
Deciphering Molecular Aspects of Potential α-Glucosidase Inhibitors within Aspergillus terreus: A Computational Odyssey of Molecular Docking-Coupled Dynamics Simulations and Pharmacokinetic Profiling

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structure Preparation
2.2. Molecular Docking-Driven Virtual Screening
2.3. Molecular Dynamics Simulations
3. Results and Discussion
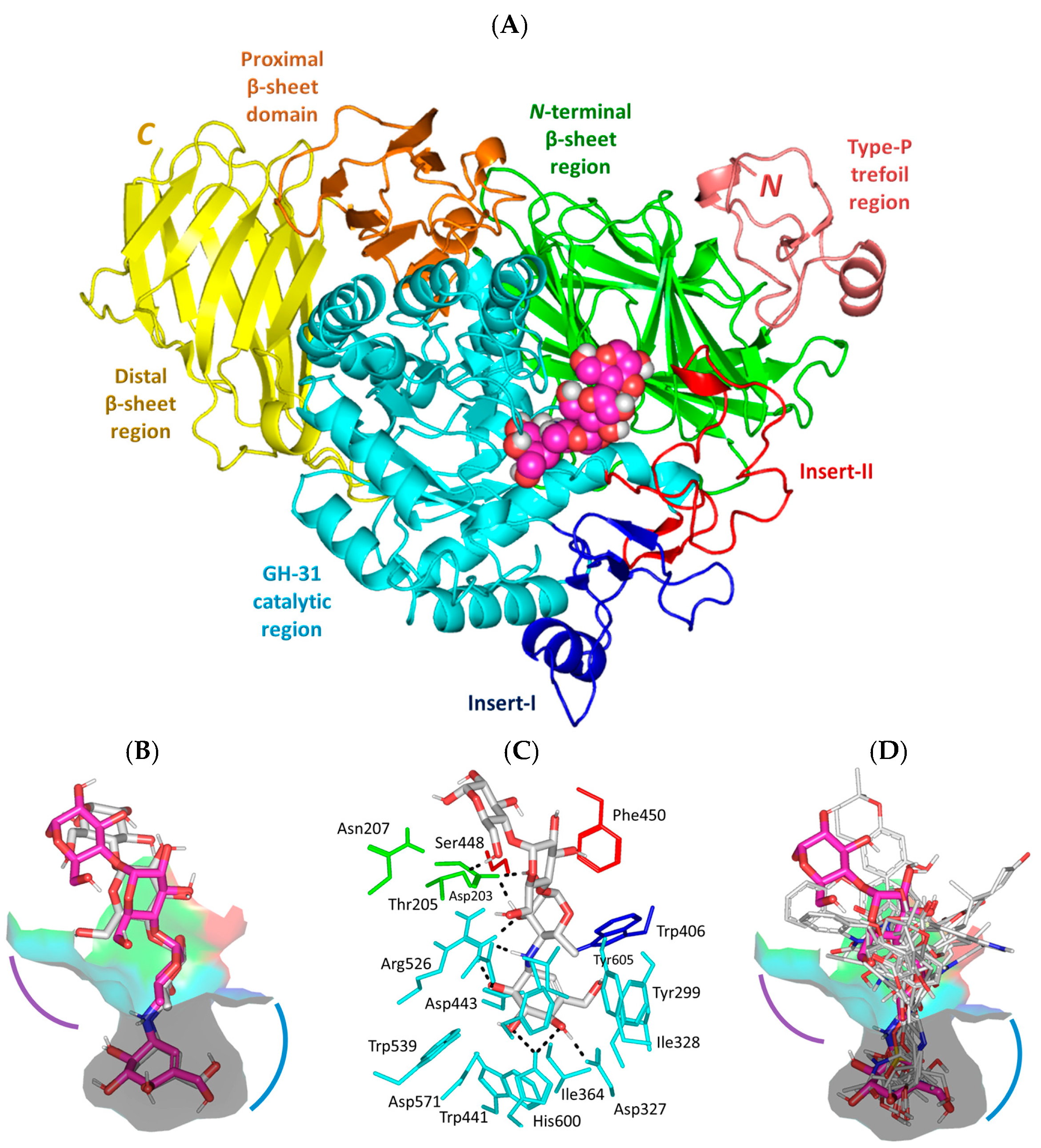
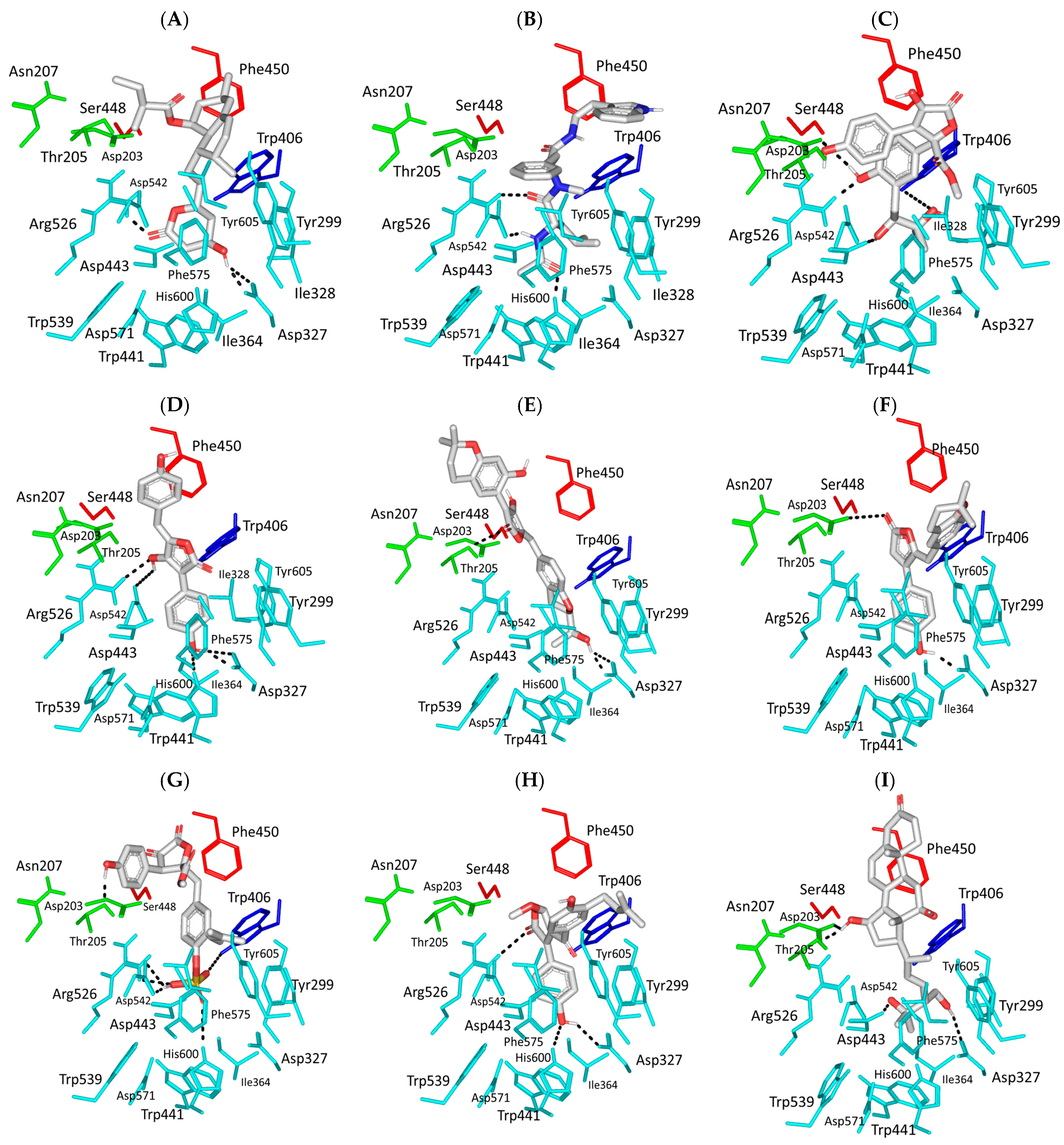
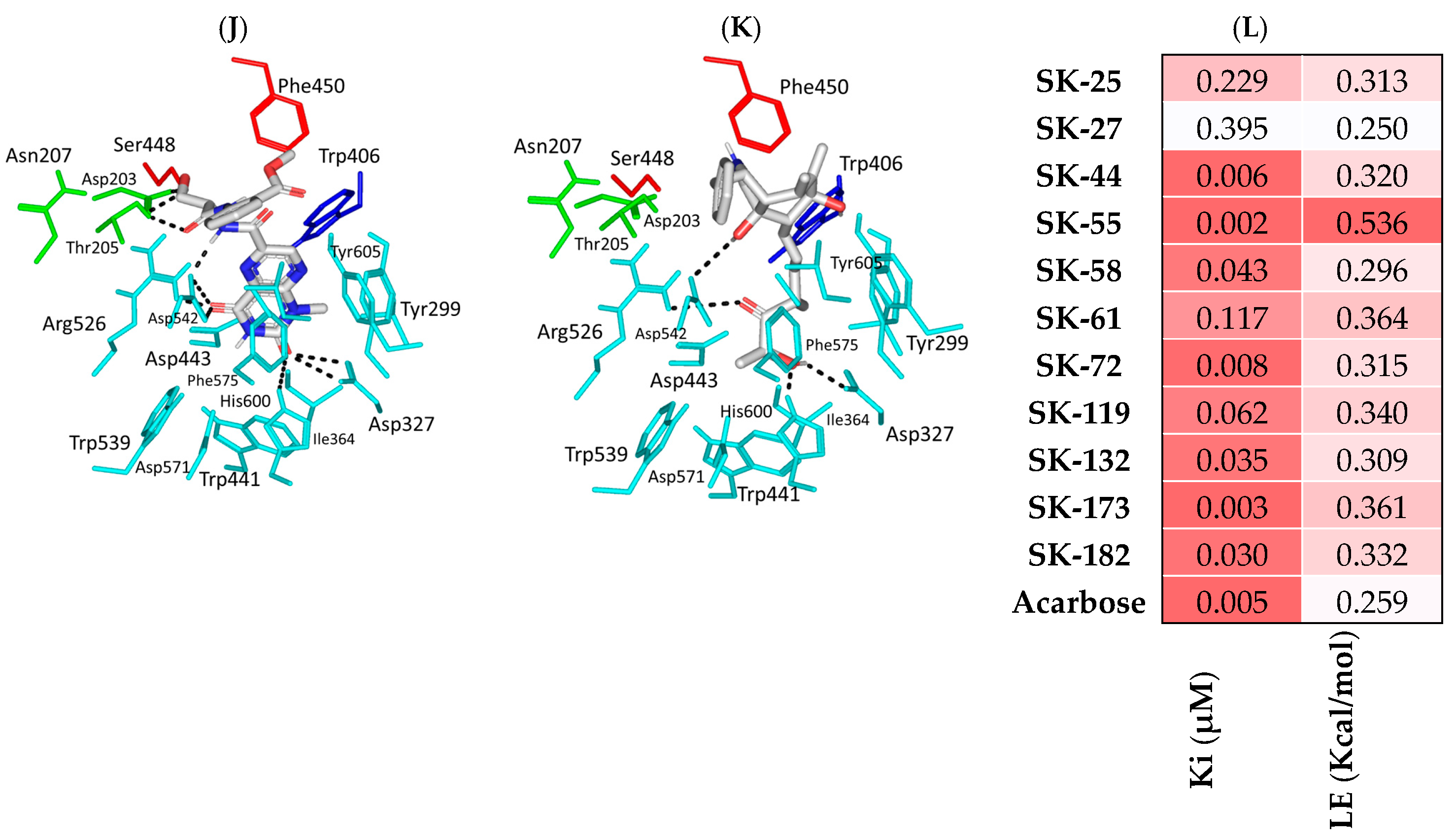
3.1. Molecular Docking Analysis

3.2. Pharmacokinetics Profiling and Biological-Activity Prediction
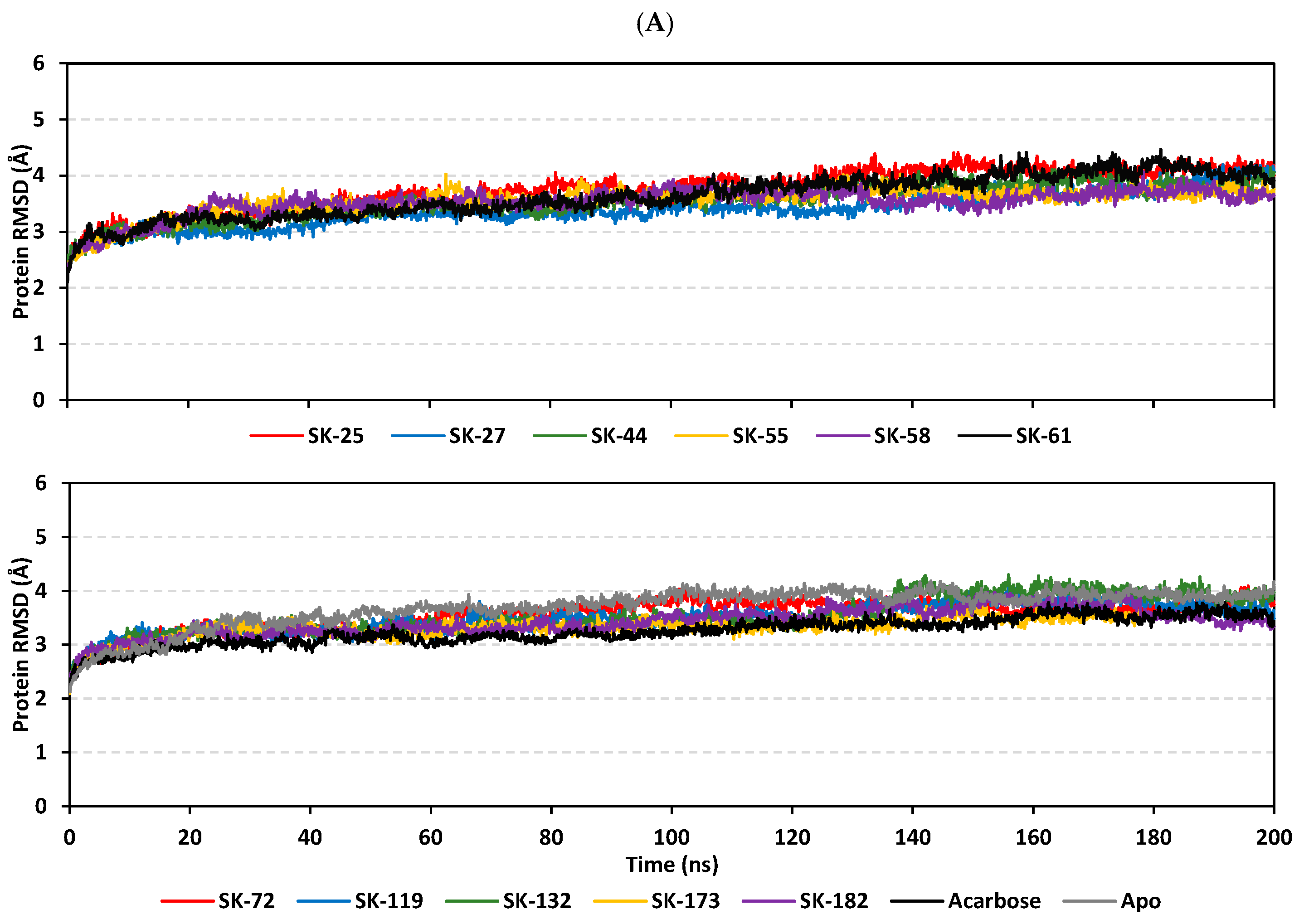
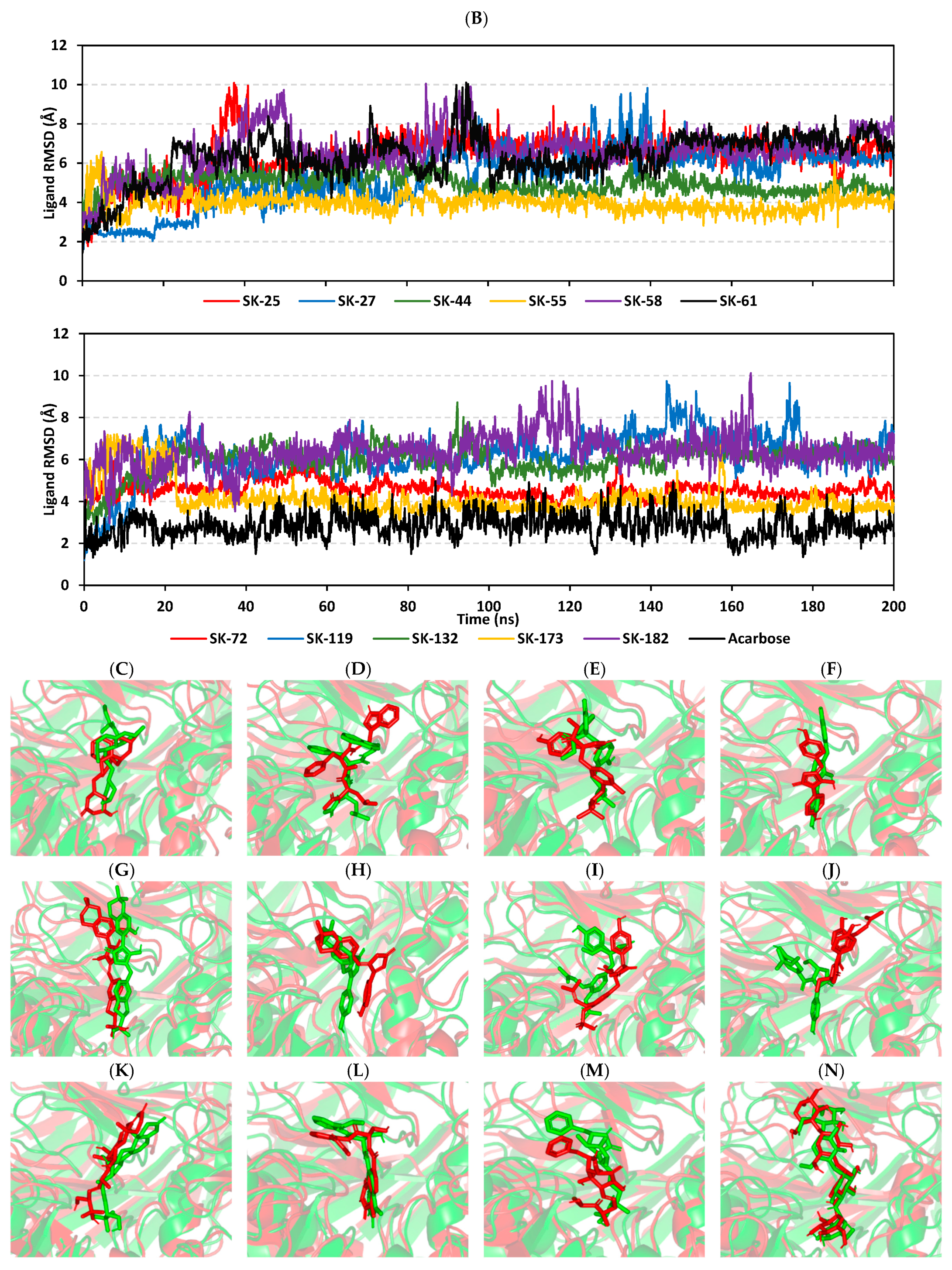
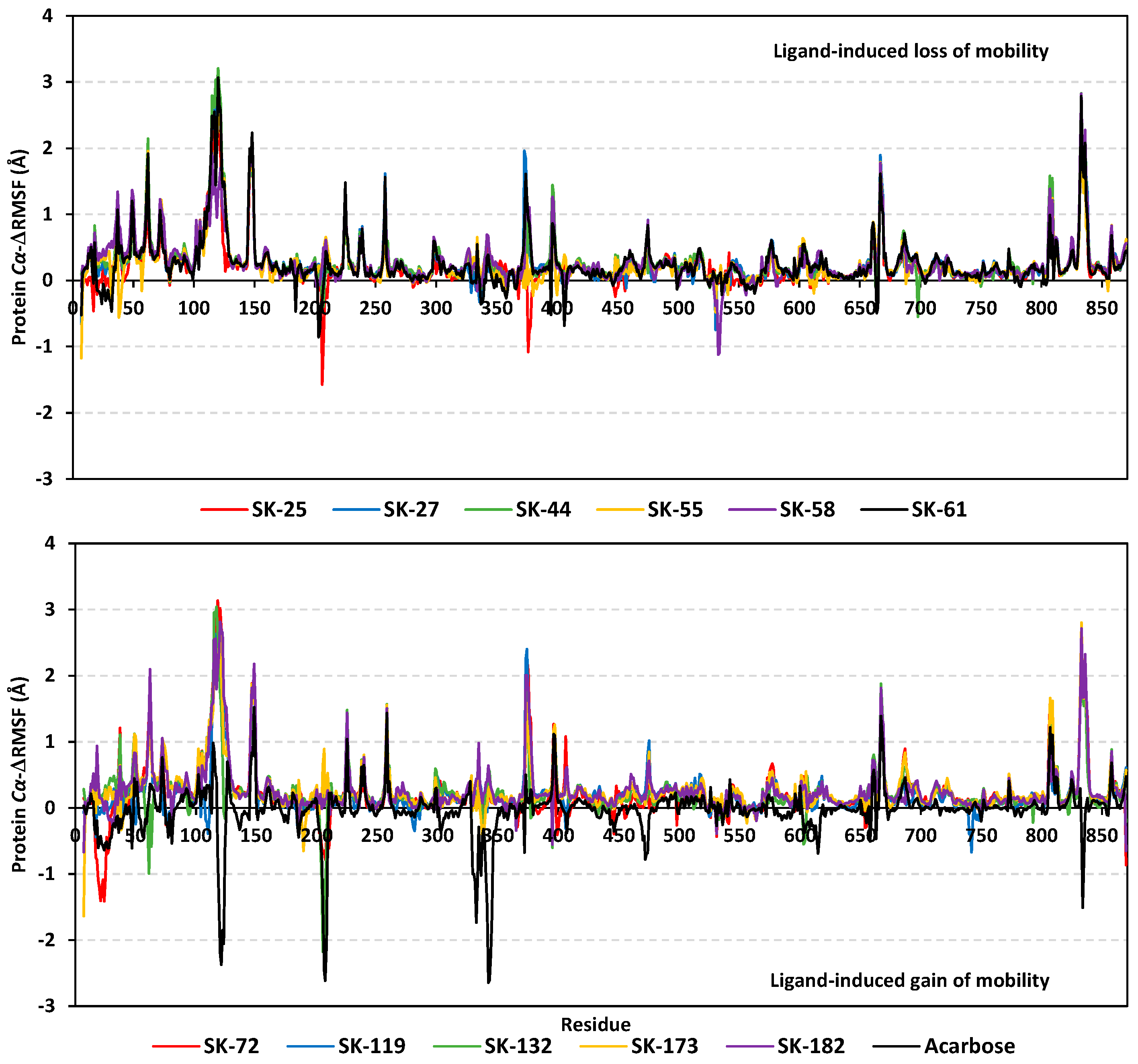
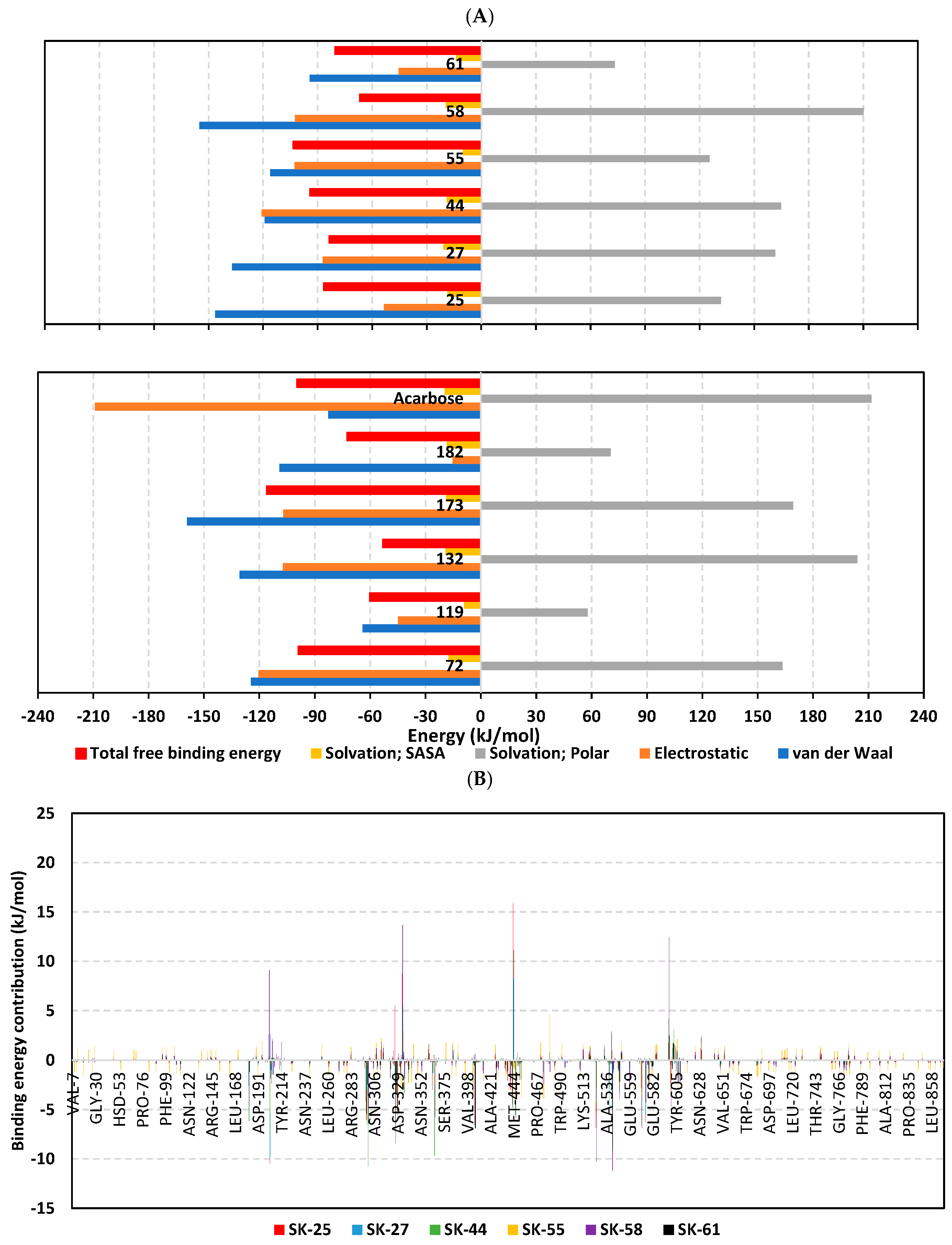
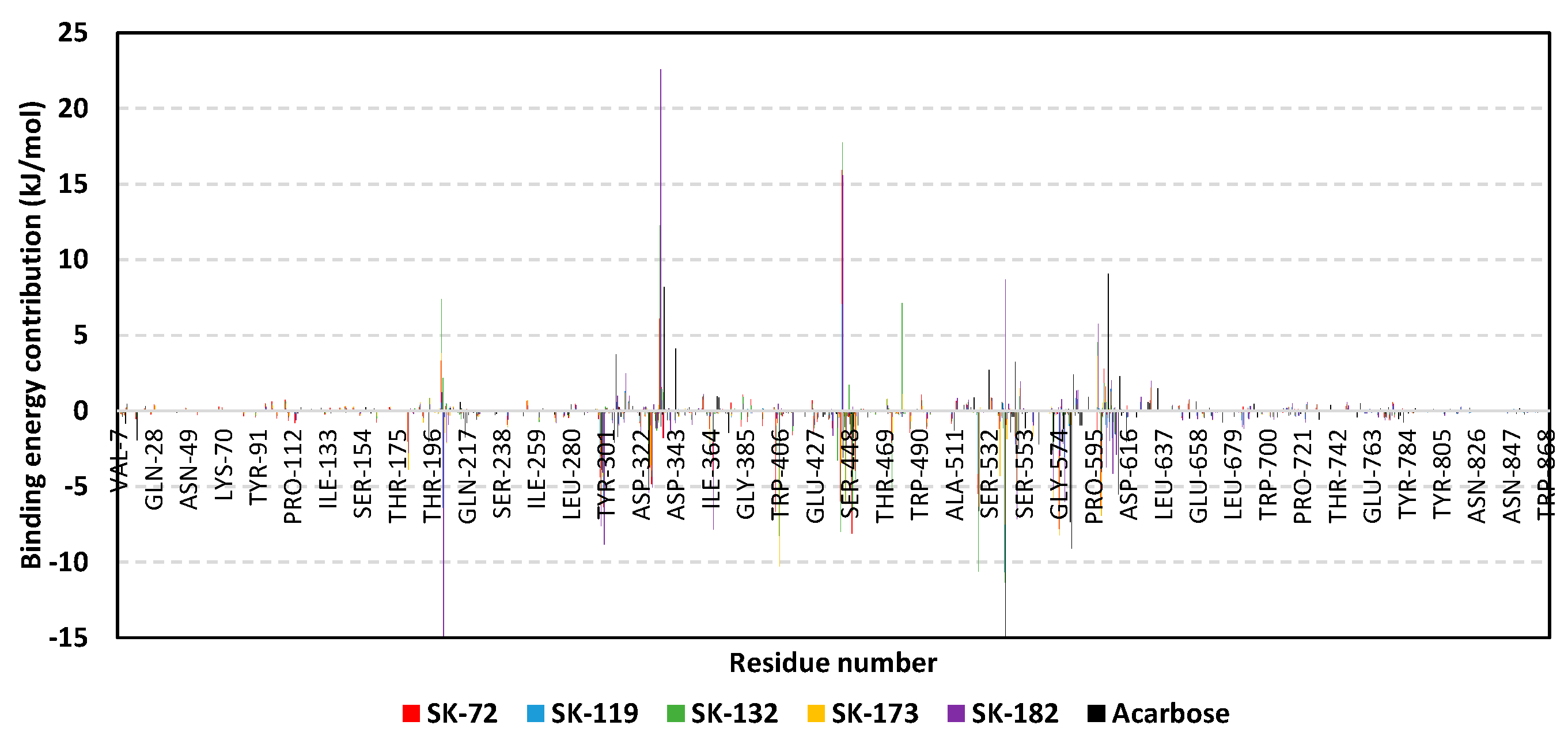
3.3. Molecular Dynamics Simulation Analysis




4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petersmann, A.; Nauck, M.; Müller-Wieland, D.; Kerner, W.; Müller, U.A.; Landgraf, R.; Freckmann, G.; Heinemann, L. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2018, 126, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Ngugi, M.; Makenzi, N.; Njagi, J. Diabetes mellitus—A devastating metabolic disorder. Asian J. Biomed. Pharm. Sci. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Ogurtsova, K.; Guariguata, L.; Barengo, N.C.; Ruiz, P.L.; Sacre, J.W.; Karuranga, S.; Sun, H.; Boyko, E.J.; Magliano, D.J. IDF diabetes Atlas: Global estimates of undiagnosed diabetes in adults for 2021. Diabetes Res. Clin. Pract. 2022, 183, 109118. [Google Scholar] [CrossRef]
- Zeng, Z.; Huang, S.Y.; Sun, T. Pharmacogenomic Studies of Current Antidiabetic Agents and Potential New Drug Targets for Precision Medicine of Diabetes. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2020, 11, 2521–2538. [Google Scholar] [CrossRef]
- Geronikaki, A. Recent Trends in Enzyme Inhibition and Activation in Drug Design. Molecules 2020, 26, 17. [Google Scholar] [CrossRef]
- Zhai, X.; Wu, K.; Ji, R.; Zhao, Y.; Lu, J.; Yu, Z.; Xu, X.; Huang, J. Structure and Function Insight of the α-Glucosidase QsGH13 from Qipengyuania seohaensis sp. SW-135. Front. Microbiol. 2022, 13, 849585. [Google Scholar] [CrossRef]
- Azam, S.S.; Uddin, R.; Wadood, A. Structure and dynamics of alpha-glucosidase through molecular dynamics simulation studies. J. Mol. Liq. 2012, 174, 58–62. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P. α-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Yoon, J.H.; Hahn, S.; Cho, Y.M. Efficacy and safety of combination therapy with an α-glucosidase inhibitor and a dipeptidyl peptidase-4 inhibitor in patients with type 2 diabetes mellitus: A systematic review with meta-analysis. J. Diabetes Investig. 2018, 9, 893–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.K.; Lee, S.H.; Shin, J.; Choi, Y.H.; Ahn, Y.B.; Lee, B.W.; Rhee, E.J.; Min, K.W.; Yoon, K.H. Acarbose Add-on Therapy in Patients with Type 2 Diabetes Mellitus with Metformin and Sitagliptin Failure: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. Diabetes Metab. J. 2019, 43, 287–301. [Google Scholar] [CrossRef]
- Kim, M.K.; Suk, J.H.; Kwon, M.J.; Chung, H.S.; Yoon, C.S.; Jun, H.J.; Ko, J.H.; Kim, T.K.; Lee, S.H.; Oh, M.K.; et al. Nateglinide and acarbose for postprandial glucose control after optimizing fasting glucose with insulin glargine in patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2011, 92, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Reuser, A.J.; Wisselaar, H.A. An evaluation of the potential side-effects of alpha-glucosidase inhibitors used for the management of diabetes mellitus. Eur. J. Clin. Investig. 1994, 24 (Suppl. S3), 19–24. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J.; Matsumoto, K.; White, S.L.; Olden, K. Inhibition of experimental metastasis by castanospermine in mice: Blockage of two distinct stages of tumor colonization by oligosaccharide processing inhibitors. Cancer Res. 1986, 46, 5215–5222. [Google Scholar]
- Sayce, A.C.; Alonzi, D.S.; Killingbeck, S.S.; Tyrrell, B.E.; Hill, M.L.; Caputo, A.T.; Iwaki, R.; Kinami, K.; Ide, D.; Kiappes, J.L.; et al. Iminosugars Inhibit Dengue Virus Production via Inhibition of ER Alpha-Glucosidases—Not Glycolipid Processing Enzymes. PLoS Negl. Trop. Dis. 2016, 10, e0004524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durantel, D.; Alotte, C.; Zoulim, F. Glucosidase inhibitors as antiviral agents for hepatitis B and C. Curr. Opin. Investig. Drugs 2007, 8, 125–129. [Google Scholar] [PubMed]
- Pili, R.; Chang, J.; Partis, R.A.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The alpha-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 1995, 55, 2920–2926. [Google Scholar]
- Tsunoda, T.; Samadi, A.; Burade, S.; Mahmud, T. Complete biosynthetic pathway to the antidiabetic drug acarbose. Nat. Commun. 2022, 13, 3455. [Google Scholar] [CrossRef]
- Zhao, Q.; Xie, H.; Peng, Y.; Wang, X.; Bai, L. Improving acarbose production and eliminating the by-product component C with an efficient genetic manipulation system of Actinoplanes sp. SE50/110. Synth. Syst. Biotechnol. 2017, 2, 302–309. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C. 3D database searching in drug design. J. Med. Chem. 1992, 35, 2145–2154. [Google Scholar] [CrossRef]
- Devi, R.; Sathya, S.S.; Coumar, M. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Dewi, R. Antidiabetic and Antioxidative Activities of Butyrolactone I from Aspergillus terreus MC751. World Acad. Sci. Eng. Technol. 2012, 6, 820–825. [Google Scholar]
- Boruta, T.; Bizukojc, M. Production of lovastatin and itaconic acid by Aspergillus terreus: A comparative perspective. World J. Microbiol. Biotechnol. 2017, 33, 34. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Qi, C.; Sun, W.; Shen, L.; Wang, J.; Liu, J.; Lai, Y.; Xue, Y.; Hu, Z.; Zhang, Y. α-Glucosidase Inhibitors From the Coral-Associated Fungus Aspergillus terreus. Front. Chem. 2018, 6, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewi, R. α-Glucosidase inhibitor compounds from Aspergillus terreus RCC1 and their antioxidant activity. Med. Chem. Res. 2014, 24, 737–743. [Google Scholar] [CrossRef]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human intestinal maltase-glucoamylase: Crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Smith, J.C. Speed vs Accuracy: Effect on Ligand Pose Accuracy of Varying Box Size and Exhaustiveness in AutoDock Vina. Mol. Inform. 2023, 42, 2200188. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Han, L.; Fu, X.; Wang, S.; Li, W.; Han, W. Targeting N-Terminal Human Maltase-Glucoamylase to Unravel Possible Inhibitors Using Molecular Docking, Molecular Dynamics Simulations, and Adaptive Steered Molecular Dynamics Simulations. Front. Chem. 2021, 9, 711242. [Google Scholar] [CrossRef] [PubMed]
- Flores-Bocanegra, L.; Pérez-Vásquez, A.; Torres-Piedra, M.; Bye, R.; Linares, E.; Mata, R. α-Glucosidase Inhibitors from Vauquelinia corymbosa. Molecules 2015, 20, 15330–15342. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, H.M.; Kashegari, A.T.; Shalabi, A.A.; Darwish, K.M.; El-Halawany, A.M.; Algandaby, M.M.; Ibrahim, S.R.M.; Mohamed, G.A.; Abdel-Naim, A.B.; Koshak, A.E.; et al. Phenolics from Chrozophora oblongifolia Aerial Parts as Inhibitors of α-Glucosidases and Advanced Glycation End Products: In-Vitro Assessment, Molecular Docking and Dynamics Studies. Biology 2022, 11, 762. [Google Scholar]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Albuquerque, S.O.; Barros, T.G.; Dias, L.R.S.; Lima, C.; Azevedo, P.; Flores-Junior, L.A.P.; Dos Santos, E.G.; Loponte, H.F.; Pinheiro, S.; Dias, W.B.; et al. Biological evaluation and molecular modeling of peptidomimetic compounds as inhibitors for O-GlcNAc transferase (OGT). Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2020, 154, 105510. [Google Scholar] [CrossRef]
- de Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorganic Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef]
- Elrayess, R.; Darwish, K.M.; Nafie, M.S.; El-Sayyed, G.S.; Said, M.M.; Yassen, A.S.A. Quinoline–hydrazone hybrids as dual mutant EGFR inhibitors with promising metallic nanoparticle loading: Rationalized design, synthesis, biological investigation and computational studies. New J. Chem. 2022, 46, 18207–18232. [Google Scholar] [CrossRef]
- Elhady, S.S.; Abdelhameed, R.F.A.; Malatani, R.T.; Alahdal, A.M.; Bogari, H.A.; Almalki, A.J.; Mohammad, K.A.; Ahmed, S.A.; Khedr, A.I.M.; Darwish, K.M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology 2021, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.H.; Abdelwaly, A.; Darwish, K.M.; Eissa, A.; Chittiboyina, A.; Helal, M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A Showing Potential Activity Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics, and SAR Studies. J. Tradit. Complement. Med. 2022, 12, 16–34. [Google Scholar] [CrossRef]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Wu, C.; Cui, X.; Sun, L.; Lu, J.; Li, F.; Song, M.; Zhang, Y.; Hao, X.; Tian, C.; Song, M.; et al. Aspulvinones Suppress Postprandial Hyperglycemia as Potent α-Glucosidase Inhibitors from Aspergillus terreus ASM-1. Front. Chem. 2021, 9, 736070. [Google Scholar] [CrossRef]
- Munasaroh, S.; Tamat, S.; Dewi, R. Isolation and Identification of α-Glucosidase Inhibitor from Aspergillus terreus F38. Indones. J. Pharm. 2018, 29, 74. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Sun, W.; Wang, J.; He, Y.; Zhang, J.; Li, F.; Qi, C.; Zhu, H.; Xue, Y.; Hu, Z.; et al. Bioactive secondary metabolites from the marine-associated fungus Aspergillus terreus. Bioorganic Chem. 2018, 80, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.G.; Wu, Z.Y.; Pang, W.W.; Ma, L.F.; Ying, Y.M.; Zhan, Z.J. α-Glucosidase Inhibitors from the Fungus Aspergillus terreus 3.05358. Chem. Biodivers. 2015, 12, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, J.; Li, L.; Gong, C.; Wang, S.; Yang, F.; Hua, H.; Lin, H. New butenolide derivatives from the marine sponge-derived fungus Aspergillus terreus. Bioorg. Med. Chem. 2018, 28, 315–318. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, Y.; Liu, W.; Liu, L.; Liu, J.; Yuan, W.; Luo, Z.; Xu, W.; Li, Q. Butenolide Derivatives with α-Glucosidase Inhibitions from the Deep-Sea-Derived Fungus Aspergillus terreus YPGA10. Mar Drugs 2019, 17, 332. [Google Scholar] [CrossRef] [Green Version]
- Dewi, R.; Darmawan, A.; Mulyani, H.; Lotulung, P.D.; Minarti; Wati, M. α-glucosidase inhibitory effect of sulochrin from aspergillusterreus and itsbrominated derivatives. Malays. J. Sci. 2018, 37, 70–81. [Google Scholar] [CrossRef]
- Wu, W.; Liu, L.; Zhu, H.; Sun, Y.; Wu, Y.; Liao, H.; Gui, Y.; Li, L.; Liu, L.; Sun, F.; et al. Butyrolactone-I, an efficient α-glucosidase inhibitor, improves type 2 diabetes with potent TNF-α-lowering properties through modulating gut microbiota in db/db mice. FASEB J. 2019, 33, 12616–12629. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Qin, X.; Cao, X.; Wang, L.; Bai, F.; Bai, G.; Shen, Y. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2011, 2, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Sim, L.; Willemsma, C.; Mohan, S.; Naim, H.Y.; Pinto, B.M.; Rose, D.R. Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J. Biol. Chem. 2010, 285, 17763–17770. [Google Scholar] [CrossRef] [Green Version]
- Lovering, A.L.; Lee, S.S.; Kim, Y.W.; Withers, S.G.; Strynadka, N.C. Mechanistic and structural analysis of a family 31 alpha-glycosidase and its glycosyl-enzyme intermediate. J. Biol. Chem. 2005, 280, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Ernst, H.A.; Lo Leggio, L.; Willemoës, M.; Leonard, G.; Blum, P.; Larsen, S. Structure of the Sulfolobus solfataricus alpha-glucosidase: Implications for domain conservation and substrate recognition in GH31. J. Mol. Biol. 2006, 358, 1106–1124. [Google Scholar] [CrossRef]
- Nagy, M.I.; Darwish, K.M.; Kishk, S.M.; Tantawy, M.A.; Nasr, A.M.; Qushawy, M.; Swidan, S.A.; Mostafa, S.M.; Salama, I. Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein. Pharmaceuticals 2021, 14, 113. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase—A guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef] [Green Version]
- Nhat Phuong, D.; Flower, D.R.; Chattopadhyay, S.; Chattopadhyay, A.K. Towards Effective Consensus Scoring in Structure-Based Virtual Screening. Interdiscip. Sci. Comput. Life Sci. 2023, 15, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput. -Aided Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Banerjee, P.; Leung, S.S.Y.; Yan, X. Application of Pharmacokinetic-Pharmacodynamic Modeling in Drug Delivery: Development and Challenges. 2020, 11, 997. Front. Pharmacol. [CrossRef]
- Edwards, M.; Price, D. Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. Annu. Rep. Med. Chem. 2010, 45, 380–391. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of Ligand Efficiency Metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef] [Green Version]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158. [Google Scholar] [CrossRef]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.-L. Cheminformatic Models to Predict Binding Affinities to Human Serum Albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar] [CrossRef] [Green Version]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Chris Lipinski discusses life and chemistry after the Rule of Five. Drug Discov. Today 2003, 8, 12–16. [Google Scholar]
- Salvatore, T.; Giugliano, D. Pharmacokinetic-Pharmacodynamic Relationships of Acarbose. Clin. Pharmacokinet. 1996, 30, 94–106. [Google Scholar] [CrossRef]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Soltan, M.A.; Eldeen, M.A.; Elbassiouny, N.; Kamel, H.L.; Abdelraheem, K.M.; El-Gayyed, H.A.; Gouda, A.M.; Sheha, M.F.; Fayad, E.; Ali, O.A.A.; et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells 2021, 10, 3014. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. -Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Manandhar, A.; Blass, B.E.; Colussi, D.J.; Almi, I.; Abou-Gharbia, M.; Klein, M.L.; Elokely, K.M. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and In Vitro Screening. J. Chem. Inf. Model. 2021, 61, 1020–1032. [Google Scholar] [CrossRef]
- Almalki, A.J.; Ibrahim, T.S.; Elhady, S.S.; Hegazy, W.A.H.; Darwish, K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals 2022, 15, 110. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, W.; Karabencheva-Christova, T.G.; Black, G.W.; Ainsley, J.; Dover, L.; Christov, C.Z. Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase. Sci. Rep. 2016, 6, 20107. [Google Scholar] [CrossRef] [Green Version]
- Fatriansyah, J.F.; Rizqillah, R.K.; Yandi, M.Y.; Fadilah; Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud Univ. Sci. 2022, 34, 101707. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H.E. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef]
- Mehraban, M.H.; Mansourian, M.; Ahrari, S.; HajiEbrahimi, A.; Odooli, S.; Motovali-Bashi, M.; Yousefi, R.; Ghasemi, Y. Maltase-glucoamylase inhibition potency and cytotoxicity of pyrimidine-fused compounds: An in silico and in vitro approach. Comput. Biol. Chem. 2019, 82, 25–36. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol Biol 2020, 2114, 257–268. [Google Scholar] [CrossRef]
- Swargiary, A.; Roy, M.K.; Mahmud, S. Phenolic compounds as α-glucosidase inhibitors: A docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2023, 41, 3862–3871. [Google Scholar] [CrossRef]
- Ahmed, S.; Ali, M.C.; Ruma, R.A.; Mahmud, S.; Paul, G.K.; Saleh, M.A.; Alshahrani, M.M.; Obaidullah, A.J.; Biswas, S.K.; Rahman, M.M.; et al. Molecular Docking and Dynamics Simulation of Natural Compounds from Betel Leaves (Piper betle L.) for Investigating the Potential Inhibition of Alpha-Amylase and Alpha-Glucosidase of Type 2 Diabetes. Molecules 2022, 27, 4526. [Google Scholar] [CrossRef] [PubMed]
- Askarzadeh, M.; Azizian, H.; Adib, M.; Mohammadi-Khanaposhtani, M.; Mojtabavi, S.; Faramarzi, M.A.; Sajjadi-Jazi, S.M.; Larijani, B.; Hamedifar, H.; Mahdavi, M. Design, synthesis, in vitro α-glucosidase inhibition, docking, and molecular dynamics of new phthalimide-benzenesulfonamide hybrids for targeting type 2 diabetes. Sci. Rep. 2022, 12, 10569. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity–shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef] [Green Version]
- Rudling, A.; Orro, A.; Carlsson, J. Prediction of Ordered Water Molecules in Protein Binding Sites from Molecular Dynamics Simulations: The Impact of Ligand Binding on Hydration Networks. J. Chem. Inf. Model. 2018, 58, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, H.; Ebadi, A.; Faramarzi, M.A.; Mojtabavi, S.; Mahdavi, M.; Najafi, Z. Synthesis, in vitro α-glucosidase inhibitory activity and molecular dynamics simulation of some new coumarin-fused 4H-pyran derivatives as potential anti-diabetic agents. J. Mol. Struct. 2023, 1284, 135349. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Affinity Energy (Kcal/mol) | H-bond Interactions [Length (Å); Angle (°); Binding Residues] | Hydrophobic Interactions | π-Driven Interactions |
|---|---|---|---|---|
| Acarbose Co-crystalline Ligand ACA  | –11.388 | 1.90; 158; Asp203 (sidechain CO−/6-deoxyglucosyl 3′-OH) 2.00; 159; Asp203 (sidechain CO−/6-deoxyglucosyl 4′-OH) 2.10; 163; Thr205 (sidechain OH/+3 maltosyl 6′-OH) 1.90; 171; Asp327 (sidechain CO−/valienamine 4′-OH) 2.00; 165; Arg526 (sidechain = NHH/6-deoxyglucosyl 3′-OH) 2.00; 145; Arg526 (sidechain = NHH/valienamine 6′−OH) 1.90; 143; Asp542 (sidechain CO−/glycosidic linker NH) 1.70; 157; Asp542 (sidechain C=O/valienamine 6′−OH) 2.30; 145; His600 (sidechain NH/valienamine 4′−OH) 2.30; 137; His600 (sidechain NH/valienamine 5′−OH) | Tyr299, Ile328, Ile364, Trp406, Trp441, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | — |
| Lovastatin CID: 53232 SK-25  | –9.071 | 2.10; 157; Asp327 (sidechain O−/4-OH) 3.10; 135; Asp327 (sidechain C=O/4-OH) 2.00; 132; Arg526 (sidechain N+HH/lactone C=O) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | — |
| Aspergillamide A CID: 6917355 SK-27  | –8.747 | 2.30; 132; Arg526 (sidechain N+HH/peptide C=O) 2.10; 125; Asp542 (sidechain C=O/peptide NH) 2.00; 167; His600 (sidechain NH/peptide C=O) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Phe575 |
| Butyrolactone VI CID: 46930025 SK-44  | –11.206 | 2.50; 171; Asp203 (sidechain O−/phenolic OH) 3.40; 129; Trp406 (sidechain NH/tail OH) 2.00; 169; Asp443 (sidechain O−/tail OH) 2.10; 160; Arg526 (sidechain N+HH/phenolic OH) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Tyr299 Trp406 |
| Aspulvinone E CID: 54675753 SK-55  | –11.789 | 2.10; 138; Asp327 (sidechain O−/phenolic OH) 2.40; 160; Asp327 (sidechain C=O/phenolic OH) 2.30; 167; Arg526 (sidechain N+HH/furan OH) 3.50; 132; Asp542 (sidechain O−/furan OH) 3.00; 139; His600 (sidechain NH/phenolic OH) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Tyr299 Trp406 Phe450 Phe575 |
| Aspulvinone F CID: 54728278 SK-58  | –10.065 | 2.10; 167; Asp327 (sidechain O−/2-propan OH) 3.10; 132; Asp327 (sidechain C=O/2-propan OH) 3.10; 139; Thr205 (sidechain OCH3/furan C=O) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Leu473, Trp539, Phe575, Ala576, Leu577, Tyr605 | Trp406 |
| Rubrolide S CID: 101885283 SK-61  | –9.469 | 3.50; 124; Asp203 (sidechain O−/tautomeric furan C=O) 2.10; 145; Asp327 (sidechain O−/phenolic OH) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Trp299 Phe575 Phe575 |
| Butyrolactone I 4′′′′-Sulfate CID: 91935887 SK-72  | –11.039 | 2.00; 173; Thr205 (sidechain OCH3/phenolic OH) 3.40; 168; Trp406 (sidechain NH/S-O−) 2.60; 129; Arg526 (sidechain N+HH/S-OH) 2.00; 126; Asp542 (sidechain O−/S-OH) 2.50; 170; Asp542 (sidechain C=O/S-OH) 3.30; 168; His600 (sidechain NH/S=O) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Trp406 |
| (+)−Asperteretone F CID: 156582453 SK-119  | –9.847 | 2.60; 157; Asp327 (sidechain O−/phenolic OH) 2.60; 171; Arg526 (sidechain N+HH/phenolic OH) 3.20; 146; His600 (sidechain NH/furan C=O) | Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Tyr299 Phe575 |
| 12,15,25,28-tetrahydroxyergosta-4,6,8(14),22-tetraen-3-one SK-132  | –10.184 | 2.10; 159; Asp203 (sidechain O−/C15 βOH) 2.90; 124; Asp203 (sidechain C=O/C15 βOH) 2.40; 122; Asp327 (sidechain C=O/C25 OH) 1.90; 159; Asp443 (sidechain O−/C26 OH) | Tyr214, Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Val451, Trp539, Phe575, Ala576, Leu577, Tyr605 | — |
| Terrelumamide B CID: 139586668 SK-173  | –11.565 | 2.70; 141; Asp203 (sidechain O−/CH2OH) 2.30; 121; Asp203 (sidechain C=O/CH2OH) 2.20; 158; Thr205 (sidechain OH/benzamide C=O) 3.10; 126; Asp327 (sidechain COO−/tautomeric 2-C=O/Enol) 1.90; 128; Arg526 (sidechain N+HH/tautomeric 4-C=O/Enol) 2.70; 126; Asp542 (sidechain COO−/tautomeric 2-C=O/Enol) 1.90; 156; Asp542 (sidechain O−/carboxamide linker NH) 3.30; 138; His600 (sidechain NH/tautomeric 2-C=O/Enol) | Pro206, Tyr214, Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Tyr299 Trp406 Phe575 |
| Cytochalasin Z11 CID: 24970396 SK-182  | –10.278 | 2.30; 133; Asp327 (sidechain O−/tail OH) 3.30; 123; Arg526 (sidechain N+HH/tail OH) 2.70; 127; Asp547 (sidechain O−/ ring junction αOH) 1.90; 153; His600 (sidechain NH/tail OH) | Pro206, Tyr299, Ile328, Ile364, Trp406, Trp441, Met444, Phe450, Trp539, Phe575, Ala576, Leu577, Tyr605 | Phe450 |
| Comp. | “RO’5” HBD; HBA; θ; M.Wt. | PlogP −2.0 to 6.5 | PlogS mol/dm3 −6.5 to 0.5 | PPCaco nm/sec <25 Poor >500 Great | %HOA <25% Poor >80% Great | PPMDCK nm/sec <25 Poor >500 Great | PlogBB −3.0 to 1.2 | PlogKHSA −1.5 to 1.5 | PlogHERG Significant Block > –5.0 | Oral Rat LD50 mg/Kg | AMES Mutagenesis (Predicted Index) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SK-25 | 1; 5; 7; 404.55 | 4.34 | −4.57 (Moderate solubility) | 717.35 | 95% | 345.47 | −1.03 | 0.71 | –4.78 | 556.97 | Negative (0.07) |
| SK-27 | 3; 7; 10; 474.61 | 3.33 | –4.91 (Moderate solubility) | 1146.17 | 100% | 792.72 | –0.79 | 0.04 | –2.47 | 779.48 | Negative (0.18) |
| SK-44 | 5; 9; 8; 401.17 | 2.43 | –3.70 (Soluble) | 356.17 | 82% | 804.20 | –0.81 | 0.65 | –4.32 | 203.09 | Negative (0.48 |
| SK-55 | 3; 5; 2; 296.28 | 2.60 | –3.61 (Soluble) | 363.07 | 83% | 921.33 | –0.77 | 0.54 | –4.98 | 110.64 | Negative (0.32) |
| SK-58 | 3; 7; 3; 464.51 | 4.81 | –5.28 (Moderate solubility) | 893.74 | 89% | 633.46 | −0.88 | 0.79 | –6.70 | 334.28 | Negative (0.32) |
| SK-61 | 1; 4; 2; 348.40 | 4.72 | –4.82 (Moderate solubility) | 623.18 | 91% | 296.72 | –0.89 | 0.78 | –5.98 | 320.74 | Negative (0.25) |
| SK-72 | 2; 10; 9; 504.51 | –0.29 | –5.00 (Moderate solubility) | 5.51 | 39% | 2.28 | –2.75 | –0.40 | –3.52 | 1520.26 | Negative (0.22) |
| SK-119 | 2; 6; 6; 394.42 | 4.23 | –4.79 (Moderate solubility) | 229.29 | 83% | 100.69 | –1.48 | 0.02 | –5.16 | 486.10 | Negative (0.04) |
| SK-132 | 4; 5; 5; 456.62 | 2.27 | –3.31 (Soluble) | 123.45 | 84% | 51.57 | –1.91 | 0.50 | –4.62 | 1900.59 | Negative (0.07) |
| SK-173 | 4; 13; 7; 442.39 | –0.82 | –1.99 (High solubility) | 16.21 | 40% | 5.75 | –3.08 | –0.46 | –6.09 | 2008.27 | Negative (0.35) |
| SK-182 | 4; 6; 7; 427.54 | 1.74 | –3.29 (Soluble) | 73.14 | 73% | 62.69 | –1.98 | –0.14 | –4.15 | 1033.63 | Negative (0.26) |
| Acarbose | 14; 19; 9; 645.61 | –5.51 | –2.13 (Extreme solubility) | 0.05 | 0% | 0.01 | –5.57 | –2.54 | –5.62 | 24,405.50 23,989.66 * | Negative (0.03) |
| Canonical Domains Comprising Substrate Pocket b | Residues | SK-25 | SK-27 | SK-44 | SK-55 | SK-58 | SK-61 | SK-72 | SK-119 | SK-132 | SK-173 | SK-182 | Acarbose |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-terminus β-sheet domain | Arg202 | 0.01 | 0.15 | 0.03 | 0.12 | –0.11 | 0.00 | –0.38 | 0.09 | –0.06 | –0.09 | 0.07 | –0.34 |
| Asp203 | −0.15 | 0.25 | 0.20 | 0.22 | –0.03 | –0.86 | –0.38 | 0.14 | –0.52 | 0.20 | 0.21 | –0.40 | |
| Thr204 | −0.06 | 0.19 | 0.17 | 0.19 | –0.07 | –0.76 | –0.60 | 0.09 | –1.18 | 0.07 | –0.01 | –0.76 | |
| Thr205 | –0.02 | 0.12 | 0.01 | 0.22 | 0.10 | –0.38 | –0.68 | 0.11 | –2.18 | 0.64 | 0.13 | –1.42 | |
| Pro206 | –1.57 | 0.02 | –0.34 | 0.27 | 0.23 | –0.29 | –0.65 | 0.17 | –2.10 | 0.89 | 0.03 | –2.47 | |
| Asn207 | –0.98 | 0.30 | –0.39 | 0.41 | 0.50 | –0.08 | –0.78 | 0.35 | –1.16 | 0.08 | 0.13 | –2.61 | |
| Asn209 | –0.14 | 0.29 | 0.15 | 0.66 | 0.61 | 0.44 | –0.15 | 0.32 | 0.25 | 0.53 | 0.27 | –0.58 | |
| Thr211 | 0.02 | 0.25 | 0.12 | 0.43 | 0.20 | 0.17 | –0.20 | 0.00 | 0.36 | 0.24 | –0.10 | 0.15 | |
| Tyr214 | 0.07 | 0.21 | 0.16 | 0.19 | 0.07 | 0.17 | 0.06 | 0.14 | 0.09 | 0.14 | 0.18 | 0.09 | |
| GH-31 catalytic domain | Arg298 | 0.31 | 0.52 | 0.52 | 0.57 | 0.66 | 0.59 | 0.47 | 0.56 | 0.59 | 0.46 | 0.27 | 0.30 |
| Tyr299 | 0.52 | 0.45 | 0.49 | 0.55 | 0.59 | 0.58 | 0.44 | 0.43 | 0.56 | 0.33 | –0.02 | 0.01 | |
| Asp327 | 0.38 | 0.13 | 0.38 | 0.46 | 0.48 | 0.26 | 0.34 | 0.35 | 0.34 | 0.33 | 0.28 | 0.40 | |
| Ile328 | 0.26 | 0.09 | 0.29 | 0.43 | 0.39 | 0.20 | 0.10 | 0.31 | 0.26 | 0.49 | 0.20 | –0.24 | |
| Ile364 | 0.08 | 0.03 | 0.09 | 0.17 | 0.15 | 0.07 | 0.06 | 0.06 | 0.08 | 0.07 | 0.03 | –0.02 | |
| Trp441 | 0.04 | 0.02 | 0.05 | 0.05 | 0.01 | 0.04 | –0.01 | –0.04 | –0.03 | 0.03 | 0.03 | –0.07 | |
| Asp443 | 0.08 | 0.11 | 0.10 | 0.06 | 0.08 | 0.12 | 0.02 | 0.12 | 0.00 | 0.15 | 0.10 | –0.25 | |
| Met444 | 0.06 | 0.12 | 0.09 | 0.14 | 0.10 | 0.10 | –0.20 | 0.11 | 0.07 | 0.16 | 0.14 | –0.19 | |
| Ser448 | –0.24 | 0.08 | 0.09 | 0.05 | 0.08 | 0.01 | –0.13 | 0.14 | 0.05 | 0.07 | 0.13 | –0.18 | |
| Arg526 | 0.31 | 0.07 | –0.11 | 0.07 | –0.05 | –0.10 | –0.15 | –0.11 | –0.05 | –0.04 | 0.04 | 0.31 | |
| Trp539 | 0.15 | –0.05 | 0.02 | 0.11 | 0.09 | 0.09 | –0.13 | 0.04 | 0.10 | 0.08 | 0.27 | 0.10 | |
| Gly541 | 0.09 | –0.21 | –0.02 | –0.22 | 0.05 | 0.10 | –0.24 | 0.09 | 0.06 | 0.16 | –0.12 | –1.04 | |
| Asp542 | 0.42 | 0.03 | 0.19 | 0.09 | 0.06 | 0.15 | 0.10 | 0.02 | 0.02 | 0.10 | 0.08 | 0.43 | |
| Asp571 | 0.08 | 0.13 | 0.16 | 0.20 | 0.16 | 0.17 | 0.19 | 0.11 | 0.13 | 0.17 | 0.18 | 0.01 | |
| Phe575 | 0.32 | 0.15 | 0.48 | 0.16 | 0.36 | 0.42 | 0.56 | 0.29 | 0.30 | 0.54 | 0.31 | 0.14 | |
| Ala576 | 0.46 | 0.47 | 0.50 | 0.33 | 0.55 | 0.50 | 0.62 | 0.48 | 0.42 | 0.47 | 0.35 | 0.11 | |
| Leu577 | 0.44 | 0.61 | 0.53 | 0.42 | 0.55 | 0.61 | 0.67 | 0.55 | 0.42 | 0.55 | 0.45 | 0.16 | |
| Arg598 | 0.01 | 0.03 | 0.03 | –0.04 | 0.10 | 0.04 | 0.02 | 0.06 | 0.00 | 0.04 | 0.06 | –0.04 | |
| His600 | 0.33 | 0.12 | 0.45 | 0.51 | 0.21 | 0.23 | 0.12 | 0.13 | 0.13 | 0.21 | 0.19 | 0.33 | |
| Gly602 | 0.45 | 0.40 | 0.40 | 0.43 | 0.37 | 0.22 | 0.36 | 0.03 | –0.09 | 0.46 | 0.27 | –0.10 | |
| Gln603 | 0.56 | 0.54 | 0.38 | 0.64 | 0.19 | 0.55 | 0.48 | 0.27 | –0.55 | 0.30 | 0.33 | –0.27 | |
| Phe605 | 0.40 | 0.50 | 0.44 | 0.45 | 0.39 | 0.44 | 0.43 | 0.23 | –0.37 | 0.55 | 0.27 | –0.27 | |
| Insert-I catalytic loop | Val405 | 0.08 | –0.02 | 0.05 | 0.26 | 0.09 | –0.09 | 0.42 | –0.01 | 0.22 | 0.30 | 0.20 | –0.07 |
| Trp406 | 0.32 | 0.06 | 0.14 | 0.39 | 0.11 | –0.68 | 1.08 | –0.31 | 0.49 | 0.44 | 0.57 | –0.10 | |
| Insert-II catalytic loop | Ser448 | –0.24 | 0.08 | 0.39 | 0.35 | 0.08 | 0.01 | 0.33 | 0.14 | 0.05 | 0.07 | 0.13 | –0.18 |
| Phe450 | –0.11 | 0.01 | 0.37 | 0.31 | 0.00 | 0.08 | 0.25 | 0.14 | 0.16 | 0.10 | 0.20 | –0.11 | |
| Leu473 | 0.21 | 0.27 | 0.18 | 0.34 | 0.44 | 0.47 | 0.37 | 0.50 | 0.12 | 0.49 | 0.28 | –0.68 | |
| Asp474 | 0.57 | 0.54 | 0.37 | 0.62 | 0.59 | 0.70 | 0.41 | 0.80 | 0.20 | 0.51 | 0.65 | –0.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elhady, S.S.; Alshobaki, N.M.; Elfaky, M.A.; Koshak, A.E.; Alharbi, M.; Abdelhameed, R.F.A.; Darwish, K.M. Deciphering Molecular Aspects of Potential α-Glucosidase Inhibitors within Aspergillus terreus: A Computational Odyssey of Molecular Docking-Coupled Dynamics Simulations and Pharmacokinetic Profiling. Metabolites 2023, 13, 942. https://doi.org/10.3390/metabo13080942
Elhady SS, Alshobaki NM, Elfaky MA, Koshak AE, Alharbi M, Abdelhameed RFA, Darwish KM. Deciphering Molecular Aspects of Potential α-Glucosidase Inhibitors within Aspergillus terreus: A Computational Odyssey of Molecular Docking-Coupled Dynamics Simulations and Pharmacokinetic Profiling. Metabolites. 2023; 13(8):942. https://doi.org/10.3390/metabo13080942
Chicago/Turabian StyleElhady, Sameh S., Noha M. Alshobaki, Mahmoud A. Elfaky, Abdulrahman E. Koshak, Majed Alharbi, Reda F. A. Abdelhameed, and Khaled M. Darwish. 2023. "Deciphering Molecular Aspects of Potential α-Glucosidase Inhibitors within Aspergillus terreus: A Computational Odyssey of Molecular Docking-Coupled Dynamics Simulations and Pharmacokinetic Profiling" Metabolites 13, no. 8: 942. https://doi.org/10.3390/metabo13080942