
Caffeine for Prevention of Alzheimer’s Disease: Is the A2A Adenosine Receptor Its Target?

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Caffeine Metabolism
3. Alzheimer’s Disease (AD)
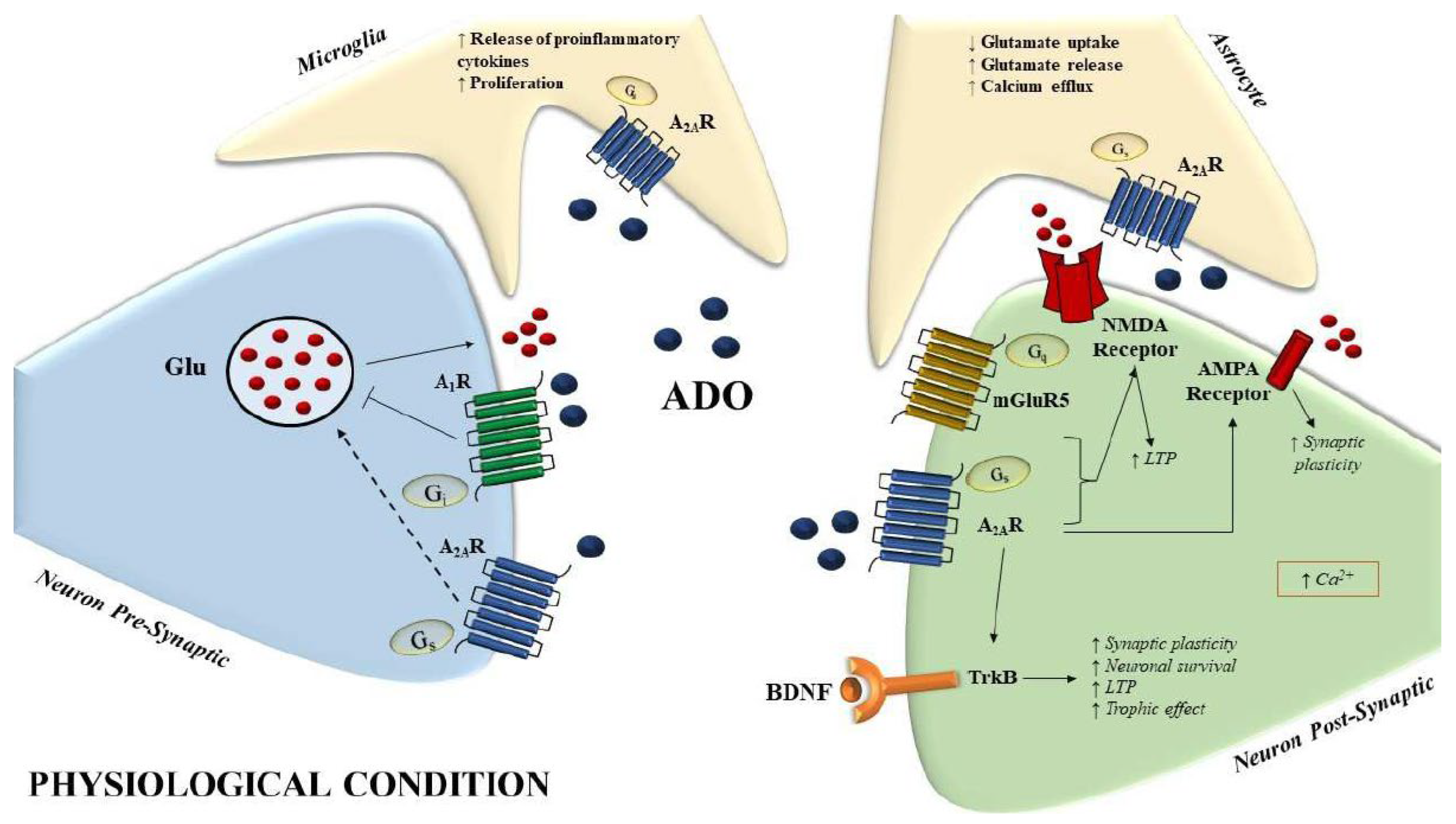
4. A2A Adenosine Receptors in AD
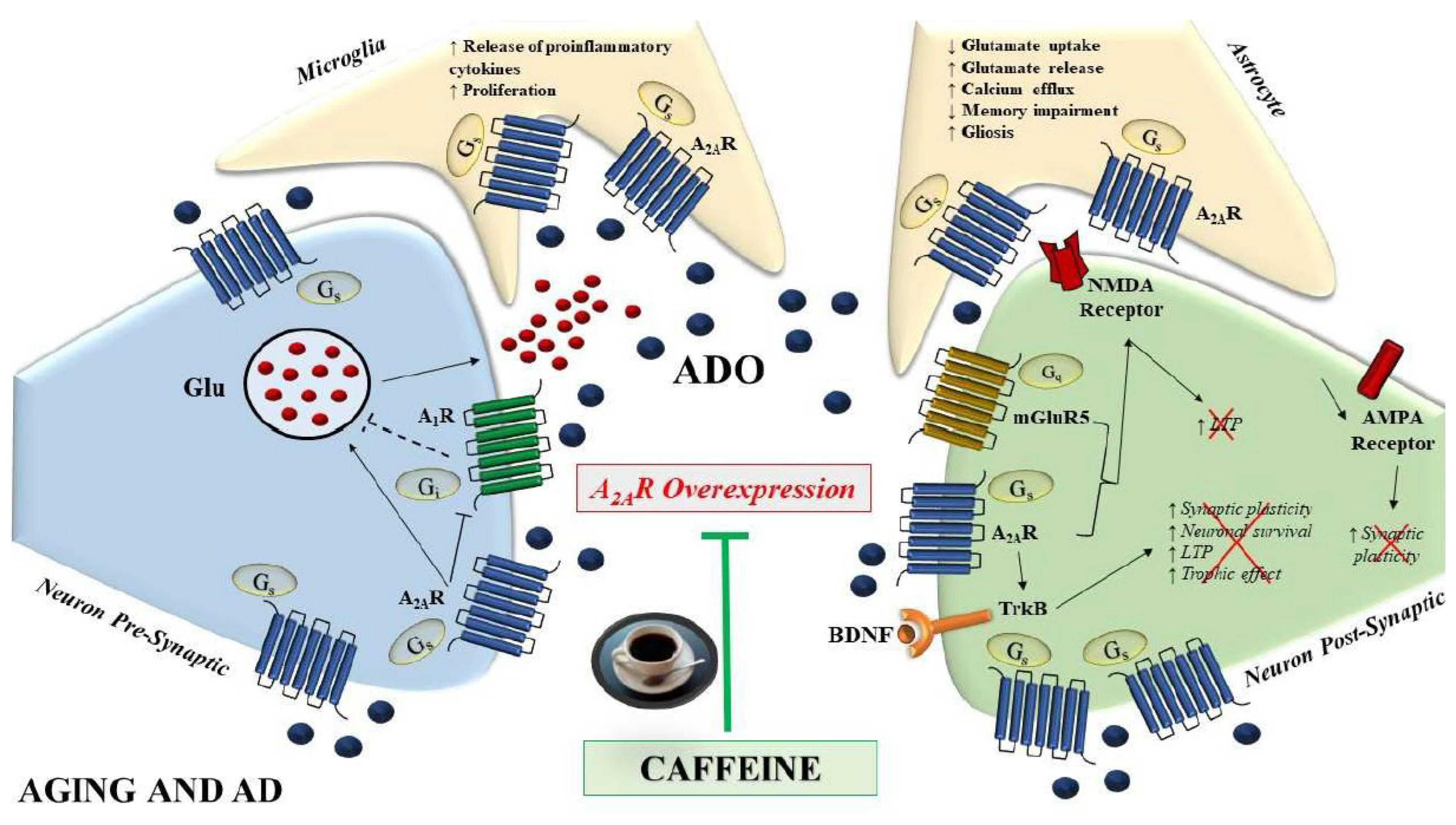
5. Caffeine and A2AR: In Vitro and In Vivo Models of AD
6. Caffeine and AD: Clinical Studies
7. AD, Caffeine, and Gender Differences
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahoney, C.R.; Giles, G.E.; Marriott, B.P.; Judelson, D.A.; Glickman, E.L.; Geiselman, P.J.; Lieberman, H.R. Intake of Caffeine from All Sources and Reasons for Use by College Students. Clin. Nutr. 2019, 38, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Heckman, M.A.; Weil, J.; de Mejia, E.G. Caffeine (1, 3, 7-Trimethylxanthine) in Foods: A Comprehensive Review on Consumption, Functionality, Safety, and Regulatory Matters. J. Food Sci. 2010, 75, R77–R87. [Google Scholar] [CrossRef] [PubMed]
- Chawla, J. Neurologic Effects of Caffeine Physiologic Effects of Caffeine. Drugs Dis. Cond. Clin. Proced. 2015, 24, 2012. [Google Scholar]
- Kolahdouzan, M.; Hamadeh, M.J. The Neuroprotective Effects of Caffeine in Neurodegenerative Diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.; Jones, P.P.; Koop, A.; Zhang, L.; Duff, H.J.; Chen, S.R.W. Caffeine Induces Ca2+ Release by Reducing the Threshold for Luminal Ca2+ Activation of the Ryanodine Receptor. Biochem. J. 2008, 414, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.; Beserra, B.; Reis, C.; Dorea, J.; Da Costa, T.; Amato, A. Consumption of Coffee or Caffeine and Serum Concentration of Inflammatory Markers: A Systematic Review. Crit. Rev. Food Sci. Nutr. 2019, 59, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Zampelas, A.; Panagiotakos, D.B.; Pitsavos, C.; Chrysohoou, C.; Stefanadis, C. Associations between Coffee Consumption and Inflammatory Markers in Healthy Persons: The ATTICA Study. Am. J. Clin. Nutr. 2004, 80, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodak, K.; Kokot, I.; Kratz, E.M. Caffeine as a Factor Influencing the Functioning of the Human Body—Friend or Foe? Nutrients 2021, 13, 3088. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastião, A.M. Caffeine and Adenosine. JAD 2010, 20, S3–S15. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.; Carta, A.R.; Kachroo, A.; Schwarzschild, M.A. Pathophysiological Roles for Purines. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2010; Volume 183, pp. 183–208. ISBN 978-0-444-53614-3. [Google Scholar]
- Polito, C.; Cai, Z.-Y.; Shi, Y.-L.; Li, X.-M.; Yang, R.; Shi, M.; Li, Q.-S.; Ma, S.-C.; Xiang, L.-P.; Wang, K.-R.; et al. Association of Tea Consumption with Risk of Alzheimer’s Disease and Anti-Beta-Amyloid Effects of Tea. Nutrients 2018, 10, 655. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, M.J. Pharmacokinetics and Metabolism of Natural Methylxanthines in Animal and Man. In Methylxanthines; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 200, pp. 33–91. ISBN 978-3-642-13442-5. [Google Scholar]
- Yelanchezian, Y.M.M.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Neuroprotective Effect of Caffeine in Alzheimer’s Disease. Molecules 2022, 27, 3737. [Google Scholar] [CrossRef] [PubMed]
- De Mejia, E.G.; Ramirez-Mares, M.V. Impact of Caffeine and Coffee on Our Health. Trends Endocrinol. Metab. 2014, 25, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.B.; Greenblatt, D.J.; Ehrenberg, B.L.; Goddard, J.E.; Cotreau, M.M.; Harmatz, J.S.; Shader, R.I. Dose-Dependent Pharmacokinetics and Psychomotor Effects of Caffeine in Humans. J. Clin. Pharmacol. 1997, 37, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015, The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; University of Cambridge: Cambridge, UK, 2015. [Google Scholar]
- Santos, C.Y.; Snyder, P.J.; Wu, W.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic Relationship between Alzheimer’s Disease, Cerebrovascular Disease, and Cardiovascular Risk: A Review and Synthesis. Alzheimer Dement. Diagn. Assess. Dis. Monit. 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Schönheit, B.; Zarski, R.; Ohm, T.G. Spatial and Temporal Relationships between Plaques and Tangles in Alzheimer-Pathology. Neurobiol. Aging 2004, 25, 697–711. [Google Scholar] [CrossRef]
- Iqbal, K.; del Alonso, A.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau Pathology in Alzheimer Disease and Other Tauopathies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L.; et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Neurology 1991, 41, 479. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. JAD 2021, 80, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Londzin, P.; Zamora, M.; Kąkol, B.; Taborek, A.; Folwarczna, J. Potential of Caffeine in Alzheimer’s Disease—A Review of Experimental Studies. Nutrients 2021, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Priefer, R. Alzheimer’s Disease Failed Clinical Trials. Life Sci. 2022, 306, 120861. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, M.; Calvello, R.; Porro, C.; Messina, G.; Cianciulli, A.; Panaro, M.A. Neurodegenerative Diseases: Can Caffeine Be a Powerful Ally to Weaken Neuroinflammation? Int. J. Mol. Sci. 2022, 23, 12958. [Google Scholar] [CrossRef]
- Chen, J.; Fan, A.; Li, S.; Xiao, Y.; Fu, Y.; Chen, J.-S.; Zi, D.; Zeng, L.-H.; Tan, J. APP Mediates Tau Uptake and Its Overexpression Leads to the Exacerbated Tau Pathology. Cell. Mol. Life Sci. 2023, 80, 123. [Google Scholar] [CrossRef]
- Kurkinen, M.; Fułek, M.; Fułek, K.; Beszłej, J.A.; Kurpas, D.; Leszek, J. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules 2023, 13, 453. [Google Scholar] [CrossRef]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and Microglia: Considerations and Approaches for Neurotoxicity Assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Rojo, L.E.; Fernández, J.A.; Kuljis, R.O. The Role of Neuroimmunomodulation in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar] [CrossRef]
- Langworth-Green, C.; Patel, S.; Jaunmuktane, Z.; Jabbari, E.; Morris, H.; Thom, M.; Lees, A.; Hardy, J.; Zandi, M.; Duff, K. Chronic Effects of Inflammation on Tauopathies. Lancet Neurol. 2023, 22, 430–442. [Google Scholar] [CrossRef]
- Pamplona, R. Membrane Phospholipids, Lipoxidative Damage and Molecular Integrity: A Causal Role in Aging and Longevity. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free. Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. In Advances in Mitochondrial Medicine; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2012; Volume 942, pp. 269–286. ISBN 978-94-007-2868-4. [Google Scholar]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 Regulates Microglial Dynamics and Neuroinflammation in Experimental Parkinson’s Disease: N RF 2 Regulates Microglial Dynamics. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schäfer, A.; Parnham, M.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549. [Google Scholar] [CrossRef] [PubMed]
- Jordan, F.; Quinn, T.J.; McGuinness, B.; Passmore, P.; Kelly, J.P.; Tudur Smith, C.; Murphy, K.; Devane, D. Aspirin and Other Non-Steroidal Anti-Inflammatory Drugs for the Prevention of Dementia. Cochrane Database Syst. Rev. 2020, 2020, CD011459. [Google Scholar] [CrossRef]
- Park, K.W.; Kim, E.-J.; Han, H.J.; Shim, Y.S.; Kwon, J.C.; Ku, B.D.; Park, K.H.; Yi, H.-A.; Kim, K.K.; Yang, D.W.; et al. Efficacy and Tolerability of Rivastigmine Patch Therapy in Patients with Mild-to-Moderate Alzheimer’s Dementia Associated with Minimal and Moderate Ischemic White Matter Hyperintensities: A Multicenter Prospective Open-Label Clinical Trial. PLoS ONE 2017, 12, e0182123. [Google Scholar] [CrossRef] [Green Version]
- Stoiljkovic, M.; Horvath, T.L.; Hajós, M. Therapy for Alzheimer’s Disease: Missing Targets and Functional Markers? Ageing Res. Rev. 2021, 68, 101318. [Google Scholar] [CrossRef]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Travagli, A.; Nigro, M.; Pasquini, S.; Suresh, R.R.; Kim, S.W.; Volkow, N.D.; et al. Pathophysiological Role and Medicinal Chemistry of A2A Adenosine Receptor Antagonists in Alzheimer’s Disease. Molecules 2022, 27, 2680. [Google Scholar] [CrossRef]
- Cummings, J. New Approaches to Symptomatic Treatments for Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA Approval for Biogen’s Aducanumab Sparks Alzheimer Disease Firestorm. Nat. Rev. Drug Discov. 2021, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does It Exert Its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.G.; Gonçalves, F.Q.; Marques, J.M.; Tomé, Â.R.; Rodrigues, R.J.; Nunes-Correia, I.; Ledent, C.; Harkany, T.; Venance, L.; Cunha, R.A.; et al. Presynaptic Adenosine A2A Receptors Dampen Cannabinoid CB1 Receptor-Mediated Inhibition of Corticostriatal Glutamatergic Transmission: A2A-CB1 Receptor Interaction in Corticostriatal Terminals. Br. J. Pharmacol. 2015, 172, 1074–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. Adenosine A2A Receptor Facilitation of Hippocampal Synaptic Transmission Is Dependent on Tonic A1 Receptor Inhibition. Neuroscience 2002, 112, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.; Augusto, E.; Machado, N.J.; dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic Adenosine A2A Receptors Control the Amyloid-β Peptide-Induced Decrease of Glutamate Uptake. J. Alzheimers Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.-Q.; Adame, A.; et al. Astrocytic Adenosine Receptor A2A and Gs-Coupled Signaling Regulate Memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sánchez de Medina, V.; et al. Potentiation of Cannabinoid Signaling in Microglia by Adenosine A 2A Receptor Antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Lillo, A.; Rivas-Santisteban, R.; Reyes-Resina, I.; Navarro, G. Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells 2021, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12990. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.R.; Cunha, R.A.; Agostinho, P. Astrocytes and Adenosine A2A Receptors: Active Players in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666710. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How Does Adenosine Control Neuronal Dysfunction and Neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Launay, A.; Nebie, O.; Vijaya Shankara, J.; Lebouvier, T.; Buée, L.; Faivre, E.; Blum, D. The Role of Adenosine A2A Receptors in Alzheimer’s Disease and Tauopathies. Neuropharmacology 2023, 226, 109379. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Pelloni, L.; Pasquini, S.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. An Open Question: Is the A2A Adenosine Receptor a Novel Target for Alzheimer’s Disease Treatment? Front. Pharmacol. 2021, 12, 652455. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Poloni, T.E.; Negro, G.; Varani, K.; Pasquini, S.; Vincenzi, F.; Borea, P.A.; Merighi, S. A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease. Cells 2021, 10, 2344. [Google Scholar] [CrossRef]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A Receptors Are Essential for Long-Term Potentiation of NMDA-EPSCs at Hippocampal Mossy Fiber Synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Costenla, A.R.; Diógenes, M.J.; Canas, P.M.; Rodrigues, R.J.; Nogueira, C.; Maroco, J.; Agostinho, P.M.; Ribeiro, J.A.; Cunha, R.A.; de Mendonça, A. Enhanced Role of Adenosine A2A Receptors in the Modulation of LTP in the Rat Hippocampus upon Ageing: Ageing, Adenosine and LTP. Eur. J. Neurosci. 2011, 34, 12–21. [Google Scholar] [CrossRef]
- Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. Enhancement of AMPA Currents and GluR1 Membrane Expression through PKA-Coupled Adenosine A2A Receptors. Hippocampus 2012, 22, 276–291. [Google Scholar] [CrossRef]
- Tebano, M.T.; Martire, A.; Rebola, N.; Pepponi, R.; Domenici, M.R.; Grò, M.C.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Popoli, P. Adenosine A2A Receptors and Metabotropic Glutamate 5 Receptors Are Co-Localized and Functionally Interact in the Hippocampus: A Possible Key Mechanism in the Modulation of N-Methyl-d-Aspartate Effects. J. Neurochem. 2005, 95, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Sarantis, K.; Tsiamaki, E.; Kouvaros, S.; Papatheodoropoulos, C.; Angelatou, F. Adenosine A2A Receptors Permit MGluR5-Evoked Tyrosine Phosphorylation of NR2B (Tyr1472) in Rat Hippocampus: A Possible Key Mechanism in NMDA Receptor Modulation. J. Neurochem. 2015, 135, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and Memory Dysfunction in a β-Amyloid Model of Early Alzheimer’s Disease Depends on Increased Formation of ATP-Derived Extracellular Adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Coelho, J.E.; Pousinha, P.A.; Lopes, L.V. Novel Players in the Aging Synapse: Impact on Cognition. J. Caffeine Adenosine Res. 2019, 9, 104–127. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Jacobson, K.A.; Gessi, S. A2A Adenosine Receptor Antagonists in Neurodegenerative Diseases. CMC 2022, 29, 4138–4151. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Diógenes, M.J.; Assaife-Lopes, N.; Pinto-Duarte, A.; Ribeiro, J.A.; Sebastião, A.M. Influence of Age on BDNF Modulation of Hippocampal Synaptic Transmission: Interplay with Adenosine A2A Receptors. Hippocampus 2007, 17, 577–585. [Google Scholar] [CrossRef]
- Diógenes, M.J.; Fernandes, C.C.; Sebastião, A.M.; Ribeiro, J.A. Activation of Adenosine A2A Receptor Facilitates Brain-Derived Neurotrophic Factor Modulation of Synaptic Transmission in Hippocampal Slices. J. Neurosci. 2004, 24, 2905–2913. [Google Scholar] [CrossRef] [Green Version]
- Fontinha, B.M.; Diógenes, M.J.; Ribeiro, J.A.; Sebastião, A.M. Enhancement of Long-Term Potentiation by Brain-Derived Neurotrophic Factor Requires Adenosine A2A Receptor Activation by Endogenous Adenosine. Neuropharmacology 2008, 54, 924–933. [Google Scholar] [CrossRef]
- Tebano, M.T.; Martire, A.; Potenza, R.L.; Grò, C.; Pepponi, R.; Armida, M.; Domenici, M.R.; Schwarzschild, M.A.; Chen, J.F.; Popoli, P. Adenosine A2A Receptors Are Required for Normal BDNF Levels and BDNF-Induced Potentiation of Synaptic Transmission in the Mouse Hippocampus. J. Neurochem. 2008, 104, 279–286. [Google Scholar] [CrossRef]
- Pagnussat, N.; Almeida, A.S.; Marques, D.M.; Nunes, F.; Chenet, G.C.; Botton, P.H.S.; Mioranzza, S.; Loss, C.M.; Cunha, R.A.; Porciúncula, L.O. Adenosine A2A Receptors Are Necessary and Sufficient to Trigger Memory Impairment in Adult Mice: A2A Receptor Controls Memory. Br. J. Pharmacol. 2015, 172, 3831–3845. [Google Scholar] [CrossRef] [Green Version]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-Related Shift in LTD Is Dependent on Neuronal Adenosine A2A Receptors Interplay with MGluR5 and NMDA Receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canas, P.M.; Duarte, J.M.N.; Rodrigues, R.J.; Köfalvi, A.; Cunha, R.A. Modification upon Aging of the Density of Presynaptic Modulation Systems in the Hippocampus. Neurobiol. Aging 2009, 30, 1877–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. Cross Talk Between A1 and A2A Adenosine Receptors in the Hippocampus and Cortex of Young Adult and Old Rats. J. Neurophysiol. 1999, 82, 3196–3203. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martín, M. Up-Regulation of Adenosine Receptors in the Frontal Cortex in Alzheimer’s Disease: Adenosine Receptors in Alzheimer’s Disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine Protects Alzheimer’s Mice against Cognitive Impairment and Reduces Brain β-Amyloid Production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by β-Amyloid Peptides via P38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef] [Green Version]
- da Viana Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early Synaptic Deficits in the APP/PS1 Mouse Model of Alzheimer’s Disease Involve Neuronal Adenosine A2A Receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine Consumption Prevents Memory Impairment, Neuronal Damage, and Adenosine A2A Receptors Upregulation in the Hippocampus of a Rat Model of Sporadic Dementia. JAD 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Faivre, E.; Coelho, J.E.; Zornbach, K.; Malik, E.; Baqi, Y.; Schneider, M.; Cellai, L.; Carvalho, K.; Sebda, S.; Figeac, M.; et al. Beneficial Effect of a Selective Adenosine A2A Receptor Antagonist in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.C.; Lemos, C.; Gonçalves, F.Q.; Pliássova, A.V.; Machado, N.J.; Silva, H.B.; Canas, P.M.; Cunha, R.A.; Lopes, J.P.; Agostinho, P. Blockade of Adenosine A2A Receptors Recovers Early Deficits of Memory and Plasticity in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2018, 117, 72–81. [Google Scholar] [CrossRef]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q Dysregulation, Synaptic Loss and Memory Deficits in Tau Pathology Linked to Neuronal Adenosine A2A Receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A.; et al. A2A Adenosine Receptor Deletion Is Protective in a Mouse Model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A. The Role of Adenosine in Alzheimers Disease. CN 2009, 7, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, P.N.H.; Baltos, J.-A.; Hellyer, S.D.; May, L.T.; Gregory, K.J. Adenosine Receptor Signalling in Alzheimer’s Disease. Purinergic Signal. 2022, 18, 359–381. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Bättig, K.; Holmén, J.; Nehlig, A.; Zvartau, E.E. Actions of Caffeine in the Brain with Special Reference to Factors That Contribute to Its Widespread Use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar]
- Lopes, J.P.; Pliássova, A.; Cunha, R.A. The Physiological Effects of Caffeine on Synaptic Transmission and Plasticity in the Mouse Hippocampus Selectively Depend on Adenosine A1 and A2A Receptors. Biochem. Pharmacol. 2019, 166, 313–321. [Google Scholar] [CrossRef]
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tan, J.; Citron, B.A.; Lin, X.; et al. Caffeine Reverses Cognitive Impairment and Decreases Brain Amyloid-β Levels in Aged Alzheimer’s Disease Mice. JAD 2009, 17, 661–680. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine Suppresses Amyloid-β Levels in Plasma and Brain of Alzheimer’s Disease Transgenic Mice. JAD 2009, 17, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial Effects of Caffeine in a Transgenic Model of Alzheimer’s Disease-like Tau Pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and Adenosine A2a Receptor Antagonists Prevent β-Amyloid (25–35)-Induced Cognitive Deficits in Mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef]
- Rebola, N.; Canas, P.M.; Oliveira, C.R.; Cunha, R.A. Different Synaptic and Subsynaptic Localization of Adenosine A2A Receptors in the Hippocampus and Striatum of the Rat. Neuroscience 2005, 132, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Dall’lgna, O.P.; Porciúncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Neuroprotection by Caffeine and Adenosine A2A Receptor Blockade of β-Amyloid Neurotoxicity: Special Report. Br. J. Pharmacol. 2003, 138, 1207–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolotto, J.W.; de Melo, G.M.; Cognato, G.P.; Vianna, M.R.M.; Bonan, C.D. Modulation of Adenosine Signaling Prevents Scopolamine-Induced Cognitive Impairment in Zebrafish. Neurobiol. Learn. Mem. 2015, 118, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botton, P.H.; Costa, M.S.; Ardais, A.P.; Mioranzza, S.; Souza, D.O.; da Rocha, J.B.T.; Porciúncula, L.O. Caffeine Prevents Disruption of Memory Consolidation in the Inhibitory Avoidance and Novel Object Recognition Tasks by Scopolamine in Adult Mice. Behav. Brain Res. 2010, 214, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Reidel, W.; Hogervorst, E.; Leboux, R.; Verhey, F.; van Praag, H.; Jolles, J. Caffeine Attenuates Scopolamine-Induced Memory Impairment in Humans. Psychopharmacology 1995, 122, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-A.; Zhao, Y.; Ning, Y.-L.; Yang, N.; Peng, Y.; Li, P.; Chen, X.-Y.; Liu, D.; Wang, H.; Chen, X.; et al. Adenosine A2A Receptor Inactivation Alleviates Early-Onset Cognitive Dysfunction after Traumatic Brain Injury Involving an Inhibition of Tau Hyperphosphorylation. Transl. Psychiatry 2017, 7, e1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcez, M.L.; Damiani, A.P.; Pacheco, R.; Rodrigues, L.; De Abreu, L.L.; Alves, M.C.; De Andrade, V.M.; Boeck, C.R. Caffeine Neuroprotection Decreases A2A Adenosine Receptor Content in Aged Mice. Neurochem. Res. 2019, 44, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.R.; Oliveira, A.; Gaspar, I.; Rodrigues, M.S.; Santos, J.; Szabó, E.; Silva, H.B.; Tomé, Â.R.; Canas, P.M.; Agostinho, P.; et al. Effects of Chronic Caffeine Consumption on Synaptic Function, Metabolism and Adenosine Modulation in Different Brain Areas. Biomolecules 2023, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.H.; Boia, R.; Ambrósio, A.F.; Santiago, A.R. Having a Coffee Break: The Impact of Caffeine Consumption on Microglia-Mediated Inflammation in Neurodegenerative Diseases. Mediat. Inflamm. 2017, 2017, 4761081. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Jia, N.; Li, J.; Yang, L.; Min, L.-Q. Chronic Caffeine Treatment Reverses Memory Impairment and the Expression of Brain BNDF and TrkB in the PS1/APP Double Transgenic Mouse Model of Alzheimer’s Disease. Mol. Med. Rep. 2013, 8, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, F.M.; Khalaf, H.A.; Elsamanoudy, A.Z.; El-khair, S.M.A.; Helaly, A.M.; Mahmoud, E.-H.M.; Elshafey, S.H. Protective Effect of Chronic Caffeine Intake on Gene Expression of Brain Derived Neurotrophic Factor Signaling and the Immunoreactivity of Glial Fibrillary Acidic Protein and Ki-67 in Alzheimer’s Disease. Int. J. Clin. Exp. Pathol. 2015, 8, 7710–7728. [Google Scholar] [PubMed]
- Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules 2019, 9, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and Neuroprotective Effects of Caffeine against Alzheimer’s and Parkinson’s Disease: Insight into the Role of Nrf-2 and A2AR Signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine Prevents D-Galactose-Induced Cognitive Deficits, Oxidative Stress, Neuroinflammation and Neurodegeneration in the Adult Rat Brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Maia, L.; de Mendonca, A. Does Caffeine Intake Protect from Alzheimer’s Disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, K.; Carriere, I.; de Mendonca, A.; Portet, F.; Dartigues, J.F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.L. The Neuroprotective Effects of Caffeine: A Prospective Population Study (the Three City Study). Neurology 2007, 69, 536–545. [Google Scholar] [CrossRef]
- van Gelder, B.M.; Buijsse, B.; Tijhuis, M.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Coffee Consumption Is Inversely Associated with Cognitive Decline in Elderly European Men: The FINE Study. Eur. J. Clin. Nutr. 2007, 61, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Coffee and Tea Drinking and the Risk of Late-Life Dementia: A Population-Based CAIDE Study. JAD 2009, 16, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, K.; Tomata, Y.; Kaiho, Y.; Honkura, K.; Sugawara, Y.; Tsuji, I. Association between Coffee Consumption and Incident Risk of Disabling Dementia in Elderly Japanese: The Ohsaki Cohort 2006 Study. JAD 2016, 50, 491–500. [Google Scholar] [CrossRef]
- Dong, X.; Li, S.; Sun, J.; Li, Y.; Zhang, D. Association of Coffee, Decaffeinated Coffee and Caffeine Intake from Coffee with Cognitive Performance in Older Adults: National Health and Nutrition Examination Survey (NHANES) 2011–2014. Nutrients 2020, 12, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camfield, D.A.; Silber, B.Y.; Scholey, A.B.; Nolidin, K.; Goh, A.; Stough, C. A Randomised Placebo-Controlled Trial to Differentiate the Acute Cognitive and Mood Effects of Chlorogenic Acid from Decaffeinated Coffee. PLoS ONE 2013, 8, e82897. [Google Scholar] [CrossRef] [PubMed]
- Haskell-Ramsay, C.; Jackson, P.; Forster, J.; Dodd, F.; Bowerbank, S.; Kennedy, D. The Acute Effects of Caffeinated Black Coffee on Cognition and Mood in Healthy Young and Older Adults. Nutrients 2018, 10, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borota, D.; Murray, E.; Keceli, G.; Chang, A.; Watabe, J.M.; Ly, M.; Toscano, J.P.; Yassa, M.A. Post-Study Caffeine Administration Enhances Memory Consolidation in Humans. Nat. Neurosci. 2014, 17, 201–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, I.; Cellai, L.; Meriaux, C.; Poncelet, L.; Nebie, O.; Saliou, J.-M.; Lacoste, A.-S.; Papegaey, A.; Drobecq, H.; Le Gras, S.; et al. Caffeine Intake Exerts Dual Genome-Wide Effects on Hippocampal Metabolism and Learning-Dependent Transcription. J. Clin. Investig. 2022, 132, e149371. [Google Scholar] [CrossRef] [PubMed]
- Jee, H.J.; Lee, S.G.; Bormate, K.J.; Jung, Y.-S. Effect of Caffeine Consumption on the Risk for Neurological and Psychiatric Disorders: Sex Differences in Human. Nutrients 2020, 12, 3080. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2014 Alzheimer’s Disease Facts and Figures. Alzheimer Dement. 2014, 10, e47–e92. [Google Scholar] [CrossRef] [Green Version]
- Pankratz, V.S.; Roberts, R.O.; Mielke, M.M.; Knopman, D.S.; Jack, C.R.; Geda, Y.E.; Rocca, W.A.; Petersen, R.C. Predicting the Risk of Mild Cognitive Impairment in the Mayo Clinic Study of Aging. Neurology 2015, 84, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Seidlitz, J.; Blumenthal, J.D.; Clasen, L.S.; Raznahan, A. Integrative Structural, Functional, and Transcriptomic Analyses of Sex-Biased Brain Organization in Humans. Proc. Natl. Acad. Sci. USA 2020, 117, 18788–18798. [Google Scholar] [CrossRef]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. JAD 2018, 64, 1077–1083. [Google Scholar] [CrossRef]
- Nebel, R.A.; Aggarwal, N.T.; Barnes, L.L.; Gallagher, A.; Goldstein, J.M.; Kantarci, K.; Mallampalli, M.P.; Mormino, E.C.; Scott, L.; Yu, W.H.; et al. Understanding the Impact of Sex and Gender in Alzheimer’s Disease: A Call to Action. Alzheimer Dement. 2018, 14, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, I.; Shumaker, S.A.; Snively, B.M.; Margolis, K.L.; Manson, J.E.; Vitolins, M.Z.; Rossom, R.C.; Espeland, M.A. Relationships Between Caffeine Intake and Risk for Probable Dementia or Global Cognitive Impairment: The Women’s Health Initiative Memory Study. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1596–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Experimental Model of AD | Main Results | Mechanisms of Neuroprotection | References |
|---|---|---|---|
| APPsw transgenic (Tg) mice SweAPP N2a neuronal cultures | Caffeine induces cognitive protection in spatial learning/reference memory, working memory, and recognition/identification tests | ↓PS1 ↓BACE ↓Aβ ↓Aβ | [79] |
| Adult male Wistar rats with sporadic AD | Caffeine prevents Streptozotocin-induced memory decline, neuronal damage, and A2AR upregulation | ND | [82] |
| 18–19 month-old APPsw mice | Caffeine improves working memory | ↓PS1 ↓BACE1 ↓Aβ | [91] |
| AD Tg mice | Therapeutic value of caffeine against AD | ↓Aβ | [92] |
| THY-Tau22 Tg mouse | Caffeine improves spatial memory | ↓ Tau phosphorylation ↓ Proinflammatory and oxidative stress markers | [93] |
| CF1 adult mice injected with Aβ | Caffeine or selective A2AR treatment showed a protective effect against cognitive decline | ND | [94] |
| Primary rat cerebellar neurons | Caffeine shows neuroprotective effects through A2AR antagonism | ↓Aβ-induced neuronal cell death | [96] |
| WT Zebrafish | Caffeine pre-treatment prevented scopolamine-induced amnesia | A2AR antagonism | [97] |
| Male CF1 mice | Caffeine increases memory and cognitive functions scopolamine-decreased | ND | [98] |
| A2AR KO mice | Caffeine normalizes memory impairment induced by TBI-mediated A2AR activation | ↓Tau phosphorylation | [100] |
| Young and aged male Swiss mice | Caffeine decreases A2AR and the number of pyknotic aged neurons | ND | [101] |
| Adult male C57bl\6j mice | Caffeine increases the metabolic competence of synapses | ND | [102] |
| PS1/APP double Tg mice | Caffeine increases memory capability | ↑BDNF ↑TrkB | [104] |
| Adult male albino Sprague-Dawley rats | Caffeine has a potentially good protective effect against AD induced by AlCl3 | ↑BDNF ↑TrkB | [105] |
| C57BL/6N male mice | Caffeine may regulate oxidative stress, neuroinflammation, and synaptic dysfunctions in LPS-injected mouse brains | ↑Nrf2/TLR4 | [106] |
| Adult male Sprague-Dawley rats | Caffeine has a beneficial effect against artificial senescence in mice induced by D-galactose | ↓p-JNK, COX-2, NOS-2, TNFα and IL-1β | [108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merighi, S.; Travagli, A.; Nigro, M.; Pasquini, S.; Cappello, M.; Contri, C.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. Caffeine for Prevention of Alzheimer’s Disease: Is the A2A Adenosine Receptor Its Target? Biomolecules 2023, 13, 967. https://doi.org/10.3390/biom13060967
Merighi S, Travagli A, Nigro M, Pasquini S, Cappello M, Contri C, Varani K, Vincenzi F, Borea PA, Gessi S. Caffeine for Prevention of Alzheimer’s Disease: Is the A2A Adenosine Receptor Its Target? Biomolecules. 2023; 13(6):967. https://doi.org/10.3390/biom13060967
Chicago/Turabian StyleMerighi, Stefania, Alessia Travagli, Manuela Nigro, Silvia Pasquini, Martina Cappello, Chiara Contri, Katia Varani, Fabrizio Vincenzi, Pier Andrea Borea, and Stefania Gessi. 2023. "Caffeine for Prevention of Alzheimer’s Disease: Is the A2A Adenosine Receptor Its Target?" Biomolecules 13, no. 6: 967. https://doi.org/10.3390/biom13060967