
(Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments

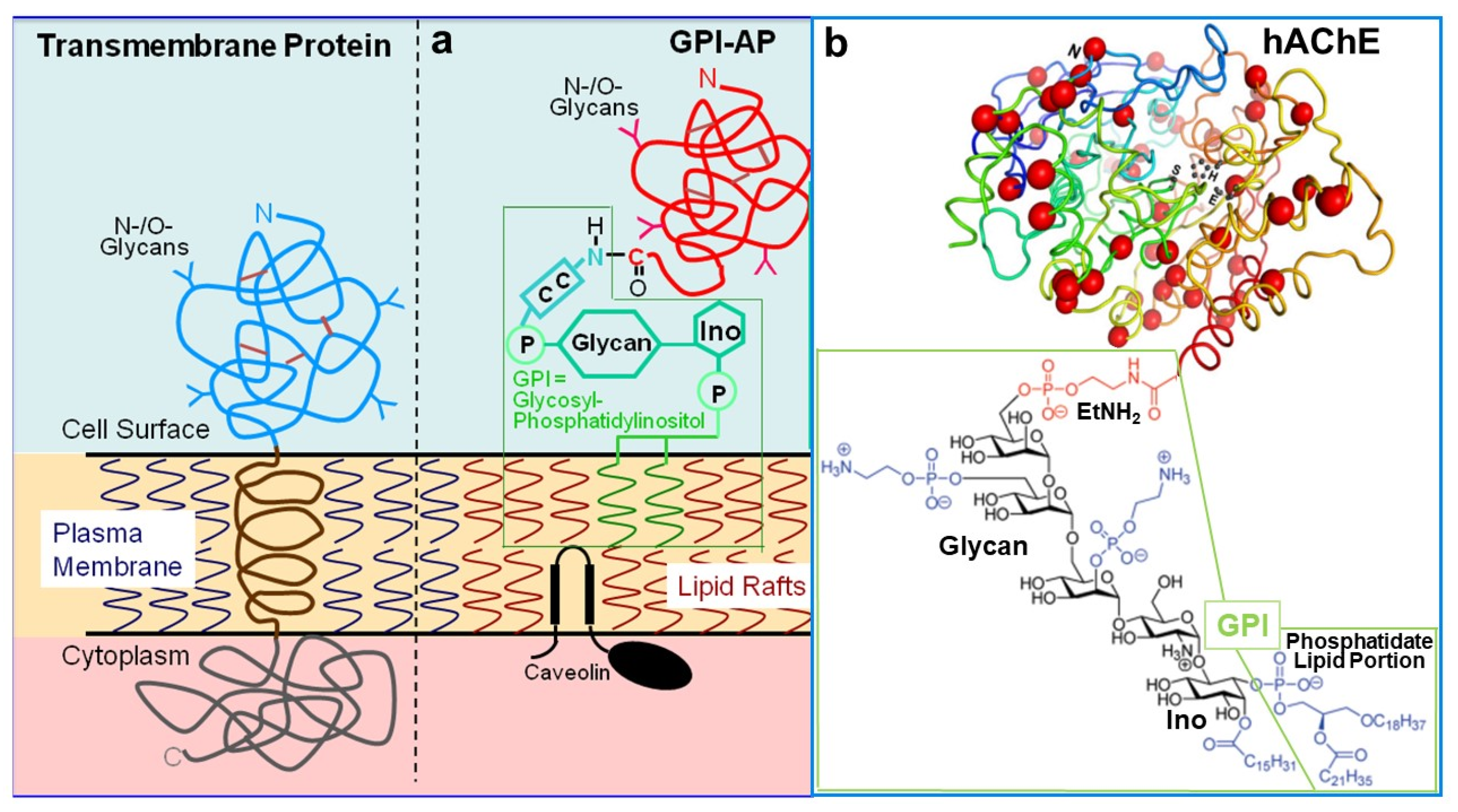{kind=link}
{kind=link}
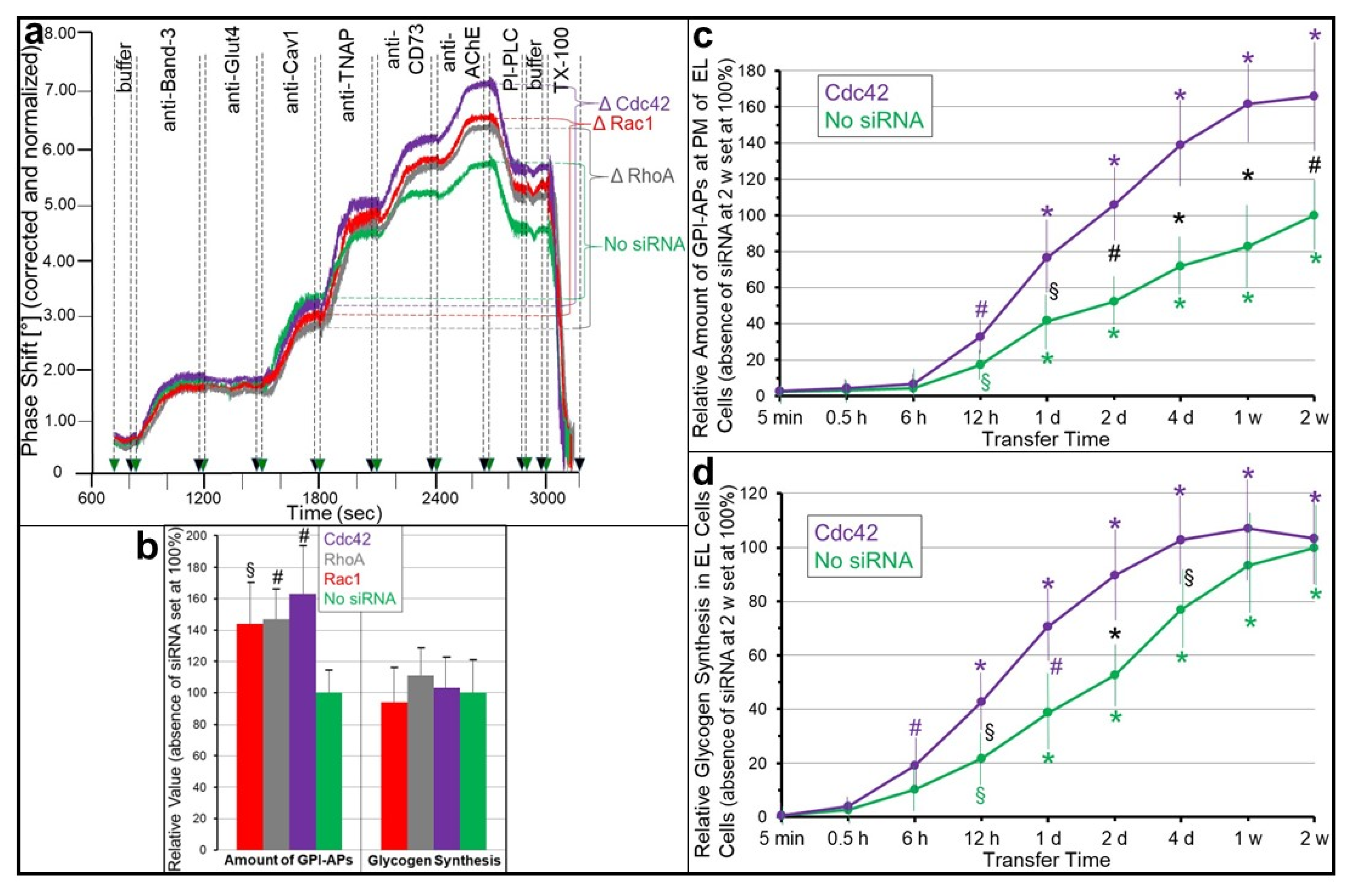{kind=link}
{kind=link}
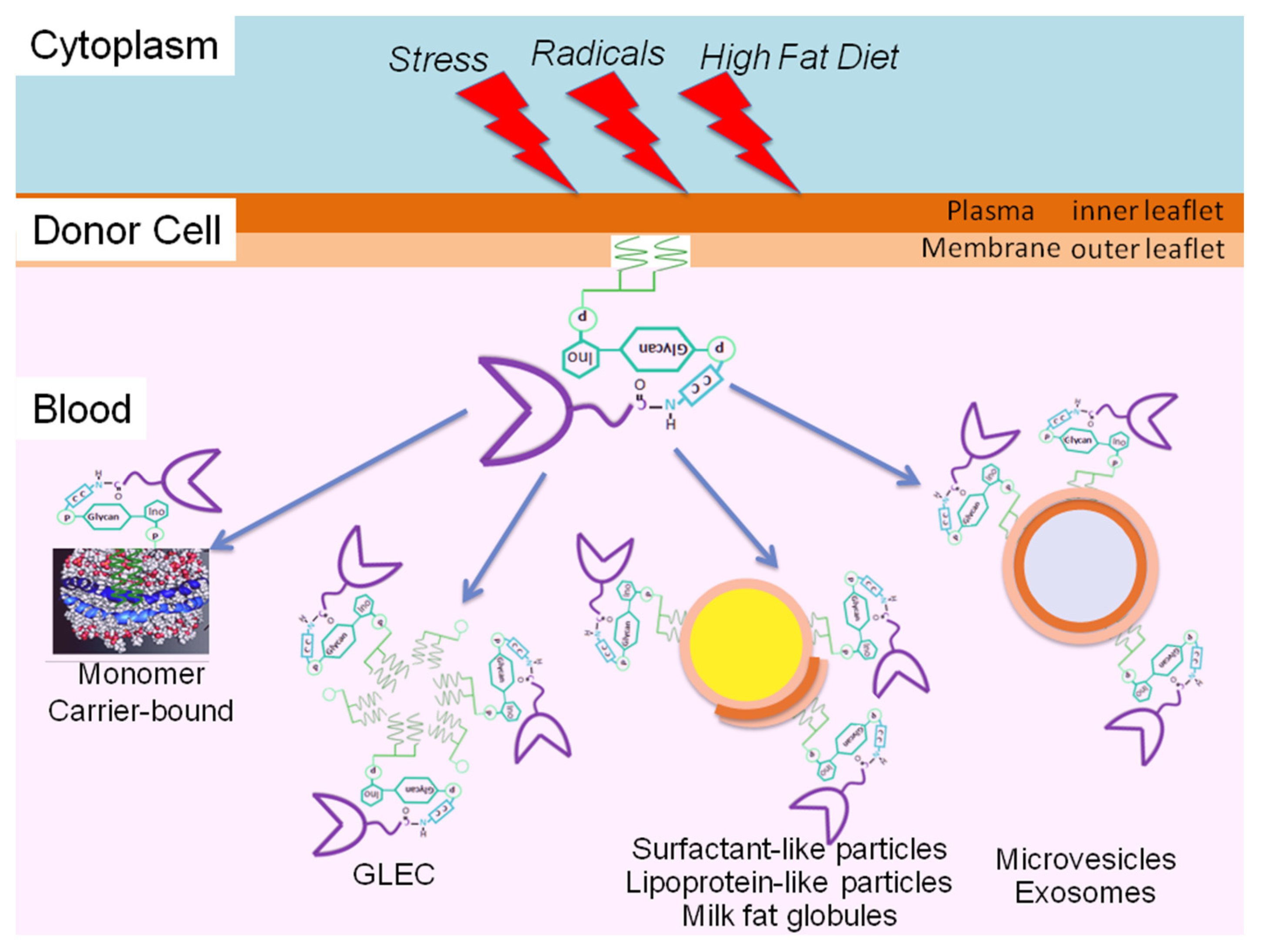{kind=link}
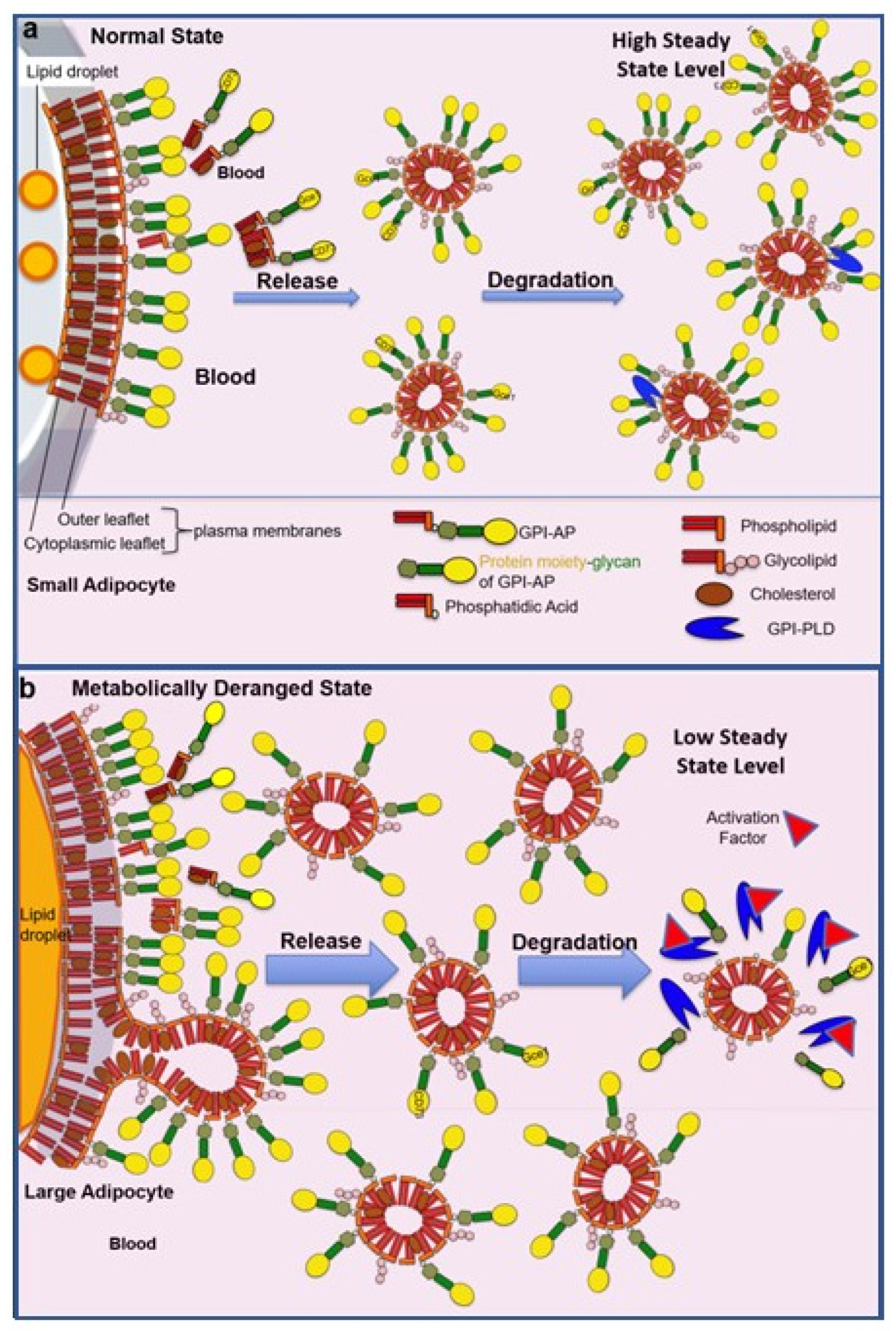{kind=link}
Abstract
:1. Introduction
2. Biogenesis and Expression at PMs of GPI-APs
3. Function, Clustering and Endocytosis of GPI-APs
4. Release of GPI-APs
4.1. Cleavage of the GPI Anchor
4.1.1. Transglucosylation of the Glycan Core
4.1.2. Proteolytic Cleavage
4.1.3. Lipolytic Cleavage
4.1.4. Physiological Roles of Proteolytic and Lipolytic Release of GPI-APs
4.2. Release of Full-Length GPI-APs
4.2.1. Release into Exosomes
4.2.2. Release into Microvesicles
4.2.3. Release into Non-Vesicular Structures and Assemblies
Release into LLPs
Release as Protein Oligomers, Multimers and Aggregates
Release for GPI-Binding Proteins
Release into Micelle-Like Complexes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eisenhaber, B.; Bork, P.; Eisenhaber, F. Prediction of potential GPI-modification sites in proprotein sequences. J. Mol. Biol. 1999, 292, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, B.; Bork, P.; Eisenhaber, B. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: Analysis of protein sequence data from complete genomes. Protein Eng. 2001, 14, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Poisson, G.; Chauve, C.; Chen, X.; Bergeron, A. FragAnchor: A large-scale predictor of glycosylphosphatidylinositol anchors in eukaryote protein sequences by qualitative scoring. Genom. Proteom. Bioinform. 2007, 5, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.J.; Haldar, K.; Cross, G.A.M. Trypanosoma brucei variant surface glycoprotein has a sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. J. Biol. Chem. 1985, 260, 4963–4968. [Google Scholar] [CrossRef] [PubMed]
- Haldar, K.; Ferguson, M.A.J.; Cross, G.A.M. Acylation of a Plasmodium falciparum merozoite surface antigen via sn-1,2-diacyl glycerol. J. Biol. Chem. 1985, 260, 4969–4974. [Google Scholar] [CrossRef]
- Schneider, P.; Ferguson, M.A.J.; McConville, M.J.; Mehlert, A.; Homans, S.W.; Bordier, C. Structure of the glycosyl-phosphatidylinositol membrane anchor of the Leishmania major Promastigote surface protease. J. Biol. Chem. 1990, 265, 16955–16964. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Homans, S.W.; Dwek, R.A.; Rademacher, T.W. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science 1988, 239, 753–759. [Google Scholar] [CrossRef]
- Ishida, M.; Maki, Y.; Ninomiya, A.; Takada, Y.; Campeau, P.; Kinoshita, T.; Murakami, Y. Ethanolamine-phosphate on the second mannose is a preferential bridge for some GPI-anchored proteins. EMBO Rep. 2022, 23, e54352. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Williams, A.F. Cell-surface anchoring of proteins via glycosylphosphatidylinositol structure. Ann. Rev. Biochem. 1988, 57, 285–320. [Google Scholar] [CrossRef]
- Englund, P.T. The structure and biosynthesis of glycosyl phosphatidylinositol protein anchors. Ann. Rev. Biochem. 1993, 62, 121–138. [Google Scholar] [CrossRef]
- Orlean, P.; Menon, A.K. Thematic review series: Lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: How we learned to stop worrying and love glycophospholipids. J. Lipid Res. 2007, 48, 993–1011. [Google Scholar]
- Fujita, M.; Kinoshita, T. GPI-anchor remodeling: Potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta 2012, 1821, 1050–1058. [Google Scholar] [CrossRef]
- Nakano, M.; Sabido-Bozo, S.; Okazaki, K.; Aguilera-Romero, A.; Rodriguez-Gallardo, S.; Cortes-Gomez, A.; Lopez, S.; Ikeda, A.; Funato, K.; Muniz, M. Structural analysis of the GPI glycan. PLoS ONE 2021, 16, e0257435. [Google Scholar] [CrossRef]
- Aguilera-Romero, A.; Sabido-Bozo, S.; Lopez, S.; Cortes-Gomez, A.; Rodriguez-Gallardo, S.; Perez-Linero, A.M.; Riezman, I.; Riezman, H.; Funato, K.; Goder, V.; et al. Determination of the lipid composition of the GPI anchor. PLoS ONE 2021, 16, e0256184. [Google Scholar] [CrossRef]
- Goldenzweig, A.; Goldsmith, M.; Hill, S.E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; et al. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 2016, 63, 337–346. [Google Scholar] [CrossRef]
- Silman, I.; Sussman, J.L. Recent developments in structural studies on acetylcholinesterase. J. Neurochem. 2017, 142 (Suppl. S2), 19–25. [Google Scholar] [CrossRef]
- Deeg, M.A.; Humphrey, D.R.; Yang, S.H.; Ferguson, T.R.; Reinhold, V.N.; Rosenberry, T.L. Glycan components in the glycoinositol phospholipid anchor of human erythrocyte acetylcholinesterase. Novel fragments produced by trifluoroacetic acid. J. Biol. Chem. 1992, 267, 18573–18580. [Google Scholar] [CrossRef]
- Manzano-Lopez, J.; Perez-Linero, A.M.; Aguilera-Romero, A.; Martin, M.E.; Okano, T.; Silva, D.V.; Seeberger, P.H.; Riezman, H.; Funato, K.; Goder, V.; et al. COPII coat composition is actively regulated by luminal cargo maturation. Curr. Biol. 2015, 25, 152–162. [Google Scholar] [CrossRef]
- Vazquez, H.M.; Vionnet, C.; Roubaty, C.; Conzelmann, A. Cdc1 removes the ethanolamine phosphate of the first mannose of GPI anchors and thereby facilitates the integration of GPI proteins into yeast cell wall. Mol. Biol. Cell 2014, 25, 3375–3388. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. GPI-anchored proteins in health and disease. In Post-Translational Modifications in Health and Disease; Springer: New York, NY, USA, 2011; pp. 39–55. [Google Scholar]
- Debierre-Grockiego, F.; Niehus, S.; Coddeville, B.; Elass, E.; Poirier, F.; Weingart, R.; Schmidt, R.R.; Mazurier, J.; Guerardel, Y.; Schwarz, R.T. Binding of Toxoplasma gondii glycosylphosphatidylinositols to galectin-3 is required for their recognition by macrophages. J. Biol. Chem. 2010, 285, 32744–32750. [Google Scholar] [CrossRef]
- Magez, S.; Stijlemans, B.; Baral, T.; De Baetselier, P. VSG-GPI anchors of African trypanosomes: Their role in macrophage activation and induction of infection-associated immunopathology. Microb. Infect. 2002, 4, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Varon Silva, D.; Lipowsky, R.; Santer, M. The importance of side branches of glycosylphosphatidylinositol anchors: A molecular dynamics perspective. Glycobiology 2022, 32, 933–948. [Google Scholar] [CrossRef]
- Duncan, S.M.; Nagar, R.; Damerow, M.; Yashunsky, D.V.; Buzzi, B.; Nikolaev, A.V.; Ferguson, M.A.J. A Trypanosoma brucei ß3 glycosyltransferase superfamily gene encodes a ß1-6 GlcNAc-transferase mediating N-glycan and GPI anchor modification. J. Biol. Chem. 2021, 297, 101153. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020, 10, 190290. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-S.; Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 2020, 48, 1129–1138. [Google Scholar] [CrossRef]
- Liu, S.-S.; Liu, Y.-S.; Guo, X.-Y.; Murakami, Y.; Yang, G.; Gao, X.-D.; Kinoshita, T.; Fujita, M. A knockout cell library of GPI biosynthetic genes for functional studies of GPI-anchored proteins. Commun. Biol. 2021, 4, 777. [Google Scholar] [CrossRef]
- Zhang, H.; Su, J.; Li, B.; Gao, Y.; Liu, M.; He, L.; Xu, H.; Dong, Y.; Zhang, X.C.; Zhao, Y. Structure of human glycosylphosphatidylinositol transamidase. Nat. Struct. Mol. Biol. 2022, 29, 203–209. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Yamamoto-Hino, M.; Matsuyama, N.; Suzuki, E.; Goto, S. Subunits of the GPI transamidase complex localize to the endoplasmic reticulum and nuclear envelope in Drosophila. FEBS Lett. 2021, 595, 960–968. [Google Scholar] [CrossRef]
- Xu, Y.; Jia, G.; Li, T.; Zhou, Z.; Luo, Y.; Chao, Y.; Bao, J.; Su, Z.; Qu, Q.; Li, D. Molecular insights into biogenesis of glycosylphosphatidylinositol anchor proteins. Nat. Commun. 2022, 13, 2617. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef]
- Liu, S.-S.; Jin, F.; Liu, Y.-S.; Murakami, Y.; Sugita, Y.; Kato, T.; Gao, X.-D.; Kinoshita, T.; Hattori, M.; Fujita, M. Functional analysis of the GPI transamidase complex by screening for amino acid mutations in each subunit. Molecules 2021, 26, 5462. [Google Scholar] [CrossRef]
- Ness, T.J.; Gamage, D.G.; Ekanayaka, S.A.; Hendrickson, T.L. A soluble, minimalistic glycosylphosphatidylinositol transamidase (GPI-T) retains transamidation activity. Biochemistry 2022, 61, 1273–1285. [Google Scholar] [CrossRef]
- Hirata, T.; Yang, J.; Tomida, S.; Tokoro, Y.; Kinoshita, T.; Fujita, M.; Kizuka, Y. ER entry pathway and glycosylation of GPI-anchored proteins are determined by N-terminal signal sequence and C-terminal GPI-attachment sequence. J. Biol. Chem. 2022, 298, 102444. [Google Scholar] [CrossRef]
- Galian, C.; Björkholm, P.; Bulleid, N.; von Heijne, G. Efficient glycosylphosphatidylinositol (GPI) modification of membrane proteins requires a C-terminal anchoring signal of marginal hydrophobicity. J. Biol. Chem. 2012, 287, 16399–16409. [Google Scholar] [CrossRef]
- Fankhauser, N.; Mäser, P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics 2005, 21, 1846–1852. [Google Scholar] [CrossRef]
- Lemus, L.; Matic, Z.; Gal, L.; Fadel, A.; Schuldiner, M.; Goder, V. Post-ER degradation of misfolded GPI-anchored proteins is linked with microautophagy. Curr. Biol. 2021, 31, 4025–4037. [Google Scholar] [CrossRef]
- Sikorska, N.; Lemus, L.; Aguilera-Romero, A.; Manzano-Lopez, J.; Riezman, H.; Muniz, M.; Goder, V. Limited ER quality control for GPI-anchored proteins. J. Cell Biol. 2016, 213, 693–704. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Wang, Y.; Zhou, X.; Zhang, L.; Yang, G.; Gao, X.-D.; Murakami, Y.; Fujita, M.; Kinoshita, T. Accumulated precursors of specific GPI-anchored proteins upregulate GPI biosynthesis with ARV1. J. Cell Biol. 2023, 222, e202208159. [Google Scholar] [CrossRef]
- Satpute-Krishnan, P.; Ajinkya, M.; Bhat, S.; Itakura, E.; Hegde, R.S.; Lippincott-Schwartz, J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 2014, 158, 522–533. [Google Scholar] [CrossRef]
- Penalva, M.A.; Moscoso-Romero, E.; Hernandez-Gonzales, M. Tracking exocytosis of a GPI-anchored protein in Aspergillus nidulans. Traffic 2020, 21, 675–688. [Google Scholar] [CrossRef]
- Ast, T.; Cohen, G.; Schuldiner, M. A network of cytosolic factors targets SRP-independent proteins to the endoplasmic reticulum. Cell 2013, 152, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Buscaglia, C.A.; Campo, V.A.; Di Noia, J.M.; Torrecilhas, A.C.; De Marchi, C.R.; Ferguson, M.A.; Frasch, A.C.; Almeida, I.C. The surface coat of the mammal-dwelling infective trypomastigote stage of Trypanosoma cruzi is formed by highly diverse immunogenic mucins. J. Biol. Chem. 2004, 279, 15860–15869. [Google Scholar] [CrossRef] [PubMed]
- Macrae, J.L.; Acosta-Serrano, A.; Morrice, N.A.; Mehlert, A.; Ferguson, M.A. Structural characterization of NETNES, a novel glycoconjugate in Trypanosoma cruzi epimastigotes. J. Biol. Chem. 2005, 280, 12201–12211. [Google Scholar] [CrossRef] [PubMed]
- Omaetxebarria, M.J.; Hagglund, P.; Elortza, F.; Hooper, N.M.; Arizmendi, J.M.; Jensen, O.N. Isolation and characterization of glycosylphosphatidylinositol-anchored peptides by hydrophilic interaction chromatography and MALDI tandem mass spectrometry. Anal. Chem. 2006, 78, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Nakayasu, E.S.; Yashunsky, D.V.; Nohara, L.L.; Torrecilhas, A.C.; Nikolaev, A.V.; Almeida, I.C. GPIomics: Global analysis of glycosylphosphatidylinositol-anchored molecules of Trypanosoma cruzi. Mol. Syst. Biol. 2009, 5, 261. [Google Scholar] [CrossRef]
- Yokoyama, M.; Yagyu, H.; Hu, Y.; Seo, T.; Hirata, K.; Homma, S.; Goldberg, I.J. Apolipoprotein B production reduces lipotoxic cardiomyopathy: Studies in heart-specific lipoprotein lipase transgenic mouse. J. Biol. Chem. 2004, 279, 4204–4211. [Google Scholar] [CrossRef]
- Gamage, D.G.; Hendrickson, T.L. GPI transamidase and GPI anchored proteins: Oncogenes and biomarkers for cancer. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 446–464. [Google Scholar] [CrossRef]
- Fujita, M.; Yoko-o, T.; Jigami, Y. Inositol deacylation by Bst1p is required for the quality control of glycosylphosphatidylinositol-anchored proteins. Mol. Biol. Cell 2006, 17, 834–850. [Google Scholar] [CrossRef]
- Bosson, R.; Jaquenoud, M.; Conzelman, A. GUP1 of Saccharomyces cerevisiae encodes an O-acetyltransferase involved in remodeling of the GPI anchor. Mol. Biol. Cell 2006, 17, 2636–2645. [Google Scholar] [CrossRef]
- Ghugtyal, V.; Vionnet, C.; Roubaty, C.; Conzelmann, A. CWH43 is required for the introduction of ceramides into GPI anchors in Saccharomyces cerevisiae. Mol. Microbiol. 2007, 65, 1493–1502. [Google Scholar] [CrossRef]
- Umemura, M.; Fujita, M.; Yoko-o, T.; Fukamizu, A.; Jigami, Y. Saccharomyces cerevisiae CWH43 is involved in the remodeling of the lipid moiety of GPI anchors to ceramides. Mol. Biol. Cell 2007, 18, 4304–4316. [Google Scholar] [CrossRef]
- Rodriguez-Gallardo, S.; Kurokawa, K.; Sabido-Bozo, S.; Cortes-Gomez, A.; Ikeda, A.; Zoni, V.; Aguilera-Romero, A.; Perez-Linero, A.M.; Lopez, S.; Waga, M.; et al. Ceramide chain length-dependent protein sorting into selective endoplasmic reticulum exit sites. Sci. Adv. 2020, 6, eaba8237. [Google Scholar] [CrossRef]
- Rodriguez-Gallardo, S.; Sabido-Bozo, S.; Ikeda, A.; Araki, M.; Okazaki, K.; Nakano, M.; Aguilera-Romero, A.; Cortes-Gomez, A.; Lopez, S.; Waga, M.; et al. Quality-controlled ceramide-based GPI-anchored protein sorting into selective ER exit sites. Cell Rep. 2022, 39, 110768. [Google Scholar] [CrossRef]
- Lopez, S.; Perez-Linero, A.M.; Manzano-Lopez, J.; Sabido-Bozo, S.; Cortes-Gomez, A.; Rodriguez-Gallardo, S.; Aguilera-Romero, A.; Goder, V.; Muniz, M. Dual independent roles of the p24 complex in selectivity of secretory cargo export from the endoplasmic reticulum. Cells 2020, 9, 1295. [Google Scholar] [CrossRef]
- Fujita, M.; Watanabe, R.; Jaensch, N.; Romanova-Michaelides, M.; Satoh, T.; Kato, M.; Riezman, H.; Yamaguchi, Y.; Maeda, Y.; Kinoshita, T. Sorting of GPI-anchored proteins into ER exit sites by p24 proteins dependent on remodeled GPI. J. Cell Biol. 2011, 194, 61–75. [Google Scholar] [CrossRef]
- Lopez, S.; Rodriguez-Gallardo, S.; Sabido-Bozo, S.; Muniz, M. Endoplasmic reticulum export of GPI-anchored proteins. Int. J. Mol. Sci. 2019, 20, 3506. [Google Scholar] [CrossRef]
- Fujita, M.; Maeda, Y.; Ra, M.; Yamaguchi, Y.; Taguchi, R.; Kinoshita, T. GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell 2009, 139, 352–365. [Google Scholar] [CrossRef]
- Fujita, M.; Umemura, M.; Yoko-o, T.; Jigami, Y. PER1 is required for GPI-phospholipase A2 activity and involved in lipid remodeling of GPI-anchored proteins. Mol. Biol. Cell 2006, 17, 5253–5264. [Google Scholar] [CrossRef]
- Tashima, Y.; Taguchi, R.; Murata, C.; Ashida, H.; Kinoshita, T.; Maeda, Y. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell 2006, 17, 1410–1420. [Google Scholar] [CrossRef]
- Muniz, M.; Riezman, H. Trafficking of glycosylphosphatidylinositol anchored proteins from the endoplasmic reticulum to the cell surface. J. Lipid Res. 2016, 57, 352–360. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Caras, L.W.; Davitz, M.A.; Rodriguez-Boulan, E. A glycophospholipid membrane anchor acts as an apical targeting signal in polarized epithelial cells. J. Cell Biol. 1989, 109, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef] [PubMed]
- de Hoop, M.J.; Dotti, C.G. Membrane traffic in polarized neurons in culture. J. Cell Sci. Suppl. 1993, 17, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Paladino, S.; Pocard, T.; Catino, M.A.; Zurzolo, C. GPI-anchored proteins are directly targeted to the apical surface in fully polarized MDCK cells. J. Cell Biol. 2006, 172, 1023–1034. [Google Scholar] [CrossRef]
- Paladino, S.; Lebreton, S.; Zurzolo, C. Trafficking and membrane organization of GPI-anchored proteins in health and diseases. Curr. Top. Membr. 2015, 75, 269–303. [Google Scholar]
- Maeda, Y.; Tashima, Y.; Houjou, T.; Fujita, M.; Yoko-o, T.; Jigami, Y.; Taguchi, R.; Kinoshita, T. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell 2007, 18, 1497–1506. [Google Scholar] [CrossRef]
- Benting, J.H.; Rietveld, A.G.; Simons, K. N-glycans mediate the apical sorting of a GPI-anchored, rat-associated protein in Madin-Darby canine kidney cells. J. Cell Biol. 1999, 146, 313–320. [Google Scholar] [CrossRef]
- McGwire, G.B.; Becker, R.P.; Skidgel, R.A. Carboxypeptidase M, a glycosylphosphatidylinositol-anchored protein, is localized on both the apical and basolateral domains of polarized Madin-Darby canine kidney cells. J. Biol. Chem. 1999, 274, 31632–31640. [Google Scholar] [CrossRef]
- Sarnataro, D.; Paladino, S.; Campana, V.; Grassi, J.; Nitsch, L.; Zurzolo, C. PrPC is sorted to the basolateral membrane of epithelial cells independently of its association with rafts. Traffic 2002, 3, 810–821. [Google Scholar] [CrossRef]
- Lebreton, S.; Paladino, S.; Zurzolo, C. Clustering in the Golgi apparatus governs sorting and function of GPI-APs in polarized epithelial cells. FEBS Lett. 2019, 593, 2351–2365. [Google Scholar] [CrossRef]
- Zurzolo, C.; Simons, K. Glycosylphosphatidylinositol-anchored proteins: Membrane organization anf transport. Biochim. Biophys. Acta 2016, 1858, 632–639. [Google Scholar] [CrossRef]
- Dudeja, P.K.; Kode, A.; Alnounou, M.; Tyagi, S.; Torania, S.; Subramanian, V.S.; Said, H.M. Mechanism of folate transport across the human colonic basolateral membrane. Am. J. Physiol. Liver Physiol. 2001, 281, G54–G60. [Google Scholar] [CrossRef]
- Bridges, C.C.; El-Sherbeny, A.; Ola, M.S.; Ganapathy, V.; Smith, S.B. Transcellular transfer of folate across the retinal pigment epithelium. Curr. Eye Res. 2002, 24, 129–138. [Google Scholar] [CrossRef]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood-brain barrier. Mol. Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef]
- Müller, G. Oral protein therapy for the future? Transport of glycolipid-modified therapeutic proteins. Pharmacology 2010, 86, 92–116. [Google Scholar] [CrossRef]
- Leidlich, S.D.; Drapp, D.A.; Orlean, P. A conditionally lethal yeast mutant blocked at the first step in glycosyl phosphatidylinositol anchor synthesis. J. Biol. Chem. 1994, 269, 10193–10196. [Google Scholar] [CrossRef]
- Nagamune, K.; Nozaki, T.; Maeda, Y.; Ohishi, K.; Fukuma, T.; Hara, T.; Schwarz, R.T.; Sutterlin, C.; Brun, R.; Riezman, H.; et al. Critical roles of glycosylphosphatidylinositol for Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2000, 97, 10336–10341. [Google Scholar] [CrossRef]
- Lallanne, E.; Honys, D.; Johnson, A.; Borner, G.H.; Lilley, K.S.; Dupree, P.; Grossniklaus, U.; Twell, D. SETH1 and SETH2, two components of the glycosylphosphatidylinositol anchor biosynthetic pathway, are required for pollen germination and tube growth in Arabidopsis. Plant Cell 2004, 16, 229–240. [Google Scholar] [CrossRef]
- Gillmor, C.S.; Lukowitz, W.; Brininstool, G.; Sedbrook, J.C.; Hamann, T.; Poindexter, P.; Somerville, C. Glycosylphosphatidylinositol-anchored proteins are required for cell wall synthesis and morphogenesis in Arabidopsis. Plant Cell 2005, 17, 1128–1140. [Google Scholar] [CrossRef]
- Nozaki, M.; Ohishi, K.; Yamada, N.; Kinoshita, T.; Nagy, A.; Takeda, J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab. Investig. 1999, 79, 293–299. [Google Scholar]
- Hazenbos, W.L.; Clausen, B.E.; Takeda, J.; Kinoshita, T. GPI-anchor deficiency in myeloid cells causes impaired FcγR effector functions. Blood 2004, 104, 2825–2831. [Google Scholar] [CrossRef] [PubMed]
- Hazenbos, W.L.; Wu, P.; Eastham-Anderson, J.; Kinoshita, T.; Brown, E.J. Impaired FcεR1 stability, signaling and effector functions in murine mast cells lacking glycosylphosphatidylinositol-anchored proteins. Blood 2011, 118, 4377–4383. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Yamaguchi, R.; Ikawa, M.; Okabe, M.; Morii, E.; Maeda, Y.; Kinoshita, T. PGAP1 knock-out mice show otocephaly and male infertility. J. Biol. Chem. 2007, 282, 30373–30380. [Google Scholar] [CrossRef] [PubMed]
- Tarutani, M.; Itami, S.; Okabe, M.; Orlean, P.; Taron, C.H. Tissue specific knock-out of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc. Natl. Acad. Sci. USA 1997, 94, 7400–7405. [Google Scholar] [CrossRef]
- Hirata, T.; Kobayashi, A.; Furuse, T.; Yamada, I.; Tamura, M.; Tomita, H.; Tokoro, Y.; Ninomiya, A.; Fujihara, Y.; Ikawa, M.; et al. Loss of the N-acetylgalactosamine side chain of the GPI-anchor impairs bone formation and brain functions and accelerates the prion disease pathology. J. Biol. Chem. 2022, 298, 101720. [Google Scholar] [CrossRef]
- Kawagoe, K.; Kitamura, D.; Okabe, M.; Taniuchi, I.; Ikawa, M.; Watanabe, T.; Kinoshita, T.; Takeda, J. Glycosylphosphatidylinositol-anchor-deficient mice: Implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood 1996, 87, 3600–3606. [Google Scholar] [CrossRef]
- Bessler, M.; Schaefer, A.; Keller, P. Paroxysmal nocturnal hemoglobinuria: Insights from recent advances in molecular biology. Transfus. Med. Rev. 2001, 15, 255–267. [Google Scholar] [CrossRef]
- Bessler, M.; Rosti, V.; Peng, Y.; Cattoretti, G.; Notaro, R.; Ohsako, S.; Elkon, K.B.; Luzzatto, L. Glycosylphosphatidylinositol-linked proteins are required for maintenance of a normal peripheral lymphoid compartment but not for lymphocyte development. Eur. J. Immunol. 2002, 32, 2607–2616. [Google Scholar] [CrossRef]
- Almeida, A.; Murakami, Y.; Layton, D.M.; Hillmen, P.; Sellick, G.S.; Maeda, Y.; Richards, S.; Patterson, S.; Kotsianidis, I.; Mollica, L.; et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006, 12, 846–851. [Google Scholar] [CrossRef]
- Krawitz, P.M.; Schweiger, M.R.; Rodelsperger, C.; Marcelis, C.; Kölsch, U.; Meisel, C.; Stephani, F.; Kinoshita, T.; Murakami, Y.; Bauer, S.; et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010, 42, 827–829. [Google Scholar] [CrossRef]
- Maydan, G.; Noyman, I.; Har-Zahav, A.; Neriah, Z.B.; Pasmanik-Chor, M.; Yeheskel, A.; Albin-Kaplanski, A.; Maya, I.; Magal, N.; Birk, E.; et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011, 48, 383–389. [Google Scholar] [CrossRef]
- Kim, P.; Scott, M.R.; Meador-Woodruff, J.H. Abnormal ER quality control of neural GPI-anchored proteins via dysfunction in ER export processing in the frontal cortex of elderly subjects with schizophrenia. Transl. Psychiatry 2019, 9, 6. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Mahida, S.; Smith-Hicks, C.; Campeau, P.M. A PIGH mutation leading to GPI deficiency is associated with developmental delay and autism. Hum. Mutat. 2018, 39, 827–829. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Murakami, Y.; Wigby, K.M.; Baratang, N.V.; Rousseau, J.; St-Denis, A.; Rosenberg, J.A.; Laniewski, S.C.; Jones, J.; Iglesias, A.D.; et al. Mutations in PIGS, encoding a GPI transamidase, cause a neurological syndrome ranging from fetal akinesia to epileptic encephalopathy. Am. J. Hum. Genet. 2018, 103, 602–611. [Google Scholar] [CrossRef]
- Segel, R.; Aran, A.; Gulsuner, S.; Nakamura, H.; Rosen, T.; Walsh, T.; Denda, H.; Zeligson, S.; Eto, K.; Beeri, R.; et al. A defect in GPI synthesis as a suggested mechanism for the role of ARV1 in intellectual disability and seizures. Case Rep. Neurogenet. 2020, 21, 259–267. [Google Scholar] [CrossRef]
- Carmody, L.C.; Blau, H.; Danis, D.; Zhang, X.A.; Gourdine, J.-P.; Vasilevsky, N.; Krawitz, P.; Thompson, M.D.; Robinson, P.N. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase + remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J. Rare Dis. 2020, 15, 40. [Google Scholar] [CrossRef]
- Duval, R.; Nicolas, G.; Willemetz, A.; Murakami, Y.; Mikdar, M.; Vrignaud, C.; Megahed, H.; Cartron, J.-P.; Masson, C.; Wehbi, S.; et al. Inherited glycosylphosphatidylinositol defects cause the rare EMM-negative blood phenotype and developmental disorders. Blood 2021, 137, 3660–3669. [Google Scholar] [CrossRef]
- Marshall, K.E.; Hughson, A.; Vascellari, S.; Priola, S.A.; Sakudo, A.; Onodera, T.; Baron, G.S. PrP knockout cells expressing transmembrane PrP resist prion infection. J. Virol. 2017, 91, e01686-16. [Google Scholar] [CrossRef]
- Kovac, V.; Hafner-Bratkovic, I.; Curin Serbec, V. Anchorless forms of prion protein—Impact of truncation on structure destabilization and prion protein conversion. Biochem. Biophys. Res. Commun. 2016, 481, 1–6. [Google Scholar] [CrossRef]
- Kinoshita, T.; Maeda, Y.; Fujita, M. Transport of glycosylphosphatidylinositol-anchored proteins from the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2473–2478. [Google Scholar] [CrossRef]
- Lebreton, S.; Zurzolo, C.; Paladino, S. Organization of GPI-anchored proteins at the cell surface and its physiopathological relevance. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Bellai-Dussault, K.; Nguyen, T.T.M.; Baratang, N.V.; Jimenez-Cruz, D.A.; Campeau, P.M. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 2019, 95, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M. Nodal signaling: Developmental roles and regulation. Development 2007, 134, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Salomon, D.S. Intercellular transfer regulation of the paracrine activity of GPI-anchored Cripto-1 as a Nodal co-receptor. Biochem. Biophys. Res. Commun. 2010, 403, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef]
- Brodsky, R.A.; Jane, S.M.; Vanin, E.F.; Mitsuya, H.; Peters, T.R.; Shimida, T.; Medof, M.E.; Nienhuis, A.W. Purified GPI-anchored CD4DAF as a receptor for HIV-mediated gene transfer. Hum. Gene Ther. 1994, 5, 1231–1239. [Google Scholar] [CrossRef]
- Bouwens, E.A.; Stavenuiter, F.; Mosnier, L.O. Cell painting with an engineered EPCR to augment the protein C system. Thromb. Haemost. 2015, 114, 1144–1155. [Google Scholar] [CrossRef]
- Morris, R.J. Thy-1, a pathfinder protein for the post-genomic era. Front. Cell Dev. Biol. 2018, 6, 173. [Google Scholar] [CrossRef]
- Brasitus, T.A.; Schachter, D. Lipid dynamics and lipid-protein interactions in rat enterocyte basolateral and microvillus membranes. Biochemistry 1980, 19, 2763–2769. [Google Scholar] [CrossRef]
- Sharom, F.J.; Lorimer, I.; Lamb, M.P. Reconstitution of lymphocyte 5′-nucleotidase in lipid bilayers: Behaviour and interaction with concanavalin A. Can. J. Biochem. Cell Biol. 1985, 63, 1049–1057. [Google Scholar] [CrossRef]
- Lehto, M.T.; Sharom, F.J. PI-specific phospholipase C cleavage of a reconstituted GPI-anchored protein: Modulation by the lipid bilayer. Biochemistry 2002, 41, 1398–1408. [Google Scholar] [CrossRef]
- Barboni, E.; Rivero, B.P.; George, A.J.; Martin, S.R.; Renoup, D.V.; Hounsell, E.F.; Barber, P.C.; Morris, R.J. The glycophosphatidylinositol anchor affects the conformation of Thy-1 protein. J. Cell Sci. 1995, 108, 487–497. [Google Scholar] [CrossRef]
- Rademacher, T.W.; Edge, C.J.; Dwek, R.A. Dropping anchor with the lipophosphoglycans. Curr. Biol. 1991, 1, 41–42. [Google Scholar] [CrossRef]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar] [CrossRef]
- Goswami, D.; Gowrishankar, K.; Bilgrami, S.; Ghosh, S.; Raghupathy, R.; Chadda, R.; Vishwakarma, R.; Rao, M.; Mayor, S. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell 2008, 135, 1085–1097. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell. Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef]
- Sengupta, P.; Jovanovic-Talisman, T.; Skoko, D.; Renz, M.; Veatch, S.L.; Lippincott-Schwartz, J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 2011, 8, 969–975. [Google Scholar] [CrossRef]
- Kusumi, A.; Shirai, Y.M.; Koyama-Honda, I.; Suzuki, K.G.; Fujiwara, T.K. Hierarchical organization of the plasma membrane: Investigations by single-molecule tracking vs. fluorescence correlation spectroscopy. FEBS Lett. 2010, 584, 1814–1823. [Google Scholar] [CrossRef]
- Giocondi, M.-C.; Seantier, B.; Dosset, P.; Milhiet, P.-E.; Le Grimellec, C. Characterizing the interactions between GPI-anchored alkaline phosphatases and membrane domains by AFM. Pflügers Arch. Eur. J. Physiol. 2008, 456, 179–188. [Google Scholar] [CrossRef]
- Busija, A.R.; Patel, H.H.; Insel, P.A. Caveolins and cavins in the trafficking, maturation, and degradation of caveolae: Implications for cell physiology. Am. J. Physiol. Cell Physiol. 2017, 312, C459–C477. [Google Scholar] [CrossRef]
- Lamaze, C.; Tardif, N.; Dewulf, M.; Vassilopoulos, S.; Blouin, C.M. The caveolae dress code: Structure and signaling. Curr. Opin. Cell Biol. 2017, 47, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Raghupathy, R.; Anilkumar, A.A.; Polley, A.; Singh, P.P.; Yadav, M.; Johnason, C.; Suryawanshi, S.; Saikam, V.; Sawant, S.D.; Panda, A.; et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell 2015, 161, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Seycsik, E.; Brameshuber, M.; Fölser, M.; Weghuber, J.; Honigmann, A.; Schütz, G. GPI-anchored proteins do not reside in ordered domains in the live cell plasma membrane. Nat. Commun. 2015, 6, 6969. [Google Scholar] [CrossRef]
- Saha, S.; Anilkumar, A.A.; Mayor, S. GPI-anchored protein organization and dynamics at the cell surface. J. Lipid Res. 2016, 57, 159–175. [Google Scholar] [CrossRef]
- Sezgin, E.; Davis, S.J.; Eggeling, C. Membrane Nanoclusters—Tails of the unexpected. Cell 2015, 161, 433–434. [Google Scholar] [CrossRef]
- Sezgin, E.; Kaiser, H.J.; Baumgart, T.; Schwille, P.; Simons, K.; Levental, I. Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat. Protoc. 2012, 7, 1042–1051. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Balatskaya, M.N.; Tkachuk, V.A. Glycosylphosphatidylinositol-anchored proteins as regulators of cortical cytoskeleton. Biochemistry 2016, 81, 636–650. [Google Scholar] [CrossRef]
- Kalappurakkal, J.M.; Sil, P.; Mayor, S. Toward a new picture of the living plasma membrane. Protein Sci. 2020, 29, 1355–1365. [Google Scholar] [CrossRef]
- Saltukoglu, D.; Özdemir, B.; Holtmannspötter, M.; Reski, R.; Piehler, J.; Kurre, R.; Reth, M. Plasma membrane topography governs the 3D dynamic localization of IgM B cell antigen receptor clusters. EMBO J. 2023, 42, e112030. [Google Scholar] [CrossRef]
- Borges, A.R.; Link, F.; Engstler, M.; Jones, N.G. The glycosylphosphatidyl anchor: A linchpin for cell surface versatility of Trypanosomatids. Front. Cell Dev. Biol. 2021, 9, 720536. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Sabharanjak, S.; De, A. Endocytosis of glycosylphosphatidylinositol-anchored proteins. J. Biomed. Sci. 2009, 16, 93. [Google Scholar] [CrossRef]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.-A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic crosstalk: Cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014, 12, e1001832. [Google Scholar] [CrossRef]
- Truong, A.; Yip, C.; Paye, A.; Blacher, S.; Munaut, C.; Deroanne, C.; Noel, A.; Sounni, N.E. Dynamics of internalization and recycling of the prometastatic membrane type 4 matrix metalloproteinase (MT4-MMP) in breast cancer cells. FEBS J. 2016, 283, 704–722. [Google Scholar] [CrossRef]
- Ferreira, A.P.A.; Boucrot, E. Mechanisms of carrier formation during clathrin-independent endocytosis. Trends Cell Biol. 2018, 28, 188–200. [Google Scholar] [CrossRef]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Mol. Biol. Cell 2004, 15, 3542–3552. [Google Scholar] [CrossRef]
- Song, S.; Fu, H.; He, B.; Wang, D.; Qin, M.; Yang, D.; Liu, D.; Song, G.; Shi, Y.; Zhang, H.; et al. Rho GTPases in A549 and Caco-2 cells dominating the endocytic pathways of nanocarbons with different morphologies. Int. J. Nanomed. 2018, 13, 4391–4404. [Google Scholar] [CrossRef]
- Chadda, R.; Howes, M.T.; Plowman, S.J.; Hancock, J.F.; Parton, R.G.; Mayor, S. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic 2007, 8, 702–717. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Bouquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef]
- Chen, H.; Gao, Z.; He, C.; Xiang, R.; van Kuppevelt, T.H.; Belting, M.; Zhang, S. GRP75 upregulates clathrin-independent endocytosis through actin cytoskeleton reorganization mediated by the concurrent activation of Cdc42 and RhoA. Exp. Cell Res. 2016, 343, 223–236. [Google Scholar] [CrossRef]
- Guha, A.; Sriram, V.; Krishnan, K.S.; Mayor, S. shibire mutations reveal distinct dynamin-independent and -dependent endocytic pathways in primary cultures of Drosophila hemocytes. J. Cell Sci. 2003, 116, 3373–3386. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Müller, T.D. Transfer of proteins from cultured human adipose to blood cells and induction of anabolic phenotype are controlled by serum, insulin and sulfonylurea drugs. Int. J. Mol. Sci. 2023, 24, 4825. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G. The glycosyl-phosphatidylinositol anchor of membrane proteins. Biochim. Biophys. Acta 1990, 988, 427–454. [Google Scholar] [CrossRef] [PubMed]
- Censullo, P.; Davitz, M.A. How GPI-anchored proteins turnover: Or where do they go after arrival at the plasma membrane. Semin. Immunol. 1994, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G.; Saltiel, A.R. Structural and functional roles of glycosylphosphatidylinositol in membranes. Science 1988, 239, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Paulik, M.G.; Bertozzi, C.R. The glycosylphosphatidylinositol anchor: A complex membrane-anchoring structure for proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef]
- Braun-Breton, C.; Rosenberry, T.L.; da Silva, L.P. Induction of the proteolytic activity of a membrane protein in Plasmodium falciparum by phosphatidyl inositol-specific phospholipase C. Nature 1988, 332, 457–459. [Google Scholar] [CrossRef]
- Gmachl, M.; Sagan, S.; Ketter, S.; Kreil, G. The human sperm protein PH-20 has hyaluronidase activity. FEBS Lett. 1993, 336, 545–548. [Google Scholar] [CrossRef]
- Brewis, L.A.; Turner, A.J.; Hooper, N.M. Activation of the glycosyl-phosphatidylinositol-anchored membrane dipeptidase upon release from pig kidney membranes by phospholipase C. Biochem. J. 1994, 303, 633–638. [Google Scholar] [CrossRef]
- Lehto, M.T.; Sharom, F.J. Release of the glycosylphosphatidylinositol-anchored enzyme ecto-5′-nucleotidase by phospholipase C: Catalytic activation and modulation by the lipid bilayer. Biochem. J. 1998, 332, 101–109. [Google Scholar] [CrossRef]
- Walter, E.I.; Ratnoff, W.D.; Long, K.E.; Kazura, J.W.; Medof, M.E. Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J. Biol. Chem. 1992, 267, 1245–1252. [Google Scholar] [CrossRef]
- Tozeren, A.; Sung, K.L.; Sung, L.A.; Dustin, M.L.; Chan, P.Y.; Springer, T.A.; Chien, S. Micromanipulation of adhesion of a Jurkat cell to a planar bilayer membrane containing lymphocyte function-associated antigen 3 molecules. J. Cell Biol. 1992, 116, 997–1006. [Google Scholar] [CrossRef]
- Müller, G.; Bandlow, W. Lipolytic membrane release of two phosphatidylinositol-anchored cAMP receptor proteins in yeast alters their ligand-binding parameters. Arch. Biochem. Biophys. 1994, 308, 504–514. [Google Scholar] [CrossRef]
- Wang, X.; Jansen, G.; Fan, J.; Kohler, W.J.; Ross, J.F.; Schornagel, J.; Ratnam, M. Variant GPI structure in relation to membrane-associated functions of a murine folate receptor. Biochemistry 1996, 35, 16305–16312. [Google Scholar] [CrossRef]
- Li, C.; Hancock, M.A.; Sehgal, P.; Zhou, S.; Reinhardt, D.P.; Philip, A. Soluble CD109 binds TGF-ß and antagonizes TGF-ß signalling and responses. Biochem. J. 2016, 473, 537–547. [Google Scholar] [CrossRef]
- Caro, L.H.; Tettelin, H.; Vossen, J.H.; Ram, A.F.; van den Ende, H.; Klis, F.M. In silico identification of glycosyl-phosphatidylinositol-anchored plasma-membrane and cell wall proteins of Saccharomyces cerevisiae. Yeast 1997, 13, 1477–1489. [Google Scholar] [CrossRef]
- Kapteyn, J.C.; Van Den Ende, H.; Klis, F.M. The contribution of cell wall proteins to the organisation of the yeast cell wall. Biochim. Biophys. Acta 1999, 1426, 373–383. [Google Scholar] [CrossRef]
- Klis, F.M.; Boorsma, A.; De Groot, P.W. Cell wall construction in Saccharomyces cerevisiae. Yeast 2006, 23, 185–202. [Google Scholar] [CrossRef]
- Fujii, T.; Shimoi, H.; Iimura, Y. Structure of the glucan-binding sugar chain of Tip1p, a cell wall protein of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1999, 1427, 133–144. [Google Scholar] [CrossRef]
- Popolo, L.; Vai, M. The Gas1 glycoprotein, a putative wall polymer cross-linker. Biochim. Biophys. Acta 1999, 1426, 385–400. [Google Scholar] [CrossRef]
- Müller, G.; Groß, E.; Wied, S.; Bandlow, W. Glucose-induced sequential processing of a glycosyl-phosphatidylinositol-anchored ectoprotein in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 442–456. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, J.M.; Caro, L.H.; Chapman, J.W.; Klis, F.M.; Verrips, C.T. Identification of three mannoproteins in the cell wall of Saccharomyces cerevisiae. J. Bacteriol. 1995, 177, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Vossen, J.H.; Muller, W.H.; Lipke, P.N.; Klis, F.M. Restrictive glycosylphosphatidylinositol anchor synthesis in cwh6/gpi3 yeast cells causes aberrant biogenesis of cell wall proteins. J. Bacteriol. 1997, 179, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Terashima, H.; Arisawa, M.; Kitada, K. Amino acid residues in the omega-minus region participate in cellular localization of yeast glycosylphosphatidylinositol-attached proteins. J. Bacteriol. 1999, 181, 3886–3889. [Google Scholar] [CrossRef] [PubMed]
- Yoko-o, T.; Umemura, M.; Komatsuzaki, A.; Ikeda, K.; Ichikawa, D.; Takase, K.; Kanzawa, N.; Saito, K.; Kinoshita, T.; Taguchi, R.; et al. Lipid moiety of glycosylphosphatidylinositol-anchored proteins contributes to the determination of their final destination in yeast. Genes Cells 2018, 23, 880–892. [Google Scholar] [CrossRef]
- Kitagaki, H.; Wu, H.; Shimoi, H.; Ito, K. Two homologous genes, DCW1 (YKL046c) and DFG5, are essential for cell growth and encode glycosylphosphatidylinositol (GPI)-anchored membrane proteins required for cell wall biogenesis in Saccharomyces cerevisiae. Mol. Microbiol. 2002, 46, 1011–1022. [Google Scholar] [CrossRef]
- de Sampaio, G.; Bourdineaud, J.P.; Lauquin, G.J. A constitutive role for GPI anchors in Saccharomyces cerevisiae: Cell wall targeting. Mol. Microbiol. 1999, 34, 247–256. [Google Scholar] [CrossRef]
- Yoko-o, T.; Ichikawa, D.; Miyagishi, Y.; Kato, A.; Umemura, M.; Takase, K.; Ra, M.; Ikeda, K.; Taguchi, R.; Jigami, Y. Determination and physiological roles of the glycosylphosphatidylinositol lipid remodelling pathway in yeast. Mol. Microbiol. 2013, 88, 140–155. [Google Scholar] [CrossRef]
- Greening, D.W.; Kapp, E.A.; Ji, H.; Speed, T.P.; Simpson, R.J. Colon tumour secretopeptidome: Insights into endogenous proteolytic cleavage events in the colon tumour microenvironment. Biochim. Biophys. Acta 2013, 1834, 2396–2407. [Google Scholar] [CrossRef]
- Mays, C.E.; Coomaraswamy, J.; Watts, J.C.; Yang, J.; Ko, K.W.S.; Strome, B.; Mercer, R.C.C.; Wohlgemuth, S.L.; Schmitt-Ulms, G.; Westaway, D. Endoproteolytic processing of the mammalian prion glycoprotein family. FEBS Lett. 2014, 281, 862–876. [Google Scholar] [CrossRef]
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. 2009, 14, 2494–2503. [Google Scholar] [CrossRef]
- Waldhauer, I.; Goehlsdorf, B.S.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef]
- Fernandez-Messina, L.; Ashiru, O.; Boutet, P.; Agüera-González, S.; Skepper, J.N.; Reyburn, H.T.; Valés-Gómez, M. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J. Biol. Chem. 2010, 285, 8543–8551. [Google Scholar] [CrossRef]
- Cavallone, D.; Malagolini, N.; Serafini-Cessi, F. Mechanism of release of urinary Tamm-Horsfall glycoprotein from the kidney GPI-anchored counterpart. Biochim. Biophys. Res. Commun. 2001, 280, 110–114. [Google Scholar] [CrossRef]
- Bülow, R.; Nonnengässer, C.; Overath, P. Release of the variant surface glycoprotein during differentiation of bloodstream to procyclic forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 1989, 32, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kalsi, K.; Lawson, C.; Dominquez, M.; Taylor, P.; Yacoub, M.H.; Smolenski, R.T. Regulation of ecto-5′-nucleotidase by TNF-alpha in human endothelial cells. Mol. Cell. Biochem. 2002, 232, 113–119. [Google Scholar] [CrossRef]
- Airas, L.; Niemela, J.; Salmi, M.; Puurunen, T.; Smith, D.J.; Jalkanen, S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J. Cell. Biol. 1997, 136, 421–431. [Google Scholar] [CrossRef]
- Pettengill, M.; Robson, S.; Tresenriter, M.; Millán, J.L.; Usheva, A.; Bingham, T.; Belderbos, M.; Bergelson, I.; Burl, S.; Kampmann, B.; et al. Soluble ecto-5′-nucleotidase (5′-NT), alkaline phosphatase, and adenosine deaminase (ADA1) activities in neonatal blood favor elevated extracellular adenosine. J. Biol. Chem. 2013, 288, 27315–27326. [Google Scholar] [CrossRef]
- van Veen, M.; Matas-Rico, E.; van de Wetering, K.; Leyton-Puig, D.; Kedziora, K.M.; De Lorenzi, V.; Stijf-Bultsma, Y.; van den Broek, B.; Jalink, K.; Sidenius, N.; et al. Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells. eLife 2017, 6pii, e23649. [Google Scholar] [CrossRef]
- Sack, T.L.; Gum, J.R.; Lowe, M.G.; Kim, Y.S. Release of carcinoembryonic antigen from human colon cancer cells by phosphatidylinositol-specific phospholipase C. J. Clin. Investig. 1988, 82, 586–593. [Google Scholar] [CrossRef]
- Park, S.W.; Choi, K.; Kim, I.C.; Lee, H.H.; Hooper, N.M.; Park, H.S. Endogenous glycosylphosphatidylinositol-specific phospholipase C releases renal dipeptidase from kidney proximal tubules in vitro. Biochem. J. 2001, 353, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Choi, K.; Lee, H.B.; Park, S.K.; Turner, A.J.; Hooper, N.M.; Park, H.S. Glycosyl-phosphatidylinositol (GPI)-anchored renal dipeptidase is released by a phospholipase C in vivo. Kidney Blood Press. Res. 2002, 25, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ermonval, M.; Baychelier, F.; Fonta, C. TNAP, an essential player in membrane lipid rafts of neuronal cells. In Neuronal Tissue-Nonspecific Alkaline Phosphatase (TNAP); Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2015; Volume 76, pp. 167–183. [Google Scholar] [PubMed]
- Ayala-Sarmiento, A.E.; Estudillo, E.; Perez-Sanchez, G.; Sierra-Sánchez, A.; González-Mariscal, L.; Martínez-Fong, D.; Segovia, J. GAS1 is present in the cerebrospinal fluid and is expressed in the choroid plexus of the adult rat. Histochem. Cell Biol. 2016, 146, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ingham, V.; Williams, A.; Bate, C. Glimepiride reduces CD14 expression and cytokine secretion from macrophages. J. Neuroinflamm. 2014, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI proteins: Sorting, membrane residence and functions. Biochim. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Stieger, A.; Cardoso de Almeida, M.L.; Blatter, M.C.; Brodbeck, U.; Bordier, C. The membrane-anchoring systems of vertebrate acetylcholinesterase and variant surface glycoproteins of African trypanosomes share a common antigenic determinant. FEBS Lett. 1986, 199, 182–186. [Google Scholar] [CrossRef]
- Roberts, W.L.; Myyher, J.J.; Kuksis, A.; Low, M.G.; Rosenberry, T.L. Lipid analysis of the glycoinositol phospholipid membrane anchor of human erythrocyte acetylcholinesterase. Palmitoylation of inositol results in resistance to phosphatidylinositol-specific phospholipase C. J. Biol. Chem. 1988, 263, 18766–18775. [Google Scholar] [CrossRef]
- Guther, M.L.; Cardoso de Almeida, M.L.; Rosenberry, T.L.; Ferguson, M.A. The detection of phospholipase-resistant and -sensitive glycosyl-phosphatidylinositol membrane anchors by western blotting. Anal. Biochem. 1994, 219, 249–255. [Google Scholar] [CrossRef]
- Hanada, K.; Izawa, K.; Nishijima, M.; Akamatsu, Y. Sphingolipid deficiency induces hypersensitivity of CD14, a glycosyl phosphatidylinositol-anchored protein, to phosphatidylinositol-specific phospholipase C. J. Biol. Chem. 1993, 268, 13820–13823. [Google Scholar] [CrossRef]
- Moon, Y.G.; Lee, H.J.; Kim, M.R.; Myung, P.K.; Park, S.Y.; Sok, D.E. Conversion of glycosylphosphatidylinositol (GPI)-anchored alkaline phosphatase by GPI-PLD. Arch. Pharm. Res. 1999, 22, 249–254. [Google Scholar] [CrossRef]
- Brunner, G.; Gabrilove, J.; Rifkin, D.B.; Wilson, E.L. Phospholipase C release of basic fibroblast growth factor from human bone marrow cultures as a biologically active complex with a phosphatidylinositol-anchored heparan sulfate proteoglycan. J. Cell Biol. 1991, 114, 1275–1283. [Google Scholar] [CrossRef]
- Brunner, G.; Metz, C.N.; Nguyen, H.; Gabrilove, J.; Patel, S.R.; Davitz, M.A.; Rifkin, D.B.; Wilson, E.L. An endogenous glycosylphosphatidylinositol-specific phospholipase D releases basic fibroblast growth factor-heparan sulfate proteoglycan complexes from human bone marrow cultures. Blood 1994, 83, 2115–2125. [Google Scholar] [CrossRef]
- Müller, G.; Wetekam, E.-M.; Jung, C.; Bandlow, W. Membrane association of lipoprotein lipase and a cAMP-binding ectoprotein in rat adipocytes. Biochemistry 1994, 33, 12149–12159. [Google Scholar] [CrossRef]
- Staudt, E.; Ramasamy, P.; Plattner, H.; Simon, M. Differential subcellular distribution of four phospholipase C isoforms and secretion of GPI-PLC activity. Biochim. Biophys. Acta 2016, 1858, 3157–3168. [Google Scholar] [CrossRef]
- Müller, A.; Klöppel, C.; Smith-Valentine, M.; Van Houten, J.; Simon, M. Selective and programmed cleavage of GPI-anchored proteins from the surface membrane by phospholipase C. Biochim. Biophys. Acta 2012, 1818, 117–124. [Google Scholar] [CrossRef]
- Matas-Rico, E.; van Veen, M.; Leyton-Puig, D.; van den Berg, J.; Koster, J.; Kedziora, K.M.; Molenaar, B.; Weerts, M.J.A.; de Rink, I.; Medema, R.H.; et al. Glycerophosphodiesterase GDE2 promotes neuroblastome differentiation through glypican release and is a marker of clinical outcome. Cancer Cell 2016, 30, 548–562. [Google Scholar] [CrossRef]
- Matas-Rico, E.; van Veen, M.; Moolenaar, W.H. Neuronal differentiation through GPI-anchor cleavage. Cell Cycle 2016, 16, 388–389. [Google Scholar] [CrossRef]
- Chan, B.L.; Lisanti, M.P.; Rodriguez-Boulan, E.; Saltiel, A.R. Insulin-stimulated release of lipoprotein lipase by metabolism of its phosphatidylinositol anchor. Science 1988, 241, 1670–1672. [Google Scholar] [CrossRef]
- Romero, G.; Luttrell, L.; Rogol, A.; Zeller, K.; Hewlett, E.; Larner, J. Phosphatidylinositol-glycan anchors of membrane proteins: Potential precursors of insulin mediators. Science 1988, 240, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, S.; Hooper, N.M. Insulin stimulates the release of the glycosylphosphatidylinositol-anchored membrane dipeptidase from 3T3-L1 adipocytes through the action of a phospholipase C. Biochem. J. 1997, 326, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Dearey, E.A.; Pünter, J. The sulphonylurea drug, glimepiride, stimulates release of glycosylphosphatidylinositol-anchored plasma-membrane proteins from 3T3 adipocytes. Biochem. J. 1993, 289, 509–521. [Google Scholar] [CrossRef]
- Müller, G.; Dearey, E.A.; Korndörfer, A.; Bandlow, W. Stimulation of a glycosylphosphatidylinositol-specific phospholipase by insulin and the sulfonylurea, glimepiride, in rat adipocytes depends on increased glucose transport. J. Cell Biol. 1994, 126, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Schulz, A.; Wied, S.; Frick, W. Regulation of lipid raft proteins by glimepiride- and insulin-induced glycosylphosphatidylinositol-specific phospholipase C in rat adipocytes. Biochem. Pharmacol. 2005, 69, 761–780. [Google Scholar] [CrossRef]
- Müller, G.; Frick, W. Signalling via caveolin: Involvement in the cross-talk between phosphoinositolglycans and insulin. Cell. Mol. Life Sci. 1999, 56, 945–970. [Google Scholar] [CrossRef]
- Müller, G. Dynamics of plasma membrane microdomains and cross-talk to the insulin signalling cascade. FEBS Lett. 2002, 531, 81–87. [Google Scholar] [CrossRef]
- Kreuger, J.; Perez, L.; Giraldez, A.J.; Cohen, S.M. Opposing activities of Dally-like glypican at high and low levels of Wingless morphogen activity. Dev. Cell 2004, 7, 503–511. [Google Scholar] [CrossRef]
- Traister, A.; Shi, W.; Filmus, J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem. J. 2007, 410, 503–511. [Google Scholar] [CrossRef]
- Davitz, M.A.; Hereld, D.; Shak, S.; Krakow, J.; Englund, P.T.; Nussenzweig, V. A glycan-phosphatidylinositol-specific phospholipase D in human serum. Science 1987, 238, 81–84. [Google Scholar] [CrossRef]
- Low, M.G.; Prasad, A.R. A phospholipase D specific for the phosphatidylinositol anchor of cell-surface proteins is abundant in plasma. Proc. Natl. Acad. Sci. USA 1988, 85, 980–984. [Google Scholar] [CrossRef]
- Stieger, S.; Diem, S.; Jakob, A.; Brodbeck, U. Enzymatic properties of phosphatidylinositol-glycan-specific phospholipase C from rat liver and phosphatidylinositol-glycan-specific phospholipase D from rat serum. Eur. J. Biochem. 1991, 197, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Flores-Borja, F.; Kieszkievicz, J.; Church, V.; Francis-West, P.H.; Schofield, J.; Rademacher, T.W.; Lund, T. Genetic regulation of mouse glycosylphosphatidylinositol-phospholipase D. Biochimie 2004, 86, 275–282. [Google Scholar] [CrossRef]
- Hoener, M.C.; Brodbeck, U. Phosphatidylinositol-glycan-specific phospholipase D is an amphiphilic glycoprotein that in serum is associated with high density lipoproteins. Eur. J. Biochem. 1992, 206, 747–757. [Google Scholar] [CrossRef]
- Heller, M.; Bieri, S.; Brodbeck, U. A novel form of glycosylphosphatidylinositol-anchor converting activity with a specificity of a phospholipase D in mammalian liver membranes. Biochim. Biophys. Acta 1992, 1109, 109–116. [Google Scholar] [CrossRef]
- Low, M.G.; Huang, K.S. Factors affecting the ability of glycosylphosphatidylinositol-specific phospholipase D to degrade the membrane anchors of cell surface proteins. Biochem. J. 1991, 279, 483–493. [Google Scholar] [CrossRef]
- Mann, K.J.; Hepworth, M.R.; Raikwar, N.S.; Deeg, M.A.; Sevlever, D. Effect of glycosylphosphatidylinositol (GPI)-phospholipase D overexpression on GPI metabolism. Biochem. J. 2004, 378, 641–648. [Google Scholar] [CrossRef]
- Mukasa, R.; Umeda, M.; Endo, T.; Kobata, A.; Inoue, K. Characterization of glycosylphosphatidylinositol (GPI)-anchored NCAM on mouse skeletal muscle cell line C2C12: The structure of the GPI glycan and release during myogenesis. Arch. Biochem. Biophys. 1995, 318, 182–190. [Google Scholar] [CrossRef]
- Wilhelm, O.G.; Wilhelm, S.; Escott, G.M.; Lutz, V.; Magdolen, V.; Schmitt, M.; Rifkin, D.B.; Wilson, E.L.; Graeff, H.; Brunner, G. Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J. Cell Physiol. 1999, 180, 225–235. [Google Scholar] [CrossRef]
- Naghibalhossaini, F.; Ebadi, P. Evidence for CEA release from human colon cancer cells by an endogenous GPI-PLD enzyme. Cancer Lett. 2006, 28, 158–167. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 2020, 369, 167–173. [Google Scholar] [CrossRef]
- Lee, G.-H.; Fujita, M.; Nakanishi, H.; Miyata, H.; Ikawa, M.; Maeda, Y.; Murakami, Y.; Kinoshita, T. PGAP6. A GPI-specific phospholipase A2, has narrow substrate specificity against GPI-anchored proteins. J. Biol. Chem. 2020, 295, 14501–14509. [Google Scholar] [CrossRef]
- Clemente, R.; Jones, D.R.; Ochoa, P.; Romero, G.; Mato, J.M.; Varela-Nieto, I. Role of glycosyl-phosphatidylinositol hydrolysis as a mitogenic signal for epidermal growth factor. Cell. Signal. 1995, 7, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Eardley, D.D.; Koshland, M.E. Glycosylphosphatidylinositol: A candidate system for IL-2 signal transduction. Science 1991, 251, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Cozza, E.N.; Lima, C.; Ramirez, M.I.; De Lederkremer, R.M. An inositol phosphoglycan from Trypanosoma cruzi inhibits ACTH action in calf adrenocortical cells. Cell. Signal. 1995, 7, 331–339. [Google Scholar] [CrossRef]
- Jacquemin, C. Glycosyl phosphatidylinositol in thyroid: Cell signalling or protein anchor? Biochimie 1991, 73, 37–40. [Google Scholar] [CrossRef]
- Ohmichi, M.; Decker, S.J.; Saltiel, A.R. Nerve growth factor stimulates the tyrosine phosphorylation of a 38-kDa protein that specifically associates with the src homology domain of phospholipase C-gamma 1. J. Biol. Chem. 1992, 267, 21601–21606. [Google Scholar] [CrossRef]
- Kondoh, G.; Gao, X.H.; Nakano, Y.; Koike, H.; Yamada, S.; Okabe, M.; Takeda, J. Tissue-inherent fate of GPI revealed by GPI-anchored GFP transgenesis. FEBS Lett. 1999, 458, 299–303. [Google Scholar] [CrossRef]
- Kondoh, G.; Tojo, H.; Nakatani, Y.; Komazawa, N.; Murata, C.; Yamagata, K.; Maeda, Y.; Kinoshita, T.; Okabe, M.; Taguchi, R.; et al. Angiotensin-converting enzyme is a GPI-anchored protein releasing factor crucial for fertilization. Nat. Med. 2005, 11, 160–166. [Google Scholar] [CrossRef]
- Leisle, L.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. Angiotensin-converting enzyme as a GPIase: A critical reevaluation. Nat. Med. 2005, 11, 11339–11340. [Google Scholar] [CrossRef]
- Fuchs, S.; Frenzel, K.; Hubert, C.; Lyng, R.; Muller, L.; Michaud, A.; Xiao, H.D.; Adams, J.W.; Capecchi, M.R.; Corvol, P.; et al. Male fertility is dependent on dipeptidase activity of testis ACE. Nat. Med. 2005, 11, 1140–1142. [Google Scholar] [CrossRef]
- Hagaman, J.R.; Moyer, J.S.; Bachman, E.S.; Sibony, M.; Magyar, P.L.; Welch, J.E.; Smithies, O.; Krege, J.H.; O’Brien, D.A. Angiotensin-converting enzyme and male fertility. Proc. Natl. Acad. Sci. USA 1998, 95, 2552–2557. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Fox, J.A.; Sherline, P.; Cuatrecasas, P. Insulin-stimulated hydrolysis of a novel glycolipid generates modulators of cAMP phosphodiesterase. Science 1986, 233, 967–972. [Google Scholar] [CrossRef]
- Thomas, J.R.; Dwek, R.A.; Rademacher, T.W. Structure, biosynthesis, and function of glycosylphosphatidylinositols. Biochemistry 1990, 29, 5413–5422. [Google Scholar] [CrossRef]
- Saltiel, A.R. Second messengers of insulin action. Diabetes Care 1990, 13, 244–256. [Google Scholar] [CrossRef]
- Romero, G.; Larner, J. Insulin mediators and the mechanism of insulin action. Adv. Pharmacol. 1993, 24, 21–50. [Google Scholar]
- Varela-Nieto, I.; León, Y.; Caro, H.N. Cell signalling by inositol phosphoglycans from different species. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1996, 115, 223–241. [Google Scholar] [CrossRef]
- Jones, D.R.; Varela-Nieto, I. Diabetes and the role of inositol-containing lipids in insulin signaling. Mol. Med. 1999, 5, 505–514. [Google Scholar] [CrossRef]
- Acosta, J.; Hettinga, J.; Fluckiger, R.; Krumrei, N.; Goldfine, A.; Angarita, L.; Halperin, J. Molecular basis for a link between complement and the vascular complications of diabetes. Proc. Natl. Acad. Sci. USA 2000, 97, 5450–5455. [Google Scholar] [CrossRef]
- Zhang, J.; Gerhardinger, C.; Lorenzi, M. Early complement activation and decreased levels of glycosylphosphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes 2002, 51, 3499–3504. [Google Scholar] [CrossRef]
- Davies, B.S.; Beigneux, A.P.; Barnes, R.H.; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyrén, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 is responsible for the entry of lipoproein lipase into capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef]
- Hakulinen, J.; Meri, S. Shedding and enrichment of the glycoplipid-anchored complement lysis inhibitor protectin (CD59) into milk fat globules. Immunology 1995, 85, 495–501. [Google Scholar] [PubMed]
- Cocuzzi, E.; Szczotka, L.B.; Brodbeck, W.G.; Bardenstein, D.S.; Wei, T.; Medof, M.E. Tears contain the complement regulator CD59 as well as decay-accelerating factor (DAF). Clin. Exp. Immunol. 2001, 123, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.P.G.; Wilson, A.B.; Davies, A.; Lachmann, P.J.; Ferriani, V.P.; Seilly, D.J.; Assis-Pandochi, A.I. Determination of CD59 protein in normal human serum by enzyme immunoassay, using octyl-glucoside detergent to release glycosyl-phosphatidyl-CD59 from lipid complex. Immunol. Lett. 2003, 90, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Cantel, S.; Kavishwar, A.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. A specific and sensitive assay for blood levels of glycated CD59: A novel biomarker for diabetes. Am. J. Hematol. 2013, 88, 670–676. [Google Scholar] [CrossRef]
- Fletcher, C.M.; Harrison, R.A.; Lachmann, P.J.; Neuhaus, D. Structure of a soluble, glycosylated form of the human complement regulatory protein CD59. Structure 1994, 2, 185–199. [Google Scholar] [CrossRef]
- De Cat, B.; David, G. Developmental roles of the glypicans. Semin. Cell Dev. Biol. 2001, 12, 117–125. [Google Scholar] [CrossRef]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- Fico, A.; Maina, F.; Dono, R. Fine-tuning of cell signaling by glypicans. Cell. Mol. Life Sci. 2011, 68, 923–929. [Google Scholar] [CrossRef]
- Ussar, S.; Bezy, O.; Blüher, M.; Kahn, C.R. Glypican-4 enhances insulin signaling via interaction with the insulin receptor and serves as a novel adipokine. Diabetes 2012, 61, 2289–2298. [Google Scholar] [CrossRef]
- Fujihara, Y.; Ikawa, M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016, 57, 538–545. [Google Scholar] [CrossRef]
- Puig, B.; Altmeppen, H.G.; Linsenmeier, L.; Chakroun, K.; Wegwitz, F.; Piontek, U.K.; Tatzelt, J.; Bate, C.; Magnus, T.; Glatzel, M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019, 15, e1007520. [Google Scholar] [CrossRef]
- Huang, K.; Park, S. Heparan sulfated glypican-4 is released from astrocytes by proteolytic shedding and GPI-anchor cleavage mechanisms. eNeuro 2021, 8, 0069-21.2021. [Google Scholar] [CrossRef]
- Trams, E.G.; Lauter, C.J.; Salem, N.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of microvesicles. Biochim. Biophys. Acta 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Mathew, A.; Mason, A.B.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalisation of obsolete membrane proteins. J. Cell. Pathol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Johnstone, R.M. Revisiting the road to the discovery of exosomes. Blood Cells Mol. Dis. 2003, 34, 214–219. [Google Scholar] [CrossRef]
- Rollins, S.A.; Zhao, J.; Ninomiya, H.; Sims, P.J. Inhibition of homologous complement by CD59 is mediated by a species-selective recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. J. Immunol. 1991, 146, 2345–2351. [Google Scholar] [CrossRef]
- Rabesandrata, H.; Toutant, J.P.; Reggio, H.; Vidal, M. Decay-Accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood 1998, 91, 2573–2580. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Watt, N.T.; Turner, A.J.; Hooper, N.M. Dual mechanisms for shedding of the cellular prion protein. J. Biol. Chem. 2004, 279, 11170–11178. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. Glycosylphosphatidylinositol-Anchored Proteins and Their Release from Cells—From Phenomenon to Meaning, 1st ed.; Nova Science Publishers—Biochemistry Research Trends: New York, NY, USA, 2018; pp. 104–115. [Google Scholar]
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.L. Platelet secretory mechanism. Semin. Thromb. Hemost. 2004, 30, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krähenbühl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef]
- Blanchard, P.J.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef]
- Karlsson, M.; Lundin, S.; Dahlgren, U.; Kahu, H.; Pettersson, I.; Telemo, E. ‘Tolerosomes’ are produced by intestinal epithelial cells. Eur. J. Immunol. 2001, 31, 2892–2900. [Google Scholar] [CrossRef]
- van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumours using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Clayton, A.; Court, J.; Navabi, H.; Adams, M.; Mason, M.D.; Hobot, J.A.; Newman, G.R.; Jasani, B. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J. Immunol. Methods 2001, 247, 163–174. [Google Scholar] [CrossRef]
- Wang, G.H.; Zhou, X.M.; Bai, Y.; Yin, X.M.; Yang, L.F.; Zhao, D. Hsp70 binds to PrPC in the process of PrPC release via exosomes from THP-1 monocytes. Cell Biol. Int. 2011, 35, 553–558. [Google Scholar] [CrossRef]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Fernandez-Messina, L.; Reyburn, H.T.; Vales-Gomez, M. Human NKG2D-ligands cell biology strategies to ensure immune recognition. Front. Immunol. 2012, 3, 299. [Google Scholar] [CrossRef]
- Ashiru, O.; Lopez-Cobo, S.; Fernandez-Messina, L.; Pontes-Quero, S.; Pandolfi, R.; Reyburn, H.T.; Valés-Gómez, M. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*0008. Biochem. J. 2013, 454, 295–302. [Google Scholar] [CrossRef]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013, 13, 8. [Google Scholar]
- Baragano-Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef]
- Takeda, Y.; Kurota, Y.; Kato, T.; Ito, H.; Araki, A.; Nara, H.; Saitoh, S.; Tanaka, N.; Tsuchiya, N.; Asao, H. GPI-80 augments NF-κB activation in tumor cells. Int. J. Mol. Sci. 2021, 22, 12027. [Google Scholar] [CrossRef]
- Nitto, T.; Araki, Y.; Takeda, Y.; Sendo, F. Pharmacological analysis for mechanisms of GPI-80 release from tumour necrosis-α-stimulated human neutrophils. Br. J. Pharmacol. 2002, 137, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Hale, G.; Rye, P.D.; Warford, A.; Lauder, I.; Brito-Babapulle, A. The glycosylphosphatidylinositol-anchored lymphocyte antigen CDw52 is associated with the epididymal maturation of human spermatozoa. J. Reprod. Immunol. 1993, 23, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Parra, A.; Padilla, L.; Lucas, X.; Rodriguez-Martinez, H.; Barranco, I.; Roca, J. Seminal extracellular vesicles and their involvement in male (in)fertility: A systematic review. Int. J. Mol. Sci. 2023, 24, 4818. [Google Scholar] [CrossRef] [PubMed]
- Malm, J.; Birn, H.; Frohm, B.; Hansen, S.-I.; Hoier-Madsen, M.; Holm, J. A minor fraction of a high-affinity folate binding protein from the epididymis is associated with membranous vesicles and spermatozoa in human semen. Int. J. Androl. 2005, 28, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Legare, C.; Villeneuve, M.; Foliguet, B.; Bissonnette, F. Levels of P34H, a sperm protein of epididymal origin, as a predictor of conventional in vitro fertilization outcome. Fertil. Steril. 2006, 85, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Saez, F.; Girouard, J.; Frenette, G. Role of exosomes in sperm maturation during the transit along the male reproductive tract. Blood Cells Mol. Dis. 2005, 35, 1–10. [Google Scholar] [CrossRef]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113, 3365–3374. [Google Scholar] [CrossRef]
- Vidal, M.; Mangeat, P.; Hoekstra, D. Aggregation reroutes molecules from a recycling to a vesicle-mediated secretion pathway during reticulocyte maturation. J. Cell Sci. 1997, 110, 1867–1877. [Google Scholar] [CrossRef]
- MacDonald, C.; Stamnes, M.A.; Katzmann, D.J.; Piper, R.C. Tetraspan cargo adaptors usher GPI-anchored proteins into multivesicular bodies. Cell Cycle 2015, 14, 3673–3678. [Google Scholar] [CrossRef]
- Edgar, J.R.; Manna, P.T.; Nishimura, S.; Banting, G.; Robinson, M.S. Tetherin is an exosomal tether. eLife 2016, 5, e17180. [Google Scholar] [CrossRef]
- Vidal, M. Exosomes and GPI-anchored proteins: Judicious pairs for investigating biomarkers from body fluids. Adv. Drug Deliv. Rev. 2020, 161–162, 110–123. [Google Scholar] [CrossRef]
- Müller, G. Microvesicles/exosomes as potential novel biomarkers of metabolic diseases. Diabetes Metab. Syndr. Obes. 2012, 5, 247–282. [Google Scholar] [CrossRef]
- Moldovan, N.I.; Radu, A.N.; Simionescu, N. Endothelial cell plasma membrane obtained by chemically induced vesiculation. Exp. Cell Res. 1987, 170, 499–510. [Google Scholar] [CrossRef]
- De Broe, M.E.; Wieme, R.J.; Logghe, G.N.; Roels, F. Spontaneous shedding of plasma membrane fragments by human cells in vivo and in vitro. Clin. Chim. Acta 1977, 81, 237–245. [Google Scholar] [CrossRef]
- Allan, D.; Thomas, P.; Limbrick, A.R. The isolation and characterization of 60 nm vesicles (‘nanovesicles’) produced during ionophore A23187-induced budding of human erythrocytes. Biochem. J. 1980, 188, 881–887. [Google Scholar] [CrossRef]
- Allan, D.; Thomas, P. Ca2+-induced biochemical changes in human erythrocytes and their relation to microvesiculation. Biochem. J. 1981, 198, 433–440. [Google Scholar] [CrossRef]
- Carr, J.M.; Dvorak, A.M.; Dvorak, H.F. Circulating membrane vesicles in leukemic blood. Cancer Res. 1984, 45, 5944–5951. [Google Scholar]
- Kobayashi, T.; Okamoto, H.; Yamada, J.; Setaka, M.; Kwan, T. Vesiculation of platelet plasma membranes. Dilauroylglycerophosphocholine-induced shedding of a platelet plasma membrane fraction enriched in acetylcholinesterase activity. Biochim. Biophys. Acta 1984, 778, 210–218. [Google Scholar] [CrossRef]
- Masella, R.; Cantafora, A.; Guidoni, L.; Luciani, A.M.; Mariutti, G.; Rosi, A.; Viti, V. Characterization of vesicles, containing an acylated oligopeptide, released by human colon adenocarcinoma cells. NMR and biochemical studies. FEBS Lett. 1989, 246, 25–29. [Google Scholar] [CrossRef]
- Bütikofer, P.; Kuypers, F.A.; Xu, C.M.; Chiu, D.T.; Lubin, B. Enrichment of two glycosyl-phosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human red blood cells. Blood 1989, 74, 1481–1485. [Google Scholar] [CrossRef]
- Morel, O.; Hugel, B.; Jesel, L.; Lanza, F.; Douchet, M.P.; Zupan, M.; Chauvin, M.; Cazenave, J.P.; Freyssinet, J.M.; Toti, F. Sustained elevated amounts of circulating procoagulant membrane microparticles and soluble GPV after acute myocardial infarction in diabetes mellitus. Thromb. Haemost. 2004, 91, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Berckmans, R.J.; Sturk, A.; van Tienen, L.M.; Schaap, M.C.; Nieuwland, R. Cell-derived vesicles exposing coagulant tissue factor in saliva. Blood 2011, 117, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Berckmans, R.J.; Nieuwland, R.; Tak, P.P.; Böing, A.N.; Romijn, F.P.; Kraan, M.C.; Breedveld, F.C.; Hack, C.E.; Sturk, A. Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism. Arthritis Rheum. 2002, 46, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.; Boing, A.N.; Hau, C.M.; Montag, M.; Strowitzki, T.; Nieuwland, R.; Toth, B. Procoagulant tissue factor-exposing vesicles in human seminal fluid. J. Reprod. Immunol. 2013, 98, 45–51. [Google Scholar] [CrossRef]
- Bulut, D.; Tuns, H.; Mugge, A. CD31+/Annexin V+ microparticles in healthy offsprings of patients with coronary artery disease. Eur. J. Clin. Investig. 2009, 39, 17–22. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Microparticles and immunomodulation in pregnancy and pre-eclampsia. J. Reprod. Immunol. 2007, 76, 61–67. [Google Scholar] [CrossRef]
- Ayers, L.; Nieuwland, R.; Kohler, M.; Kraenkel, N.; Ferry, B.; Leeson, P. Dynamic microvesicle release and clearance within the cardiovascular system: Triggers and mechanisms. Clin. Sci. 2015, 129, 915–931. [Google Scholar] [CrossRef]
- Lawson, C.; Vicencio, J.M.; Yellon, D.M.; Davidson, S.M. Microvesicles and exosomes: New players in metabolic and cardiovascular disease. J. Endocrinol. 2016, 228, R57–R71. [Google Scholar] [CrossRef]
- Nielsen, M.H.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. The impact of lipoprotein-associated oxidative stress on cell-specific microvesicle release in patients with familial hypercholesterolemia. Oxid. Med. Cell. Longev. 2016, 2016, 2492858. [Google Scholar] [CrossRef]
- Long, K.E.; Yomtovian, R.; Kida, M.; Knez, J.J.; Medof, M.E. Time-dependent loss of surface complement regulatory activity during storage of donor blood. Transfusion 1993, 33, 294–300. [Google Scholar] [CrossRef]
- Bergman, A.S.; Carlsson, S.R. Saponin-induced release of cell-surface-anchored Thy-1 by serum glycosylphosphatidylinositol-specific phospholipase D. Biochem. J. 1994, 298, 661–668. [Google Scholar] [CrossRef]
- Xie, M.; Low, M.G. Streptolysin-O induces release of glycosylphosphatidylinositol-anchored alkaline phosphatase from ROS cells by vesiculation independently of phospholipase action. Biochem. J. 1995, 305, 529–537. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef]
- Inal, J.M.; Kosgodage, U.; Azam, S.; Stratton, D.; Antwi-Baffour, S.; Lange, S. Blood/plasma secretome and microvesicles. Biochim. Biophys. Acta 2013, 1834, 2317–2325. [Google Scholar] [CrossRef]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef]
- Lynch, S.F.; Ludlam, C.A. Plasma microparticles and vascular disorders. Br. J. Haematol. 2007, 137, 36–48. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Extracellular vesicles and atherosclerotic diseases. Cell. Mol. Life Sci. 2015, 72, 2697–2708. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Aoki, N.; Jin-No, S.; Nakagawa, Y.; Asai, N.; Arakawa, E.; Tamura, N.; Tamura, T.; Matsuda, T. Identification and characterization of microvesicles secreted by 3T3-L1 adipocytes: Redox- and hormone-dependent induction of milk fat globule-epidermal growth factor 8-associated microvesicles. Endocrinology 2007, 148, 3850–3862. [Google Scholar] [CrossRef]
- Müller, G.; Schulz, A.; Dearey, E.A.; Wetekam, E.M.; Wied, S.; Frick, W. Synthetic phosphoinositolglycans regulate lipid metabolism between rat adipocytes via release of GPI-protein-harbouring adiposomes. Arch. Physiol. Biochem. 2010, 116, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Schneider, M.; Gassenhuber, J.; Wied, S. Release of exosomes and microvesicles harbouring specific RNAs and glycosylphosphatidylinositol-anchored proteins from rat and human adipocytes is controlled by histone methylation. Am. J. Mol. Biol. 2012, 2, 187–209. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Straub, J.; Wied, S.; Kramer, W. Induced release of membrane vesicles and exosomes from rat adipocytes containing lipid droplet, lipid raft and glycosylphosphatidylinositol-anchored proteins. Cell. Signal. 2009, 21, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R.; Tanaka, C.; Sato, M.; Nagasaki, H.; Sugimura, K.; Okumura, K.; Nakagawa, Y.; Aoki, N. Adipocyte-derived microvesicles contain RNA that is transported into macrophages and might be secreted into blood circulation. Biochem. Biophys. Res. Commun. 2010, 398, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Over, S.; Wied, S.; Frick, W. Association of (c)AMP-degrading glycosylphosphatidylinositol-anchored proteins with lipid droplets is induced by palmitate, H2O2 and the sulfonylurea drug, glimepiride, in rat adipocytes. Biochemistry 2008, 47, 12774–12787. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Jung, C.; Over, S. Hydrogen peroxide-induced translocation of glycolipid-anchored (c)AMP-hydrolases to lipid droplets mediates inhibition of lipolysis in rat adipocytes. Br. J. Pharmacol. 2008, 154, 901–913. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Wied, S.; Biemer-Daub, G.; Frick, W. Transfer of the glycosylphosphatidylinositol-anchored 5′-nucleotidase CD73 from adiposomes into rat adipocytes stimulates lipid synthesis. Br. J. Pharmacol. 2010, 160, 878–891. [Google Scholar] [CrossRef]
- Müller, G.; Schneider, M.; Biemer-Daub, G.; Wied, S. Microvesicles released from rat adipocytes and harbouring glycosylphosphatidylinositol-anchored proteins transfer RNA stimulating lipid synthesis. Cell. Signal. 2010, 23, 1207–1223. [Google Scholar] [CrossRef]
- Müller, G.; Schneider, M.; Biemer-Daub, G.; Wied, S. Upregulation of lipid synthesis in small rat adipocytes by microvesicle-associated CD73 from large adipocytes. Obesity 2011, 19, 1531–1544. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Dearey, E.A.; Biemer-Daub, G. Glycosylphosphatidylinositol-anchored proteins coordinate lipolysis inhibition between large and small adipocytes. Metabolism 2011, 60, 1021–1037. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Dearey, E.A.; Wetekam, E.M.; Biemer-Daub, G. Lipid storage in large and small rat adipocytes by vesicle-associated glycosylphosphatidylinositol-anchored proteins. In Results and Problems in Cell Differentiation: Sensory and Metabolic Control of Energy Balance; Richter, W., Beisiegel, U., Joost, H., Meyerhof, P., Eds.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2010; Volume 52, pp. 27–34. [Google Scholar]
- Müller, G.; Wied, S.; Straub, J.; Jung, C. Coordinated regulation of esterification and lipolysis by palmitate, H2O2 and the anti-diabetic sulfonylurea drug, glimepiride, in rat adipocytes. Eur. J. Pharmacol. 2008, 597, 6–18. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Walz, N.; Jung, C. Translocation of glycosylphosphatidylinositol-anchored proteins from plasma membrane microdomains to lipid droplets in rat adipocytes is induced by palmitate, H2O2 and the sulfonylurea drug, glimepiride. Mol. Pharmacol. 2008, 73, 1513–1529. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Jung, C.; Over, S. Translocation of glycosylphosphatidylinositol-anchored proteins to lipid droplets and inhibition of lipolysis in rat adipocytes is mediated by reactive oxygen species. Br. J. Pharmacol. 2008, 154, 901–913. [Google Scholar]
- Cross, B.; Ronzon, F.; Roux, B.; Rieu, J.P. Measurement of the anchorage force between GPI-anchored alkaline phosphatase and supported membranes by AFM force spectroscopy. Langmuir 2005, 21, 5149–5153. [Google Scholar] [CrossRef]
- Rieu, J.P.; Ronzon, F.; Place, C.; Dekkiche, F.; Cross, B.; Roux, B. Insertion of GPI-anchored alkaline phosphatase into supported membranes: A combined AFM and fluorescence microscopy study. Acta Biochim. Pol. 2004, 51, 189–197. [Google Scholar] [CrossRef]
- Caseli, L.; Masui, D.C.; Furriel, R.P.; Leone, F.A.; Zaniquelli, M.E.; Orbulescu, J.; Leblanc, R.M. Rat osseous plate alkaline phosphatase as Langmuir monolayer—An infrared study at the air-water interface. J. Colloid Interface Sci. 2008, 320, 476–482. [Google Scholar] [CrossRef]
- Caseli, L.; Oliveira, R.G.; Masui, D.C.; Furriel, R.P.; Leone, F.A.; Maggio, B.; Zaniquelli, M.E. Effect of molecular surface packing on the enzymatic activity modulation of an anchored protein on phospholipid Langmuir monolayers. Langmuir 2005, 21, 4090–4095. [Google Scholar] [CrossRef]
- Kouzayha, A.; Besson, F. GPI-alkaline phosphatase insertion into phosphatidylcholine monolayers: Phase behavior and morphology changes. Biochem. Biophys. Res. Commun. 2005, 333, 1315–1321. [Google Scholar] [CrossRef]
- Ronzon, F.; Rieu, J.P.; Chauvet, J.P.; Roux, B. A thermodynamic study of GPI-anchored and soluble form of alkaline phosphatase films at the air-water interface. J. Colloid Interface Sci. 2006, 301, 493–502. [Google Scholar] [CrossRef]
- Rooney, I.A.; Heuser, J.E.; Atkinson, J.P. GPI-anchored complement regulatory proteins in seminal plasma. An analysis of their physical conditions and the mechanisms of their binding to exogenous cells. J. Clin. Investig. 1996, 97, 1675–1686. [Google Scholar] [CrossRef]
- Müller, G.A.; Herling, A.W.; Stemmer, K.; Lechner, A.; Tschöp, M.H. Chip-based sensing for release of unprocessed cell surface proteins in vitro and in serum and its (patho)physiological relevance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E212–E233. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Ussar, S.; Tschöp, M.H.; Müller, T.D. Age-dependent membrane release and degradation of full-length glycosylphosphatidylinositol-anchored proteins in rats. Mech. Ageing Dev. 2020, 190, 111307. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. Membrane insertion and intracellular transfer of glycosylphosphatidylinositol-anchored proteins: Potential therapeutic applications. Arch. Physiol. Biochem. 2020, 126, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Medof, M.E.; Nagarajan, S.; Tykocinski, M.L. Cell-surface engineering with GPI-anchored proteins. FASEB J. 1996, 10, 574–586. [Google Scholar] [CrossRef]
- Zaruba, M.; Roschitz, L.; Sami, H.; Ogris, M.; Gerner, W.; Metzner, C. Surface modification of E. coli outer membrane vesicles with glycosylphosphatidylinositol-anchored proteins: Generating pro/eukaryote chimera constructs. Membranes 2021, 11, 428. [Google Scholar] [CrossRef]
- Bouma, S.R.; Drislane, F.W.; Huestis, W.H. Selective extraction of membrane-bound proteins by phospholipid vesicles. J. Biol. Chem. 1977, 252, 6759–6763. [Google Scholar] [CrossRef]
- Keiding, N.R. Intestinal alkaline phosphatase in human lymph and serum. Scand. J. Clin. Lab. Investig. 1966, 18, 134–140. [Google Scholar] [CrossRef]
- Young, G.P.; Yedlin, S.T.; Alpers, D.H. Distribution of soluble and membranous forms of alkaline phosphatase in the small intestine of the rat. Biochim. Biophys. Acta 1981, 676, 257–265. [Google Scholar] [CrossRef]
- Götze, H.; Adelson, J.W.; Hadorn, H.B.; Portmann, R.; Troesch, V. Hormone-elicited enzyme release by the small intestinal wall. Gut 1972, 13, 471–476. [Google Scholar] [CrossRef]
- Dyck, W.P.; Hall, F.F.; Ratliff, C.F. Hormonal control of intestinal alkaline phosphatase secretion in the dog. Gastroenterology 1973, 65, 445–450. [Google Scholar] [CrossRef]
- Young, G.P.; Friedman, S.; Yedlin, S.T.; Allers, D.H. Effect of fat feeding on intestinal alkaline phosphatase activity in tissue and serum. Am. J. Physiol. 1981, 241, G461–G468. [Google Scholar] [CrossRef] [PubMed]
- Eliakim, R.; Mahmood, A.; Alpers, D.H. Rat intestinal alkaline phosphatase secretion into lumen and serum is coordinately regulated. Biochim. Biophys. Acta 1991, 1091, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Eliakim, R.; DeSchryver-Kecskemeti, K.; Nogee, L.; Stenson, W.F.; Alpers, D.H. Isolation and characterization of a small intestinal surfactant-like particle containing alkaline phosphatase and other digestive enzymes. J. Biol. Chem. 1989, 264, 20614–20619. [Google Scholar] [CrossRef] [PubMed]
- DeSchryver-Kecskemeti, K.; Eliakim, R.; Carroll, S.; Stenson, W.F.; Moxley, M.A.; Alpers, D.H. Intestinal surfactant-like material: A novel secretory product of the enterocyte. J. Clin. Investig. 1989, 84, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.H.; Niels-Christiansen, L.L.; Immerdal, L.; Nystrøm, B.T.; Danielsen, E.M. Intestinal alkaline phosphatase: Selective endocytosis from the enterocyte brush border during fat absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1325–G1332. [Google Scholar] [CrossRef]
- Hansen, G.H.; Rasmussen, K.; Niels-Christiansen, L.L.; Danielsen, E.M. Dietary free fatty acids form alkaline phosphatase-enriched microdomains in the intestinal brush border membrane. Mol. Membr. Biol. 2011, 28, 136–144. [Google Scholar] [CrossRef]
- Mahmood, A.; Yamagishi, F.; Eliakim, R.; DeSchryver-Kecskemeti, K.; Gramlich, T.L.; Alpers, D.H. A possible role for rat intestinal surfactant-like particles in transepithelial triacylglycerol transport. J. Clin. Investig. 1994, 93, 70–80. [Google Scholar] [CrossRef]
- Eliakim, R.; Alpers, D.H.; Oren, R.; Fich, A.; DeSchryver-Kecskemeti, K. Abundance of surfactant-like particles reflects mucosal integrity in patients with peptic ulcer disease. Gut 1996, 39, 353–359. [Google Scholar] [CrossRef]
- Dugail, I.; Hajduch, E. A new look at adipocyte lipid droplets: Towards a role in the sensing of triacylglycerol stores? Cell. Mol. Life Sci. 2007, 64, 2452–2458. [Google Scholar] [CrossRef]
- Le Lay, S.; Blouin, C.M.; Hajduch, E.; Dugail, I. Filling up adipocytes with lipids. Lessons from caveolin-1 deficiency. Biochim. Biophys. Acta 2008, 1791, 514–518. [Google Scholar] [CrossRef]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef]
- Patton, S.; Huston, G.E. A method for isolation of milk fat globules. Lipids 1986, 21, 170–174. [Google Scholar] [CrossRef]
- Welsch, U.; Schumacher, U.; Bucheim, W.; Schinko, I.; Jenness, P.; Patton, S. Histochemical and biochemical observatioins on milk-fat-globule membranes from several mammalian species. Acta Histochem. 1990, 40, 59–64. [Google Scholar]
- Hakulinen, J.; Meri, S. Expression and function of the complement membrane attack complex inhibitor CD59 protectin on human breast cancer cells. Lab. Investig. 1994, 71, 820. [Google Scholar]
- Ceriani, R.L.; Blank, E.W. Experimental therapy of human breast tumors with 131I-labeled monoclonal antibodies prepared against the human milk fat globule. Cancer Res. 1988, 48, 4664–4672. [Google Scholar]
- Sugita, Y.; Nakano, Y.; Tomita, M. Isolation from human erythrocytes of a new membrane protein which inhibits the formation of complement transmembrane channels. J. Biochem. 1988, 104, 633–637. [Google Scholar] [CrossRef]
- Davis, A.; Simmons, D.L.; Hale, G.; Harrison, R.A.; Tighe, H.; Lachmann, P.J.; Waldmann, H. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J. Exp. Med. 1989, 170, 637–654. [Google Scholar] [CrossRef]
- Holguin, M.H.; Fredrick, L.R.; Bernshaw, N.J.; Wilcox, L.A.; Parker, C.J. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J. Clin. Investig. 1989, 84, 7–17. [Google Scholar] [CrossRef]
- Okada, N.; Harada, R.; Fujita, T.; Okada, H. A novel membrane glycoprotein capable of inhibiting membrane attack by homologous complement. Int. Immunol. 1989, 1, 205–208. [Google Scholar] [CrossRef]
- Panakova, D.; Sprong, H.; Marois, E.; Thiele, C.; Eaton, S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005, 435, 58–65. [Google Scholar] [CrossRef]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Meri, S.; Morgan, B.P.; Davies, A.; Daniels, R.H.; Olavesen, M.G.; Waldmann, H.; Lachmann, P.J. Human protectin (CD59), an 18000-20000 MW complement lysis restricting factor, inhibits C5b-8 catalyzed insertion of C9 into lipid bilayer. Immunology 1990, 71, 1–9. [Google Scholar] [PubMed]
- Väkevä, A.; Jauhiainen, M.; Ehnholm, C.; Lehto, T.; Meri, S. High-density lipoproteins can act as carriers of glycophosphoinositol lipid-anchored CD59 in human plasma. Immunology 1994, 82, 28–33. [Google Scholar]
- Lehto, T.; Meri, S. Interactions of soluble CD59 with the terminal complement complexes: CD59 and C9 compete for a nascent epitope on C8. J. Immunol. 1993, 151, 4941–4949. [Google Scholar] [CrossRef]
- Olofsson, S.O.; Asp, L.; Boren, J. The assembly and secretion of apolipoprotein B-containing lipoproteins. Curr. Opin. Lipidol. 1999, 10, 341–346. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Wied, S.; Biemer-Daub, G. Induced translocation of glycosylphosphatidylinositol-anchored proteins from lipid droplets to adiposomes in rat adipocytes. Br. J. Pharmacol. 2009, 158, 749–770. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Over, S.; Frick, W. Inhibition of lipolysis by palmitate, H2O2 and the sulfonylurea drug, glimepiride, in rat adipocytes depends on cAMP degradation by lipid droplets. Biochemistry 2008, 47, 1259–1273. [Google Scholar] [CrossRef]
- Neumann, S.; Harterink, M.; Sprong, H. Hitch-hiking between cells on lipoprotein particles. Traffic 2007, 8, 331–338. [Google Scholar] [CrossRef]
- Zeng, X.; Goetz, J.A.; Suber, L.M.; Scott, W.J., Jr.; Schreiner, C.M.; Robbins, D.J. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 2001, 411, 716–720. [Google Scholar] [CrossRef]
- Mann, R.S.; Culi, J. Developmental biology: Morphogens hitch a greasy ride. Nature 2005, 435, 30–33. [Google Scholar] [CrossRef]
- Feng, J.; White, B.; Tyurina, O.V.; Guner, B.; Larson, T.; Lee, H.Y.; Karlstrom, R.O.; Kohtz, J.D. Synergistic and antagonistic roles of the Sonic hedgehog N- and C-terminal lipids. Development 2004, 131, 4357–4370. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P. Hedgehog signaling: From the Drosophila cuticle to anti-cancer drugs. Dev. Cell 2005, 8, 143–151. [Google Scholar] [CrossRef]
- Rooney, I.A.; Atkinson, J.P.; Krul, E.S.; Schonfeld, G.; Polakoski, K.; Saffitz, J.E.; Morgan, B.P. Physiologic relevance of the membrane attack complex inhibitory protein CD59 in human seminal plasma: CD59 is present on extracellular organelles (prostasomes), binds cell membranes, and inhibits complement-mediated lysis. J. Exp. Med. 1993, 177, 1409–1420. [Google Scholar] [CrossRef]
- Raymond, F.; Datta, H.; Moss, D. Alkaline phosphatase isoforms in bile and serum and their generation from cells in vitro. Biochim. Biophys. Acta 1991, 1074, 217–222. [Google Scholar] [CrossRef]
- Müller, G.A.; Müller, T.D. Biological role of the intercellular transfer of glycosylphosphatidylinositol-anchored proteins: Stimulation of lipid and glycogen synthesis. Int. J. Mol. Sci. 2022, 23, 7418. [Google Scholar] [CrossRef]
- Ronquist, G.; Brody, I. The prostasome: Its secretion and function in man. Biochim. Biophys. Acta 1985, 822, 203–218. [Google Scholar] [CrossRef]
- Hale, G.; Xia, M.Q.; Tighe, H.P.; Dyer, M.J.; Waldmann, H. The CAMPATH-1 antigen. Tissue Antigens 1990, 35, 118–127. [Google Scholar] [CrossRef]
- Hara, T.; Kuriyama, S.; Kiyohara, H.; Nagase, Y.; Matsumoto, M.; Seya, T. Soluble forms of membrane cofactor protein (CD46, MCP) are present in plasma, tears and seminal fluid in normal subject. Clin. Exp. Immunol. 1992, 89, 490–494. [Google Scholar] [CrossRef]
- Hara, T.; Matsumoto, M.; Fukumori, Y.; Miyagawa, S.; Hatanaka, M.; Kinoshita, T.; Seya, T.; Akedo, H. A monoclonal antibody against human decay accelerating factor (DAF, CD55), D17, which lacks reactivity with semen-DAF. Immunol. Lett. 1993, 37, 145–152. [Google Scholar] [CrossRef]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Upregulated phospholipase D activity toward glycosylphosphatidylinositol-anchored proteins in micelle-like serum complexes in metabolically deranged rats and humans. Am. J. Physiol. Endocrinol. Metabol. 2020, 318, E462–E479. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Herling, A.; Tschöp, M.H. Signatures of complete glycosylphosphatidylinositol-anchored proteins in serum are correlated with distinct metabolic states of obese rats. Adipositas—Mehr (als) Gewicht. Adipositas 2017, 11, A19. [Google Scholar]
- Müller, G.A.; Herling, A.; Tschöp, M.H. A chip-based biosensor for the detection of glycosylphosphatidylinositol-anchored proteins in serum as stress biomarkers. Biomed. Eng. Biomed. Tech. 2017, 62, S358. [Google Scholar]
- Müller, G.A.; Tschöp, M.H. Biosensing of intact glycosylphosphatidylinositol-anchored proteins in serum as biomarkers for stress-induced diseases. FEBS J. 2015, 282, 274–275. [Google Scholar]
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Nicolson, G.L.; de Mattos, G.F. Fifty years of the fluid-mosaic model of biomembrane structure and organization and its importance in biomedicine with particular emphasis on membrane lipid replacement. Biomedicines 2022, 10, 1711. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Qi, X.; Chen, C. Insulin receptor trafficking: Consequences for insulin sensitivity and diabetes. Int. J. Mol. Sci. 2019, 20, 5007. [Google Scholar] [CrossRef]
- Schwihla, M.; Korbei, B. The beginning of the end: Initial steps in the degradation of plasma membrane proteins. Front. Plant Sci. 2020, 11, 680. [Google Scholar] [CrossRef]
- Tsumagari, K.; Chang, C.-H.; Ishihama, Y. Exploring the landscape of ectodomain shedding by quantitative protein terminomics. iScience 2021, 24, 102259. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals—Hardware, concepts, and recent development. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef]
- Hanneken, A.; Mercado, M.; Maher, P. Constitutive and regulated shedding of soluble FGF receptors releases biologically active inhibitors of FGF-2. Int. J. Mol. Sci. 2021, 22, 2712. [Google Scholar] [CrossRef]
- Koo, C.Z.; Matthews, A.L.; Harrison, N.; Szyroka, J.; Nieswandt, B.; Gardiner, E.E.; Poulter, N.S.; Tomlinson, M.G. The platelet collagen receptor GPVI is cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 molecular scissors. Int. J. Mol. Sci. 2022, 23, 2440. [Google Scholar] [CrossRef]
- Huang, D.; Chen, J.; Hu, D.; Xie, F.; Yang, T.; Li, Z.; Wang, X.; Xiao, Y.; Zhong, J.; Jiang, Y.; et al. Advances in biological function and clinical application of small extracellular vesicle membrane proteins. Front. Oncol. 2021, 11, 675940. [Google Scholar] [CrossRef]
- Ko, S.Y.; Naora, H. Extracellular vesicle membrane-associated proteins: Emerging roles in tumor angiogenesis and anti-angiogenesis therapy resistance. Int. J. Mol. Sci. 2020, 21, 5418. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell. Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Wu, S.; Luo, M.; To, K.K.W.; Zhang, J.; Su, C.; Zhang, H.; An, S.; Wang, F.; Chen, D.; Fu, L. Intercellular transfer of exosomal wild type EGFR triggers Osimertinib resistance in non-small cell lung cancer. Mol. Cancer 2021, 20, 17. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef]
- Hargett, L.A.; Bauer, N.N. On the origin of microparticles: From “platelet dust” to mediators of intercellular communication. Pulm. Circ. 2013, 3, 329–340. [Google Scholar] [CrossRef]
- Couch, Y.; Buzas, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Thery, C.; et al. A brief history of nearly EV-erything—The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.A.; Müller, T.D. (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments. Biomolecules 2023, 13, 855. https://doi.org/10.3390/biom13050855
Müller GA, Müller TD. (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments. Biomolecules. 2023; 13(5):855. https://doi.org/10.3390/biom13050855
Chicago/Turabian StyleMüller, Günter A., and Timo D. Müller. 2023. "(Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments" Biomolecules 13, no. 5: 855. https://doi.org/10.3390/biom13050855