

A Distinctive Metabolomics Profile and Potential Biomarkers for Very Long Acylcarnitine Dehydrogenase Deficiency (VLCADD) Diagnosis in Newborns


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
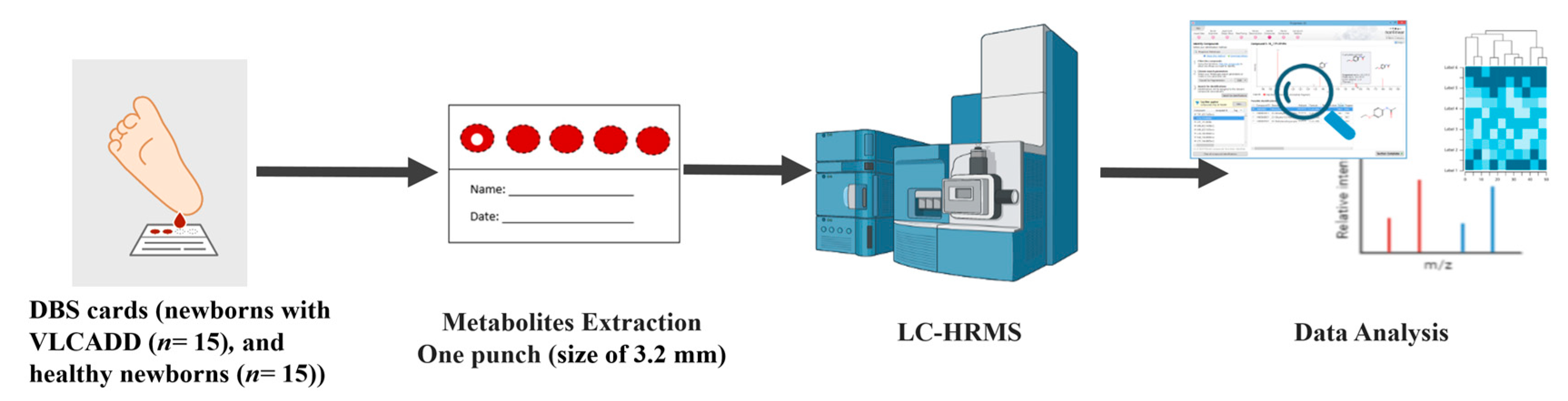
2.2. Patient Inclusion and DBS Collection
2.3. Chemicals and Materials
2.4. Sample Preparation
2.5. LC-MS Metabolomics
2.6. Data Processing and Statistical Analyses
2.7. Peak Annotation (Metabolite Identification)
3. Results
3.1. Demographic and Clinical Characteristics of Study Participants
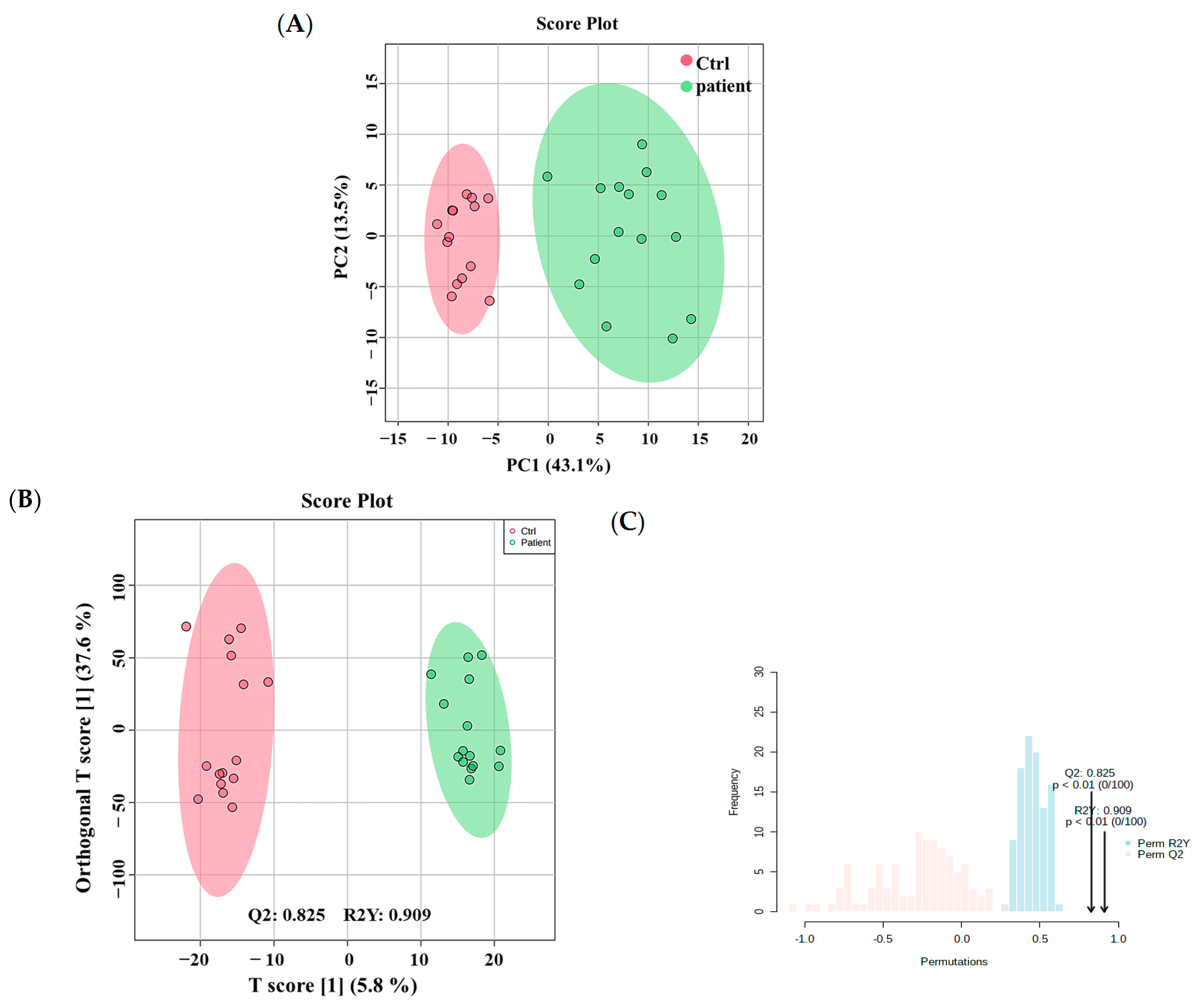
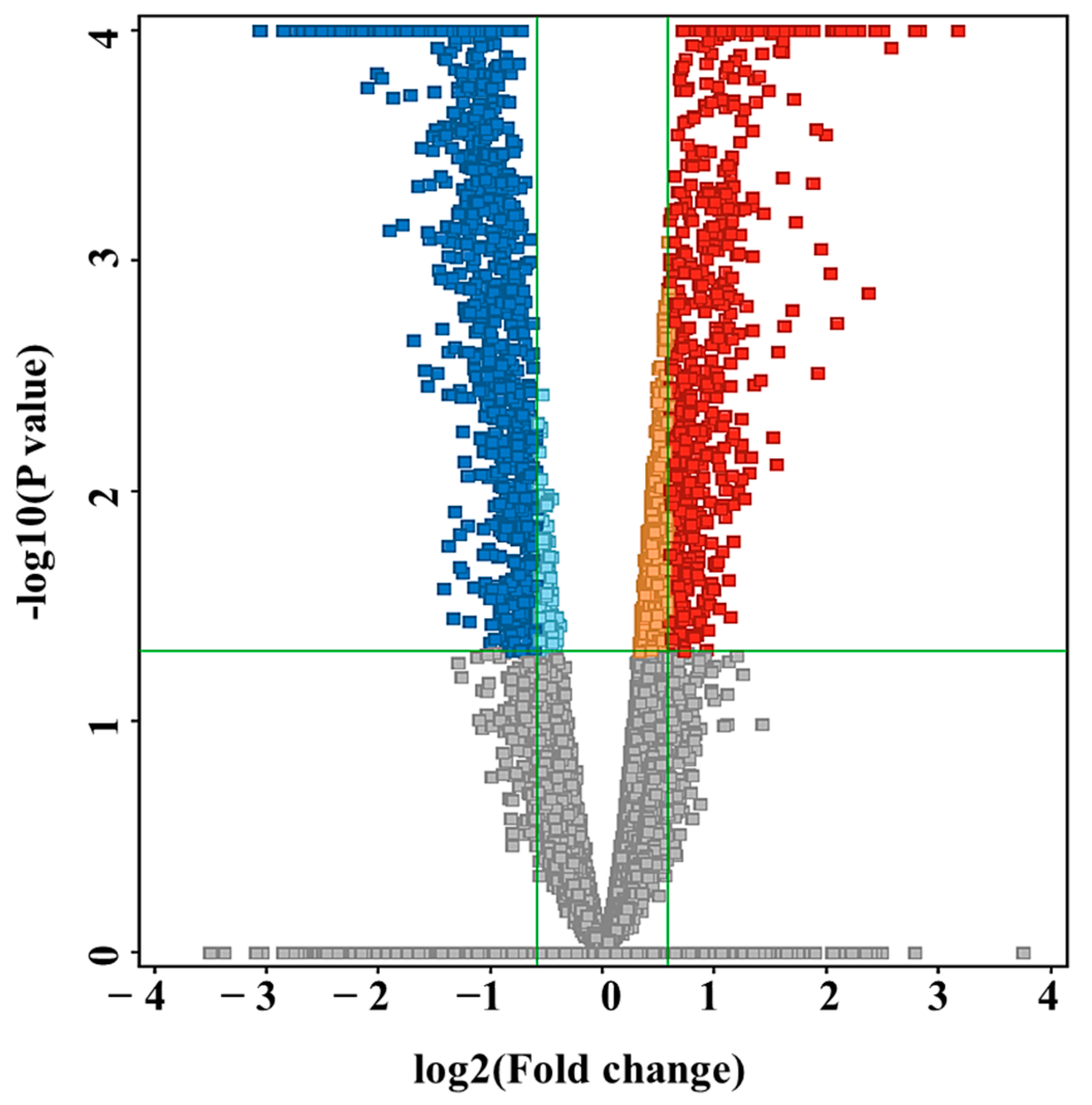
3.2. Metabolomics Profiling of VLCADD Newborns
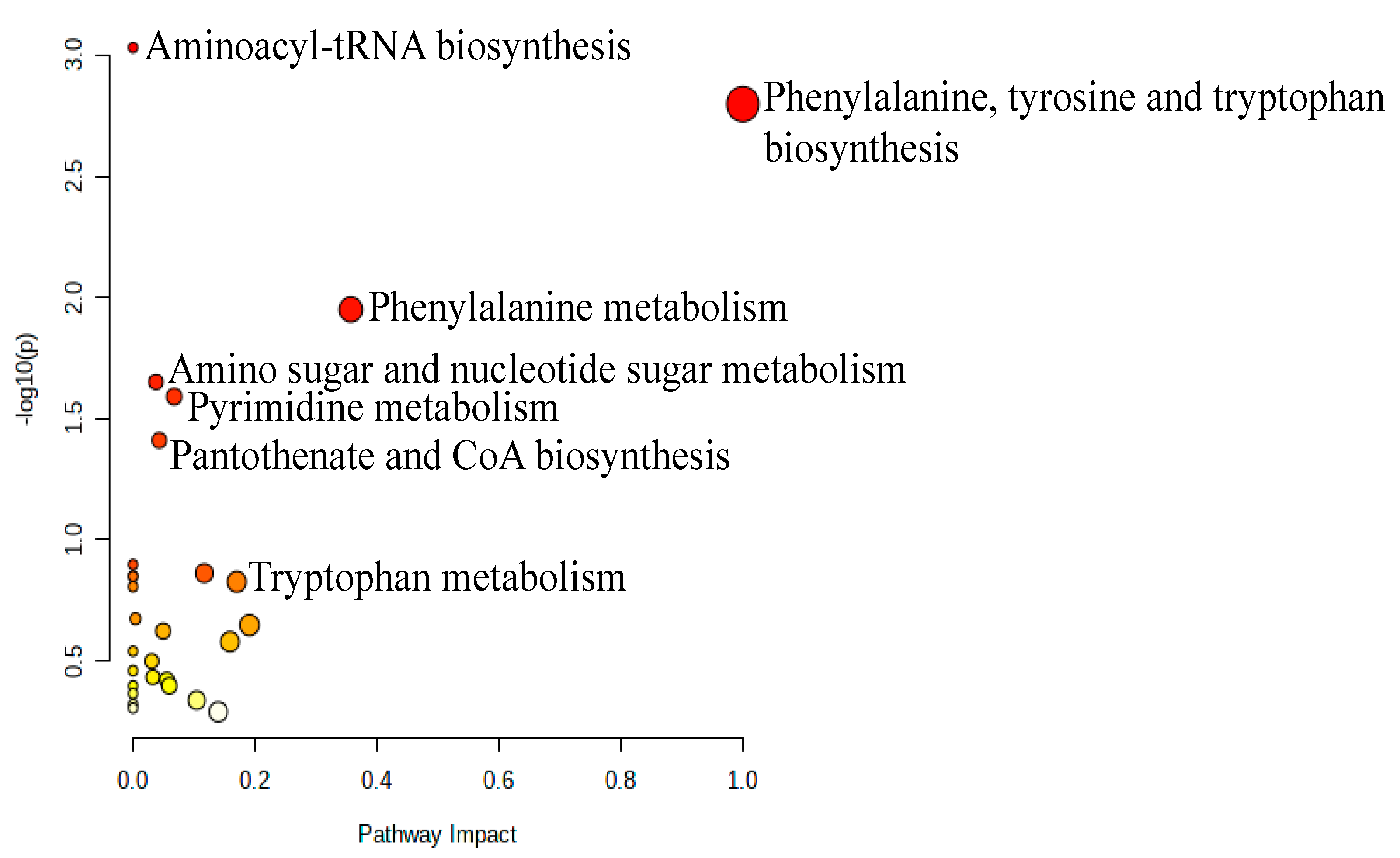
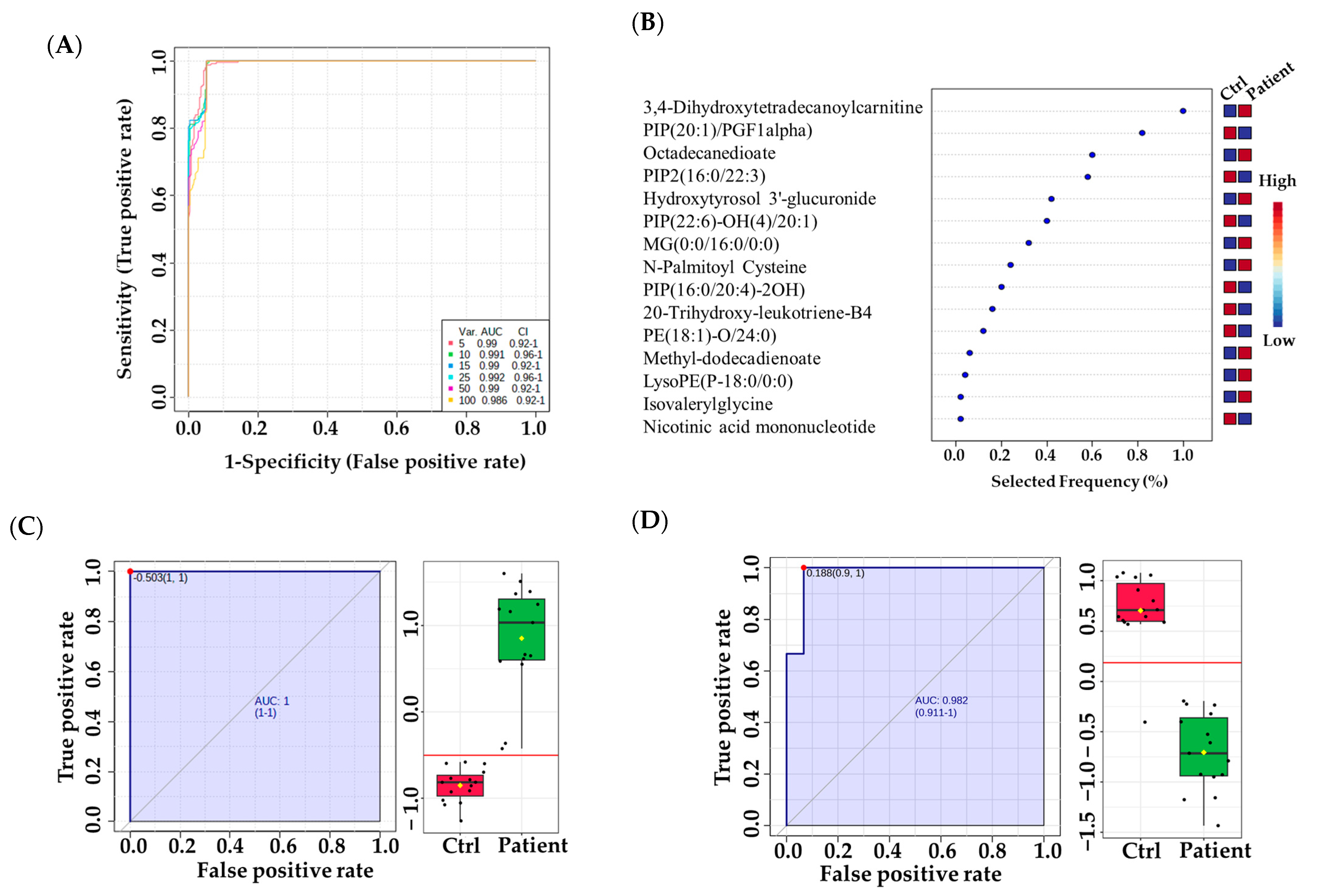
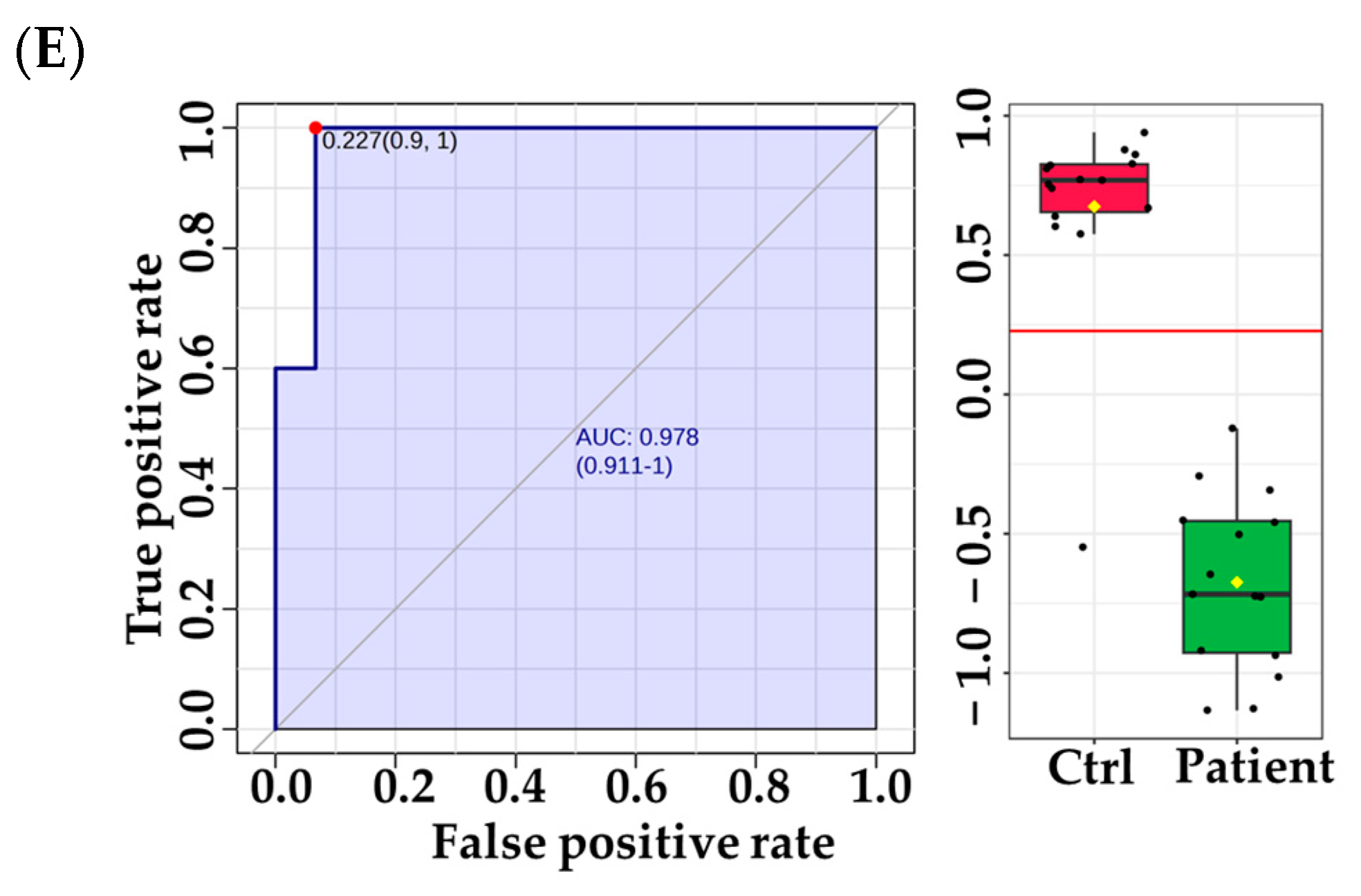
3.3. Metabolomic Pathway and Biomarker Analyses
4. Discussion
4.1. Untargeted Metabolomics as a Diagnostic Tool for VLCADD Newborns
4.2. Distinctive Metabolomics Profile of VLCADD Newborns
4.3. Distinctive Metabolic Biomarkers for VLCADD Newborns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Aoyama, T.; Yamaguchi, S.; Hashimoto, T. Relationship between Structure and Substrate-Chain-Length Specificity of Mitochondrial Very-Long-Chain Acyl-Coenzyme A Dehydrogenase. Eur. J. Biochem. 1998, 257, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; Bennett, M.J. Chapter 4-Disorders of Mitochondrial Fatty Acid β-Oxidation. In Biomarkers in Inborn Errors of Metabolism; Garg, U., Smith, L.D., Eds.; Clinical Aspects and Laboratory Determination; Elsevier: San Diego, CA, USA, 2017; pp. 87–101. ISBN 978-0-12-802896-4. [Google Scholar]
- Ruiz-Sala, P.; Peña-Quintana, L. Biochemical Markers for the Diagnosis of Mitochondrial Fatty Acid Oxidation Diseases. J. Clin. Med. 2021, 10, 4855. [Google Scholar] [CrossRef] [PubMed]
- Guerra, I.M.S.; Ferreira, H.B.; Melo, T.; Rocha, H.; Moreira, S.; Diogo, L.; Domingues, M.R.; Moreira, A.S.P. Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review. Int. J. Mol. Sci. 2022, 23, 13933. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Acquaviva-Bourdain, C.; Rigal, O.; Brivet, M.; Penisson-Besnier, I.; Chabrol, B.; Chaigne, D.; Boespflug-Tanguy, O.; Laroche, C.; Bedat-Millet, A.-L.; et al. Diagnostic Assessment and Long-Term Follow-up of 13 Patients with Very Long-Chain Acyl-Coenzyme A Dehydrogenase (VLCAD) Deficiency. Neuromuscul. Disord. 2009, 19, 324–329. [Google Scholar] [CrossRef]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old Actors Auditioning for New Roles in Metabolic Physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Kiyosue, T.; Arita, M. Inhibitory Effects of Palmitoylcarnitine and Lysophosphatidylcholine on the Sodium Current of Cardiac Ventricular Cells. Pflugers Arch. 1992, 420, 94–100. [Google Scholar] [CrossRef]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.-E. Acylcarnitines: Potential Implications for Skeletal Muscle Insulin Resistance. FASEB J. 2014, 29, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Rutkowsky, J.M.; Knotts, T.A.; Ono-Moore, K.D.; McCoin, C.S.; Huang, S.; Schneider, D.; Singh, S.; Adams, S.H.; Hwang, D.H. Acylcarnitines Activate Proinflammatory Signaling Pathways. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1378–E1387. [Google Scholar] [CrossRef] [Green Version]
- McCoin, C.S.; Knotts, T.A.; Ono-Moore, K.D.; Oort, P.J.; Adams, S.H. Long-Chain Acylcarnitines Activate Cell Stress and Myokine Release in C2C12 Myotubes: Calcium-Dependent and -Independent Effects. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E990–E1000. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.D.; Saenz-Ayala, S. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Marsden, D.; Bedrosian, C.L.; Vockley, J. Impact of Newborn Screening on the Reported Incidence and Clinical Outcomes Associated with Medium- and Long-Chain Fatty Acid Oxidation Disorders. Genet. Med. 2021, 23, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Fatehi, F.; Okhovat, A.A.; Nilipour, Y.; Mroczek, M.; Straub, V.; Töpf, A.; Palibrk, A.; Peric, S.; Stojanovic, V.R.; Najmabadi, H.; et al. Adult-Onset Very Long-Chain Acyl-CoA Dehydrogenase Deficiency (VLCADD). Eur. J. Neurol. 2020, 27, 2257. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Hasegawa, Y.; Yamada, K.; Kobayashi, H.; Purevsuren, J.; Yang, Y.; Dung, V.C.; Khanh, N.N.; Verma, I.C.; Bijarnia-Mahay, S.; et al. Diversity in the Incidence and Spectrum of Organic Acidemias, Fatty Acid Oxidation Disorders, and Amino Acid Disorders in Asian Countries: Selective Screening vs. Expanded Newborn Screening. Mol. Genet. Metab. Rep. 2018, 16, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Benmeakel, M.; Hossain, M.A.; Al Mutairi, F.; Al Othaim, A.; Alfares, A.A.; Al Balwi, M.; Alzaben, A.; Eyaid, W. Thirteen Year Retrospective Review of the Spectrum of Inborn Errors of Metabolism Presenting in a Tertiary Center in Saudi Arabia. Orphanet J. Rare Dis. 2016, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Pena, L.D.M.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.-W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and Genotype-Phenotype Correlations in 52 Individuals with VLCAD Deficiency Diagnosed by NBS and Enrolled in the IBEM-IS Database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, J.C.; Kok, I.L.; Ferdinandusse, S.; van der Pol, W.L.; Cuppen, I.; Bosch, A.M.; Langeveld, M.; Derks, T.G.J.; Williams, M.; de Vries, M.; et al. Impact of Newborn Screening for Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency on Genetic, Enzymatic, and Clinical Outcomes. J. Inherit. Metab. Dis. 2019, 42, 414–423. [Google Scholar] [CrossRef] [Green Version]
- D’Annibale, O.M.; Koppes, E.A.; Sethuraman, M.; Bloom, K.; Mohsen, A.-W.; Vockley, J. Characterization of Exonic Variants of Uncertain Significance in Very Long-Chain Acyl-CoA Dehydrogenase Identified through Newborn Screening. J. Inherit. Metab. Dis. 2022, 45, 529–540. [Google Scholar] [CrossRef]
- Zybert, K.; Borawska-Kowalczyk, U.; Wozniacki, L.; Dawidziuk, M.; Ołtarzewski, M.; Sands, D. Clinical Complications in Children with False-Negative Results in Cystic Fibrosis Newborn Screening. J. Pediatr. 2021, 98, 419–424. [Google Scholar] [CrossRef]
- Kwon, C.; Farrell, P.M. The Magnitude and Challenge of False-Positive Newborn Screening Test Results. Arch. Pediatr. Adolesc. Med. 2000, 154, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Lipstein, E.A.; Perrin, J.M.; Waisbren, S.E.; Prosser, L.A. Impact of False-Positive Newborn Metabolic Screening Results on Early Health Care Utilization. Genet. Med. 2009, 11, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Spiekerkoetter, U.; Mueller, M.; Sturm, M.; Hofmann, M.; Schneider, D.T. Lethal Undiagnosed Very Long-Chain Acyl-CoA Dehydrogenase Deficiency with Mild C14-Acylcarnitine Abnormalities on Newborn Screening. JIMD Rep. Case Res. Rep. 2012, 6, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Bo, R.; Awano, H.; Nishida, K.; Fujioka, K.; Nishiyama, A.; Miyake, O.; Iijima, K. False Positive Cases of Elevated Tetradecenoyl Carnitine in Newborn Mass Screening Showed Significant Loss of Body Weight. Mol. Genet. Metab. Rep. 2020, 24, 100634. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Indo, Y.; Coates, P.M.; Hashimoto, T.; Tanaka, K. Identification of Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency in Three Patients Previously Diagnosed with Long-Chain Acyl-CoA Dehydrogenase Deficiency. Pediatr. Res. 1993, 34, 111–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobazzi, V.; Pasquali, M.; Singh, R.; Matern, D.; Rinaldo, P.; di San Filippo, C.A.; Palmieri, F.; Longo, N. Response to Therapy in Carnitine/Acylcarnitine Translocase (CACT) Deficiency Due to a Novel Missense Mutation. Am. J. Med. Genet. Part. A 2004, 126A, 150–155. [Google Scholar] [CrossRef]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of Carnitine Transport and the Carnitine Cycle. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-Generation Metabolic Screening: Targeted and Untargeted Metabolomics for the Diagnosis of Inborn Errors of Metabolism in Individual Patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Abdel Rahman, A.M. A Targeted Metabolomics Approach for Clinical Diagnosis of Inborn Errors of Metabolism. Anal. Chim. Acta 2018, 1025, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xiao, J.; Gijavanekar, C.; Pappan, K.L.; Glinton, K.E.; Shayota, B.J.; Kennedy, A.D.; Sun, Q.; Sutton, V.R.; Elsea, S.H. Comparison of Untargeted Metabolomic Profiling vs Traditional Metabolic Screening to Identify Inborn Errors of Metabolism. JAMA Netw. Open. 2021, 4, e2114155. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Mass Spectrometry-Based Metabolomics: Applications to Biomarker and Metabolic Pathway Research. Biomed. Chromatogr. 2016, 30, 7–12. [Google Scholar] [CrossRef]
- Alodaib, A.; Nimer, R.; AlMalki, R.; Alhumaidy, R.; Alhenaky, A.; Abdel Jabar, M.; Abdel Raman, A.M. Biomarker Discovery in Galactosemia: Metabolomics with UPLC/HRMS in Dried Blood Spots. Front. Mol. Biosci. 2023, 10, 1154149. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.D.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted Metabolomic Analysis for the Clinical Screening of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, S.J.G.; Pras-Raves, M.L.; van der Ham, M.; Ferdinandusse, S.; Houtkooper, R.H.; Schielen, P.C.J.I.; Visser, G.; Wijburg, F.A.; de Sain-van der Velden, M.G.M. Prediction of VLCAD Deficiency Phenotype by a Metabolic Fingerprint in Newborn Screening Bloodspots. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165725. [Google Scholar] [CrossRef] [PubMed]
- Almontashiri, N.A.M.; Zha, L.; Young, K.; Law, T.; Kellogg, M.D.; Bodamer, O.A.; Peake, R.W.A. Clinical Validation of Targeted and Untargeted Metabolomics Testing for Genetic Disorders: A 3 Year Comparative Study. Sci. Rep. 2020, 10, 9382. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.A.; Benabdelkamel, H.; Dahabiyeh, L.A.; Masood, A.; AlMalki, R.H.; Musambil, M.; Alfadda, A.A.; Abdel Rahman, A.M. The Metabolomics Approach Revealed a Distinctive Metabolomics Pattern Associated with Hyperthyroidism Treatment. Front. Endocrinol. 2022, 13, 1050201. [Google Scholar] [CrossRef]
- Dahabiyeh, L.A.; Malkawi, A.K.; Wang, X.; Colak, D.; Mujamammi, A.H.; Sabi, E.M.; Li, L.; Dasouki, M.; Abdel Rahman, A.M. Dexamethasone-Induced Perturbations in Tissue Metabolomics Revealed by Chemical Isotope Labeling LC-MS Analysis. Metabolites 2020, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Al Dubayee, M.; Alshahrani, A.; Masood, A.; Benabdelkamel, H.; Zahra, M.; Li, L.; Abdel Rahman, A.M.; Aljada, A. Distinctive Metabolomics Patterns Associated With Insulin Resistance and Type 2 Diabetes Mellitus. Front. Mol. Biosci. 2020, 7, 411. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2021, 50, D622–D631. [Google Scholar] [CrossRef]
- Tarini, B.A.; Clark, S.J.; Pilli, S.; Dombkowski, K.J.; Korzeniewski, S.J.; Gebremariam, A.; Eisenhandler, J.; Grigorescu, V. False-Positive Newborn Screening Result and Future Health Care Use in a State Medicaid Cohort. Pediatrics 2011, 128, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Boneh, A.; Andresen, B.; Gregersen, N.; Ibrahim, M.; Tzanakos, N.; Peters, H.; Yaplito-Lee, J.; Pitt, J. VLCAD Deficiency: Pitfalls in Newborn Screening and Confirmation of Diagnosis by Mutation Analysis. Mol. Genet. Metab. 2006, 88, 166–170. [Google Scholar] [CrossRef]
- Alatibi, K.I.; Hagenbuchner, J.; Wehbe, Z.; Karall, D.; Ausserlechner, M.J.; Vockley, J.; Spiekerkoetter, U.; Grünert, S.C.; Tucci, S. Different Lipid Signature in Fibroblasts of Long-Chain Fatty Acid Oxidation Disorders. Cells 2021, 10, 1239. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T. Ganglioside Biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutillo, G.; Saariaho, A.-H.; Meri, S. Physiology of Gangliosides and the Role of Antiganglioside Antibodies in Human Diseases. Cell. Mol. Immunol. 2020, 17, 313–322. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Limphong, P.; Pieper, J.; Liu, Q.; Rodesch, C.K.; Christians, E.; Benjamin, I.J. Glutathione-Dependent Reductive Stress Triggers Mitochondrial Oxidation and Cytotoxicity. FASEB J. 2012, 26, 1442–1451. [Google Scholar] [CrossRef] [Green Version]
- Corteselli, E.M.; Gibbs-Flournoy, E.; Simmons, S.O.; Bromberg, P.; Gold, A.; Samet, J.M. Long Chain Lipid Hydroperoxides Increase the Glutathione Redox Potential Through Glutathione Peroxidase 4. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Kabuyama, Y.; Suzuki, T.; Nakazawa, N.; Yamaki, J.; Homma, M.K.; Homma, Y. Dysregulation of Very Long Chain Acyl-CoA Dehydrogenase Coupled with Lipid Peroxidation. Am. J. Physiol. Cell. Physiol. 2010, 298, C107–C113. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Sergi, C. Biochemistry, Amino Acid Synthesis and Degradation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and Cellular Mechanisms for Managing Lipid Excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef] [Green Version]
- Dambrova, M.; Makrecka-Kuka, M.; Kuka, J.; Vilskersts, R.; Nordberg, D.; Attwood, M.M.; Smesny, S.; Sen, Z.D.; Guo, A.C.; Oler, E.; et al. Acylcarnitines: Nomenclature, Biomarkers, Therapeutic Potential, Drug Targets, and Clinical Trials. Pharmacol. Rev. 2022, 74, 506–551. [Google Scholar] [CrossRef] [PubMed]
- Vissing, C.R.; Dunø, M.; Wibrand, F.; Christensen, M.; Vissing, J. Hydroxylated Long-Chain Acylcarnitines Are Biomarkers of Mitochondrial Myopathy. J. Clin. Endocrinol. Metab. 2019, 104, 5968–5976. [Google Scholar] [CrossRef]
- Mejillano, M.; Yamamoto, M.; Rozelle, A.L.; Sun, H.Q.; Wang, X.; Yin, H.L. Regulation of Apoptosis by Phosphatidylinositol 4,5-Bisphosphate Inhibition of Caspases, and Caspase Inactivation of Phosphatidylinositol Phosphate 5-Kinases. J. Biol. Chem. 2001, 276, 1865–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as Membrane Organizers. Nat. Rev. Mol. Cell. Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Saltiel, A.R. Phosphoinositides in Insulin Action and Diabetes. Curr. Top. Microbiol. Immunol. 2012, 362, 61–85. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward Personalized Medicine. Mass. Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic and Clinical Characteristics | VLCADD Newborns (n = 15) | Healthy Newborns (n = 15) | p-Value | |||
|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | |||
| Age (Day) | 6.20 | 1.19 | 5.66 | 2.5 | 0.7518 | |
| Female (%) | 53.3 | NA | 53.3 | NA | NA | |
| Data | C14:1-carnitine (Cutoff: <0.75 μM) | 2.30 | 0.51 | <0.75 | NA | 0.005 ** |
| C14:1/C16-carnitine ratio (Cutoff: <0.25 μM) | 0.44 | 0.05 | <0.25 | NA | 0.002 ** | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebaa, R.; AlMalki, R.H.; Alseraty, W.; Abdel Rahman, A.M. A Distinctive Metabolomics Profile and Potential Biomarkers for Very Long Acylcarnitine Dehydrogenase Deficiency (VLCADD) Diagnosis in Newborns. Metabolites 2023, 13, 725. https://doi.org/10.3390/metabo13060725
Sebaa R, AlMalki RH, Alseraty W, Abdel Rahman AM. A Distinctive Metabolomics Profile and Potential Biomarkers for Very Long Acylcarnitine Dehydrogenase Deficiency (VLCADD) Diagnosis in Newborns. Metabolites. 2023; 13(6):725. https://doi.org/10.3390/metabo13060725
Chicago/Turabian StyleSebaa, Rajaa, Reem H. AlMalki, Wafaa Alseraty, and Anas M. Abdel Rahman. 2023. "A Distinctive Metabolomics Profile and Potential Biomarkers for Very Long Acylcarnitine Dehydrogenase Deficiency (VLCADD) Diagnosis in Newborns" Metabolites 13, no. 6: 725. https://doi.org/10.3390/metabo13060725