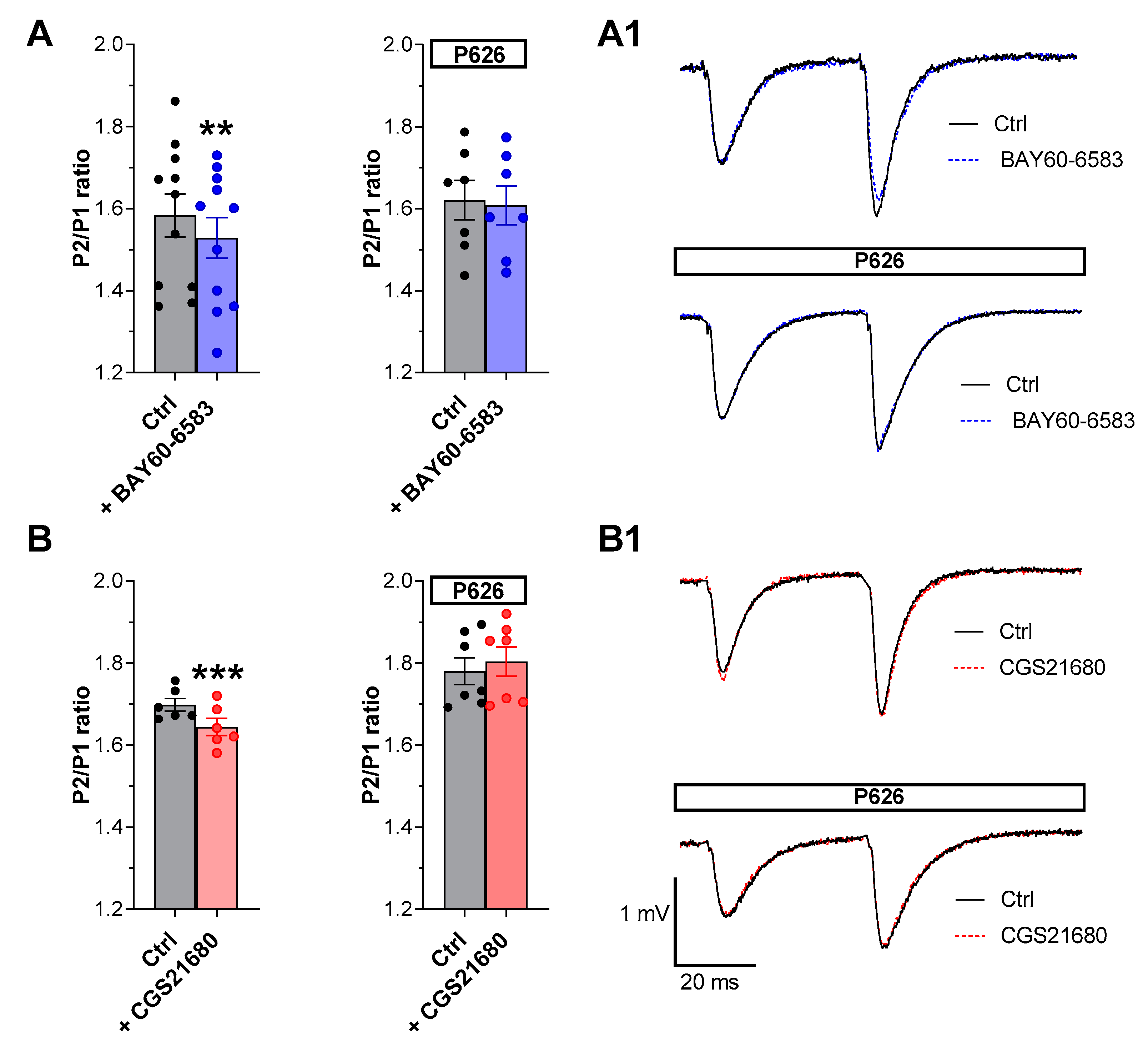
3.1. The New A2AAR/A2BAR Antagonist, P626, Prevented the Effects of Selective A2AAR or A2BAR Agonists on PPF in the CA1 Hippocampal Slices
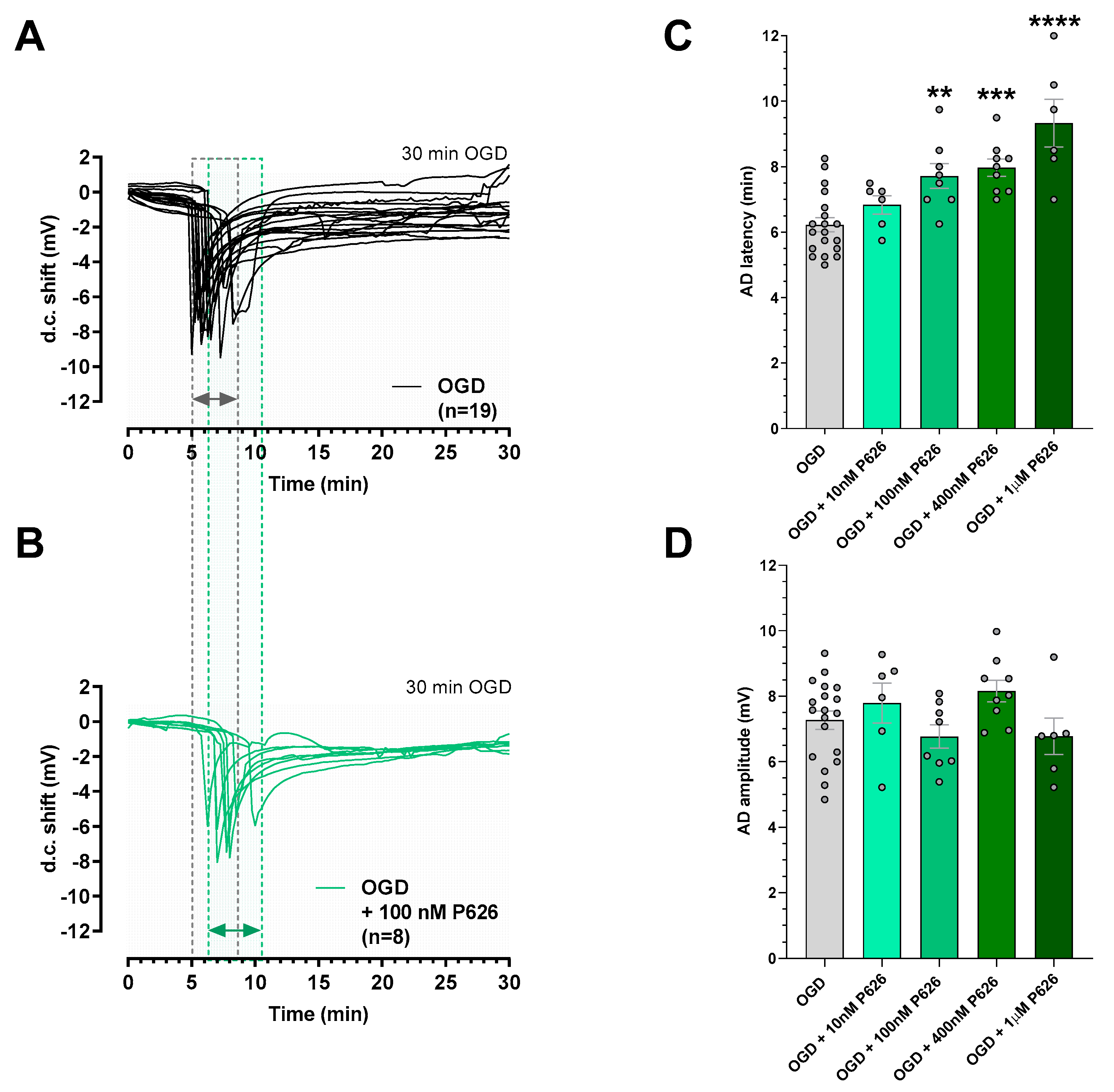
3.2. P626 Delayed AD Onset Induced by Irreversible OGD in the CA1 Rat Hippocampus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Millan, M.J. Dual- and Triple-Acting Agents for Treating Core and Co-Morbid Symptoms of Major Depression: Novel Concepts, New Drugs. Neurotherapeutics 2009, 6, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Favia, A.D.; Recanatini, M.; Cavalli, A. The Role of Fragment-Based and Computational Methods in Polypharmacology. Drug Discov. Today 2012, 17, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef]
- Anthony, S.; Cabantan, D.; Monsour, M.; Borlongan, C.V. Neuroinflammation, Stem Cells, and Stroke. Stroke 2022, 53, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Boltze, J.; Perez-Pinzon, M.A. Focused Update on Stroke Neuroimmunology: Current Progress in Preclinical and Clinical Research and Recent Mechanistic Insight. Stroke 2022, 53, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Bordoni, F.; Corradetti, R.; Pepeu, G.; Pedata, F. Effect of A2A Adenosine Receptor Stimulation and Antagonism on Synaptic Depression Induced by in Vitro Ischaemia in Rat Hippocampal Slices. Br. J. Pharmacol. 1999, 128, 1035–1044. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Diao, L. Extracellular Adenosine Concentrations in Hippocampal Brain Slices and the Tonic Inhibitory Modulation of Evoked Excitatory Responses. J. Pharmacol. Exp. Ther. 1994, 268, 537–545. [Google Scholar] [PubMed]
- Latini, S.; Bordoni, F.; Corradetti, R.; Pepeu, G.; Pedata, F. Temporal Correlation between Adenosine Outflow and Synaptic Potential Inhibition in Rat Hippocampal Slices during Ischemia-like Conditions. Brain Res. 1998, 794, 325–328. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic Signalling in Brain Ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Fredholm, B.B. Adenosine Receptors Mediating Inhibitory Electrophysiological Responses in Rat Hippocampus Are Different from Receptors Mediating Cyclic AMP Accumulation. Naunyn. Schmiedebergs. Arch. Pharmacol. 1984, 326, 294–301. [Google Scholar] [CrossRef]
- Cunha, R.A.; Johansson, B.; van der Ploeg, I.; Sebastião, A.M.; Alexandre Ribeiro, J.; Fredholm, B.B. Evidence for Functionally Important Adenosine A2A Receptors in the Rat Hippocampus. Brain Res. 1994, 649, 208–216. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Ribeiro, J.A. Neuromodulation and Metamodulation by Adenosine: Impact and Subtleties upon Synaptic Plasticity Regulation. Brain Res. 2015, 1621, 102–113. [Google Scholar] [CrossRef]
- Fernández-Fernández, D.; Rosenbrock, H.; Kroker, K.S. Inhibition of PDE2A, but Not PDE9A, Modulates Presynaptic Short-Term Plasticity Measured by Paired-Pulse Facilitation in the CA1 Region of the Hippocampus. Synapse 2015, 69, 484–496. [Google Scholar] [CrossRef]
- Fusco, I.; Cherchi, F.; Catarzi, D.; Colotta, V.; Varano, F.; Pedata, F.; Pugliese, A.M.; Coppi, E. Functional Characterization of a Novel Adenosine A2B Receptor Agonist on Short-Term Plasticity and Synaptic Inhibition during Oxygen and Glucose Deprivation in the Rat CA1 Hippocampus. Brain Res. Bull. 2019, 151, 174–180. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. Adenosine A2A Receptor Facilitation of Hippocampal Synaptic Transmission Is Dependent on Tonic A1 Receptor Inhibition. Neuroscience 2002, 112, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Polenzani, L.; Magnani, M.; Zianni, E.; Stocca, G.; Gardoni, F.; Pellegrini-Giampietro, D.E.; Corradetti, R. Antidepressant-Induced Increase in GluA2 Expression Does Not Translate in Changes of AMPA Receptor-Mediated Synaptic Transmission at CA3/CA1 Synapses in Rats. Neuropharmacology 2023, 223, 109307. [Google Scholar] [CrossRef] [PubMed]
- Regehr, W.G. Short-Term Presynaptic Plasticity. Cold Spring Harb. Perspect. Biol. 2012, 4, a005702. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.Q.; Pires, J.; Pliassova, A.; Beleza, R.; Lemos, C.; Marques, J.M.; Rodrigues, R.J.; Canas, P.M.; Köfalvi, A.; Cunha, R.A.; et al. Adenosine A2b Receptors Control A1 Receptor-Mediated Inhibition of Synaptic Transmission in the Mouse Hippocampus. Eur. J. Neurosci. 2015, 41, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. Cross Talk between A1 and A2A Adenosine Receptors in the Hippocampus and Cortex of Young Adult and Old Rats. J. Neurophysiol. 1999, 82, 3196–3203. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxic Cell Death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Mody, I.; MacDonald, J.F. NMDA Receptor-Dependent Excitotoxicity: The Role of Intracellular Ca2+ Release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Somjen, G.G. Mechanisms of Spreading Depression and Hypoxic Spreading Depression-like Depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [PubMed]
- Joshi, I.; Andrew, R.D. Imaging Anoxic Depolarization during Ischemia-like Conditions in the Mouse Hemi-Brain Slice. J. Neurophysiol. 2001, 85, 414–424. [Google Scholar] [CrossRef]
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical Evoked Potential and Extracellular K+ and H+ at Critical Levels of Brain Ischemia. Stroke 1977, 8, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Symon, L. The relationship between CBF, evoked potentials and the clinical features in cerebral ischaemia. Acta Neurol. Scand. 1980, 62, 175–190. [Google Scholar] [CrossRef]
- Astrup, J.; Siesjö, B.K.; Symon, L. Thresholds in Cerebral Ischemia—The Ischemic Penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R. Four Decades of Ischemic Penumbra and Its Implication for Ischemic Stroke. Transl. Stroke Res. 2021, 12, 937–945. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, Z.; Wang, M.; Sun, M.; Jiang, X.; Ma, C.; Xie, F.; Ma, X. Penumbra in Acute Ischemic Stroke. Curr. Neurovasc. Res. 2021, 18, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, A.M.; Traini, C.; Cipriani, S.; Gianfriddo, M.; Mello, T.; Giovannini, M.G.; Galli, A.; Pedata, F. The Adenosine A 2A Receptor Antagonist ZM241385 Enhances Neuronal Survival after Oxygen-Glucose Deprivation in Rat CA1 Hippocampal Slices. Br. J. Pharmacol. 2009, 157, 818–830. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Coppi, E.; Spalluto, G.; Corradetti, R.; Pedata, F. A3 Adenosine Receptor Antagonists Delay Irreversible Synaptic Failure Caused by Oxygen and Glucose Deprivation in the Rat CA1 Hippocampus in Vitro. Br. J. Pharmacol. 2006, 147, 524–532. [Google Scholar] [CrossRef]
- Maraula, G.; Traini, C.; Mello, T.; Coppi, E.; Galli, A.; Pedata, F.; Pugliese, A.M. Effects of Oxygen and Glucose Deprivation on Synaptic Transmission in Rat Dentate Gyrus: Role of A2A Adenosine Receptors. Neuropharmacology 2013, 67, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Merighi, S.; Varani, K.; Borea, P.A.; Spalluto, G. A3 Adenosine Receptor Ligands: History and Perspectives. Med. Res. Rev. 2000, 20, 103–128. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Bar-Yehuda, S.; Fishman, P. A3 Adenosine Receptor: Pharmacology and Role in Disease. Handb. Exp. Pharmacol. 2009, 193, 297–327. [Google Scholar] [CrossRef]
- Cunha, R.A. How Does Adenosine Control Neuronal Dysfunction and Neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.K.; Widdowson, L.; Richardson, P.J. Desensitisation of the Adenosine A1 Receptor by the A2A Receptor in the Rat Striatum. J. Neurochem. 1997, 69, 315–321. [Google Scholar] [CrossRef]
- Perez-Buira, S.; Barrachina, M.; Rodriguez, A.; Albasanz, J.L.; Martín, M.; Ferrer, I. Expression Levels of Adenosine Receptors in Hippocampus and Frontal Cortex in Argyrophilic Grain Disease. Neurosci. Lett. 2007, 423, 194–199. [Google Scholar] [CrossRef]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The Selective Antagonism of Adenosine A2B Receptors Reduces the Synaptic Failure and Neuronal Death Induced by Oxygen and Glucose Deprivation in Rat CA1 Hippocampus in Vitro. Front. Pharmacol. 2018, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Tatini, F.; Pugliese, A.M.; Traini, C.; Niccoli, S.; Maraula, G.; Ed Dami, T.; Mannini, B.; Scartabelli, T.; Pedata, F.; Casamenti, F.; et al. Amyloid-β Oligomer Synaptotoxicity Is Mimicked by Oligomers of the Model Protein HypF-N. Neurobiol. Aging 2013, 34, 2100–2109. [Google Scholar] [CrossRef]
- Anderson, W.W.; Collingridge, G.L. The LTP Program: A Data Acquisition Program for on-Line Analysis of Long-Term Potentiation and Other Synaptic Events. J. Neurosci. Methods 2001, 108, 71–83. [Google Scholar] [CrossRef]
- Coppi, E.; Pugliese, A.M.; Stephan, H.; Müller, C.E.; Pedata, F. Role of P2 Purinergic Receptors in Synaptic Transmission under Normoxic and Ischaemic Conditions in the CA1 Region of Rat Hippocampal Slices. Purinergic Signal. 2007, 3, 203–219. [Google Scholar] [CrossRef]
- Dettori, I.; Fusco, I.; Bulli, I.; Gaviano, L.; Coppi, E.; Cherchi, F.; Venturini, M.; Di Cesare Mannelli, L.; Ghelardini, C.; Nocentini, A.; et al. Protective Effects of Carbonic Anhydrase Inhibition in Brain Ischaemia in Vitro and in Vivo Models. J. Enzyme Inhib. Med. Chem. 2021, 36, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Pratt, R.; Sengpiel, F.; Obrenovitch, T.P. Direct, Live Imaging of Cortical Spreading Depression and Anoxic Depolarisation Using a Fluorescent, Voltage-Sensitive Dye. J. Cereb. Blood Flow Metab. 2008, 28, 251. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Pasquini, S.; Pelletier, J.; Lopes Rangel Fietto, J.; Espindola Gelsleichter, N.; Sarlandie, M.; Guilbaud, A.; Sévigny, J.; et al. Structural Investigation on Thiazolo[5,4-d]Pyrimidines to Obtain Dual-Acting Blockers of CD73 and Adenosine A2A Receptor as Potential Antitumor Agents. Bioorg. Med. Chem. Lett. 2020, 30, 127067. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Pedata, F. Adenosine in the Central Nervous System: Release Mechanisms and Extracellular Concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Manabe, T.; Wyllie, D.J.A.; Perkel, D.J.; Nicoll, R.A. Modulation of Synaptic Transmission and Long-Term Potentiation: Effects on Paired Pulse Facilitation and EPSC Variance in the CA1 Region of the Hippocampus. J. Neurophysiol. 1993, 70, 1451–1459. [Google Scholar] [CrossRef]
- Debanne, D.; Guérineau, N.C.; Gähwiler, B.H.; Thompson, S.M. Paired-Pulse Facilitation and Depression at Unitary Synapses in Rat Hippocampus: Quantal Fluctuation Affects Subsequent Release. J. Physiol. 1996, 491, 163–176. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-Term Synaptic Plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Dos Santos-Rodrigues, A.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Agostinho, P. Adenosine A2A Receptors Modulate Glutamate Uptake in Cultured Astrocytes and Gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Agostinho, P.; Cunha, R.A.; Chen, J.F. Antagonistic Interaction between Adenosine A2A Receptors and Na+/K+-ATPase-A2 Controlling Glutamate Uptake in Astrocytes. J. Neurosci. 2013, 33, 18492–18502. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A Receptors Are Essential for Long-Term Potentiation of NMDA-EPSCs at Hippocampal Mossy Fiber Synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-Related Shift in LTD Is Dependent on Neuronal Adenosine A2A Receptors Interplay with MGluR5 and NMDA Receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.S.; Richardson, P.J.; Freeman, T.C. Tissue Distribution of Adenosine Receptor MRNAs in the Rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Lana, D.; Giovannini, M.G.; Pedata, F.; Pugliese, A.M. A2B Adenosine Receptors: When Outsiders May Become an Attractive Target to Treat Brain Ischemia or Demyelination. Int. J. Mol. Sci. 2020, 21, 9697. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Ribeiro, J.A. Evidence for the Presence of Excitatory A2 Adenosine Receptors in the Rat Hippocampus. Neurosci. Lett. 1992, 138, 41–44. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Hoffer, B.J.; Fredholm, B.B. Alkylxanthines Elevate Hippocampal Excitability. Evidence for a Role of Endogenous Adenosine. Naunyn. Schmiedebergs Arch. Pharmacol. 1981, 316, 326–330. [Google Scholar] [CrossRef]
- Li, X.; Sun, W.; An, L. Nano-CuO Impairs Spatial Cognition Associated with Inhibiting Hippocampal Long-Term Potentiation via Affecting Glutamatergic Neurotransmission in Rats. Toxicol. Ind. Health 2018, 34, 409–421. [Google Scholar] [CrossRef]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA Receptors Are Expressed in Oligodendrocytes and Activated in Ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef]
- Al-Majed, A.A.; Al-Omar, F.A.; Nagi, M.N. Neuroprotective Effects of Thymoquinone against Transient Forebrain Ischemia in the Rat Hippocampus. Eur. J. Pharmacol. 2006, 543, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, K.; Toth, A.; Deres, P.; Kalai, T.; Hideg, K.; Gallyas, F.; Sumegi, B. Critical Role of PI3-Kinase/Akt Activation in the PARP Inhibitor Induced Heart Function Recovery during Ischemia-Reperfusion. Biochem. Pharmacol. 2006, 71, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Frenguelli, B.G.; Wigmore, G.; Llaudet, E.; Dale, N. Temporal and Mechanistic Dissociation of ATP and Adenosine Release during Ischaemia in the Mammalian Hippocampus. J. Neurochem. 2007, 101, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Corti, F.; Stephan, H.; Müller, C.E.; Donati, C.; Bruni, P.; Vannucchi, M.G.; Pedata, F. Ecto-ATPase Inhibition: ATP and Adenosine Release under Physiological and Ischemic in Vivo Conditions in the Rat Striatum. Exp. Neurol. 2012, 233, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In Vitro Oxygen-Glucose Deprivation to Study Ischemic Cell Death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar] [CrossRef]
- Zur Nedden, S.; Doney, A.S.; Frenguelli, B.G. Modulation of Intracellular ATP Determines Adenosine Release and Functional Outcome in Response to Metabolic Stress in Rat Hippocampal Slices and Cerebellar Granule Cells. J. Neurochem. 2014, 128, 111–124. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | n | Before (Ctrl) (mV/ms) | After (Drugs) (mV/ms) |
|---|---|---|---|
| 200 nM BAY60-6583 | 11 | 0.39 ± 0.03 | 0.40 ± 0.04 |
| 50 nM CGS21680 | 6 | 0.49 ± 0.03 | 0.53 ± 0.03 * |
| 200 nM P626 | 13 | 0.40 ± 0.03 | 0.40 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturini, M.; Cherchi, F.; Santalmasi, C.; Frulloni, L.; Dettori, I.; Catarzi, D.; Pedata, F.; Colotta, V.; Varano, F.; Coppi, E.; et al. Pharmacological Characterization of P626, a Novel Dual Adenosine A2A/A2B Receptor Antagonist, on Synaptic Plasticity and during an Ischemic-like Insult in CA1 Rat Hippocampus. Biomolecules 2023, 13, 894. https://doi.org/10.3390/biom13060894
Venturini M, Cherchi F, Santalmasi C, Frulloni L, Dettori I, Catarzi D, Pedata F, Colotta V, Varano F, Coppi E, et al. Pharmacological Characterization of P626, a Novel Dual Adenosine A2A/A2B Receptor Antagonist, on Synaptic Plasticity and during an Ischemic-like Insult in CA1 Rat Hippocampus. Biomolecules. 2023; 13(6):894. https://doi.org/10.3390/biom13060894
Chicago/Turabian StyleVenturini, Martina, Federica Cherchi, Clara Santalmasi, Lucia Frulloni, Ilaria Dettori, Daniela Catarzi, Felicita Pedata, Vittoria Colotta, Flavia Varano, Elisabetta Coppi, and et al. 2023. "Pharmacological Characterization of P626, a Novel Dual Adenosine A2A/A2B Receptor Antagonist, on Synaptic Plasticity and during an Ischemic-like Insult in CA1 Rat Hippocampus" Biomolecules 13, no. 6: 894. https://doi.org/10.3390/biom13060894