
Data Independent Acquisition Reveals In-Depth Serum Proteome Changes in Canine Leishmaniosis
 and
and
Abstract
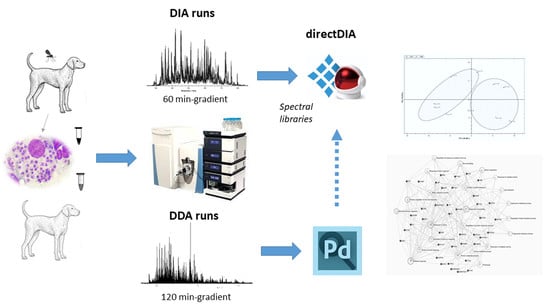
:
1. Introduction
2. Materials and Methods
2.1. Sample Description
2.2. Sample Collection
2.3. Sample Preparation for Spectral Library Generation
2.3.1. In-Solution Digestion
2.3.2. Strong Cation Exchange (SCX) Chromatography
2.3.3. Combinatorial Peptide Ligand Library (ProteoMiner)
2.4. MS-Based Proteomic Analysis
DDA Proteomic Analysis for Spectral Library Generation
2.5. DIA Proteomic Analysis
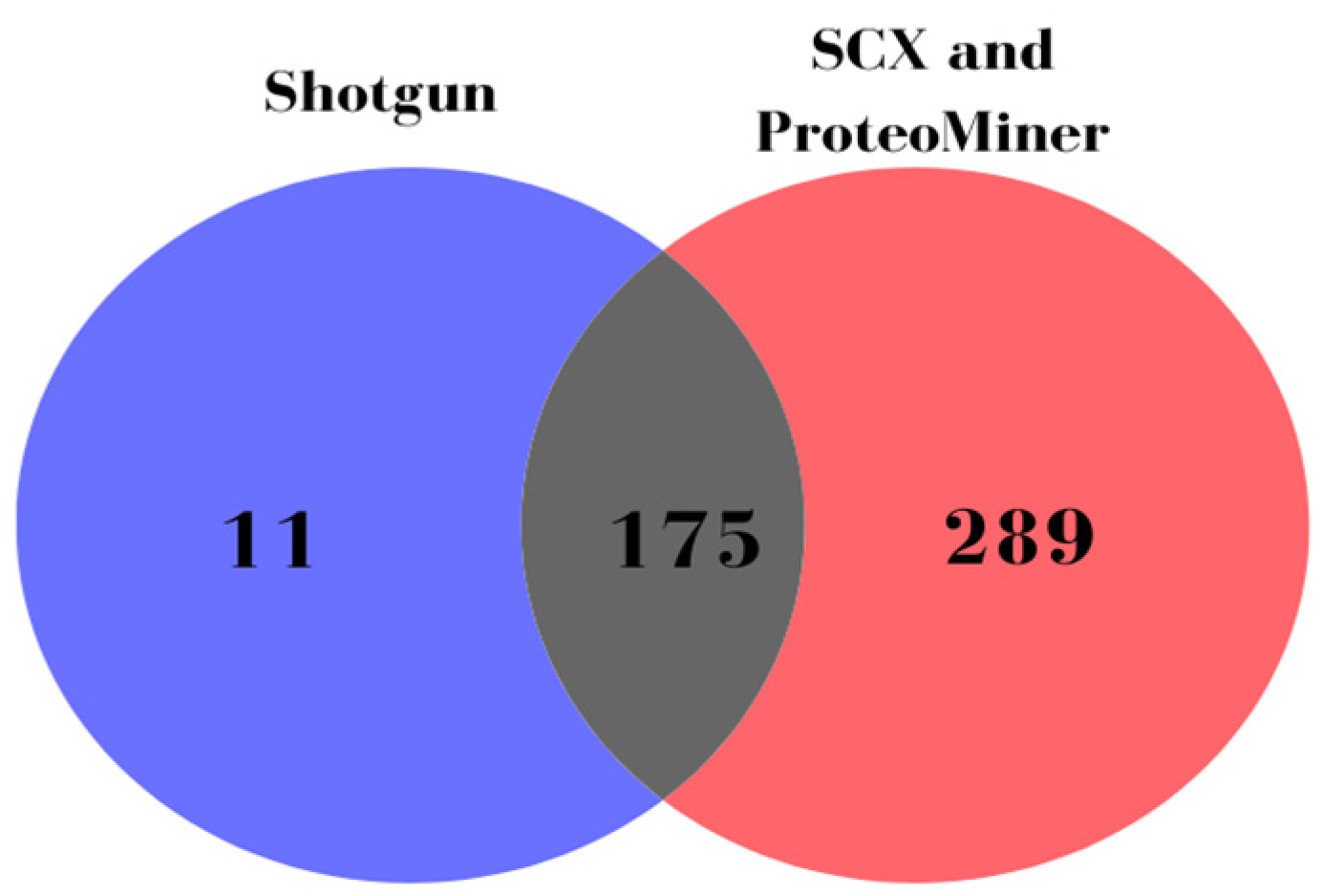
2.6. Spectral Library Generation
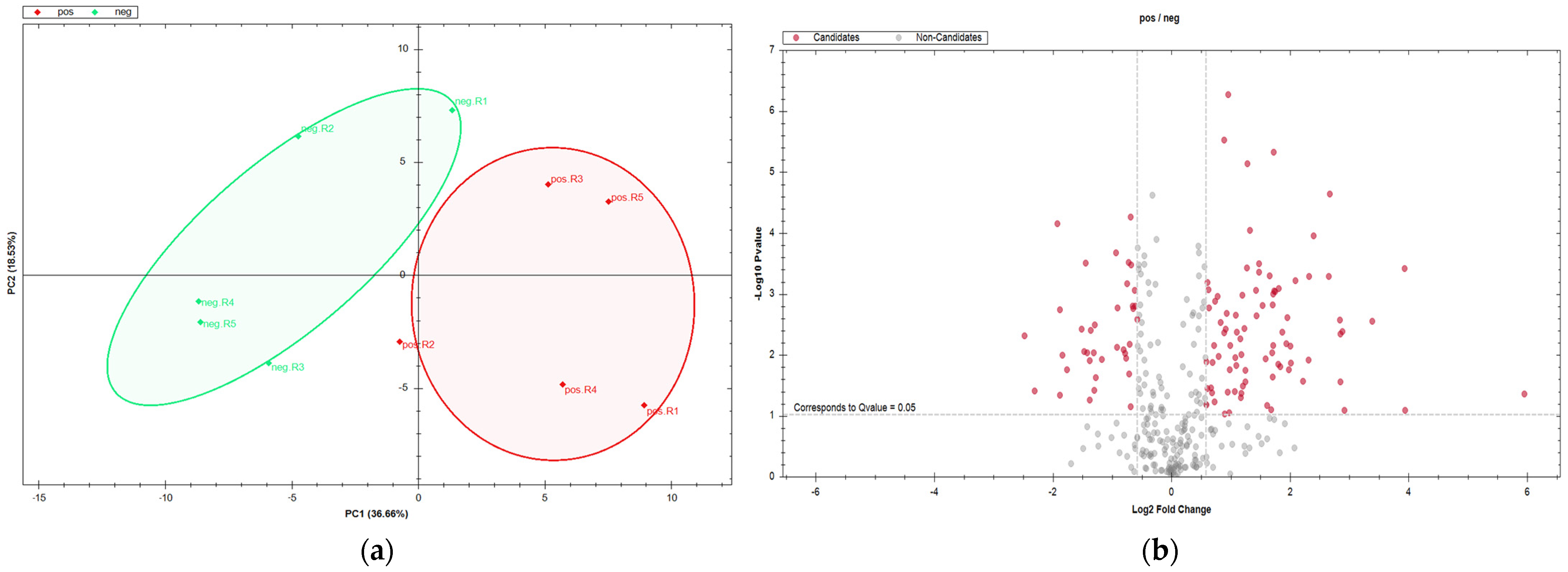
3. Results
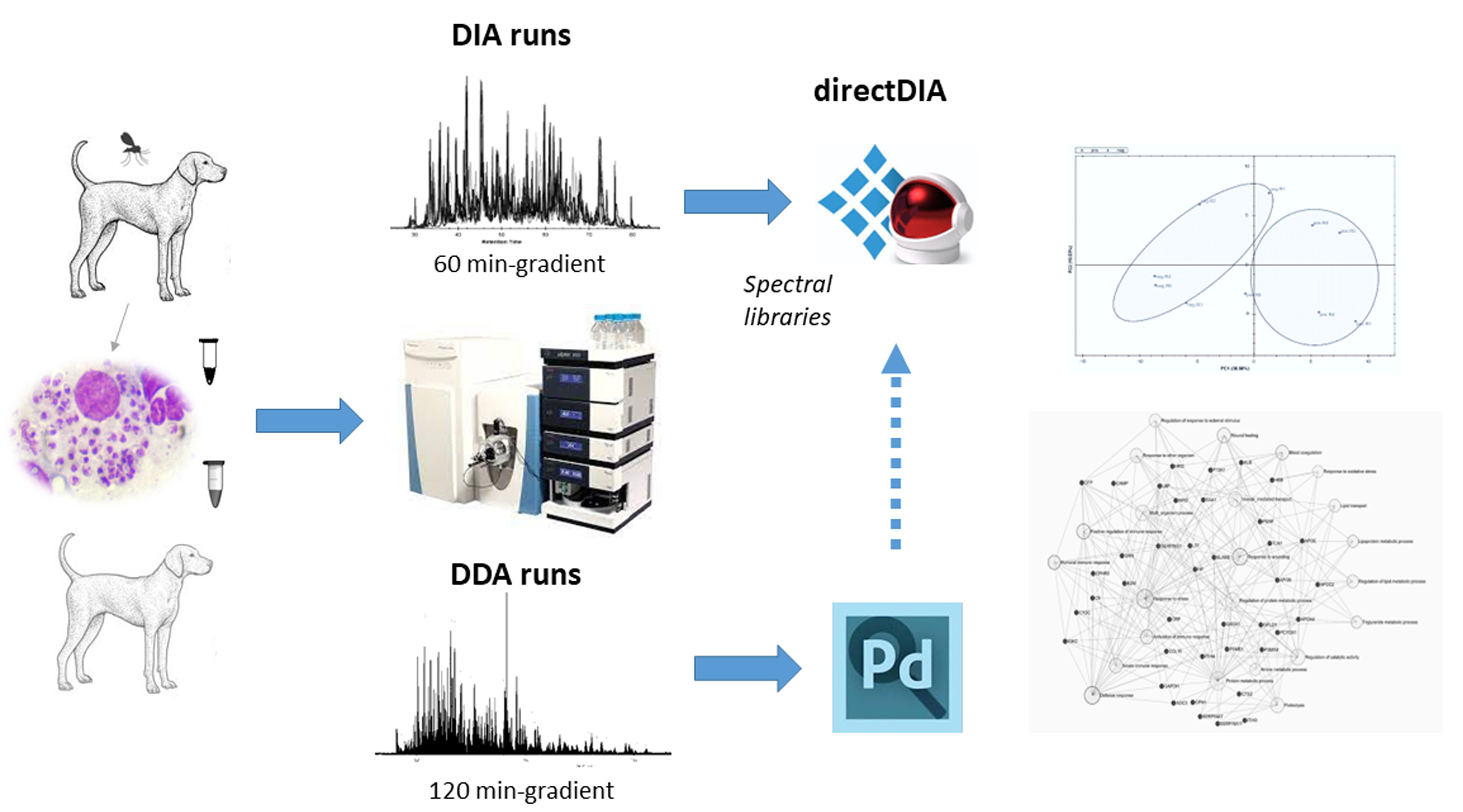
3.1. DDA-Based Proteomic Results
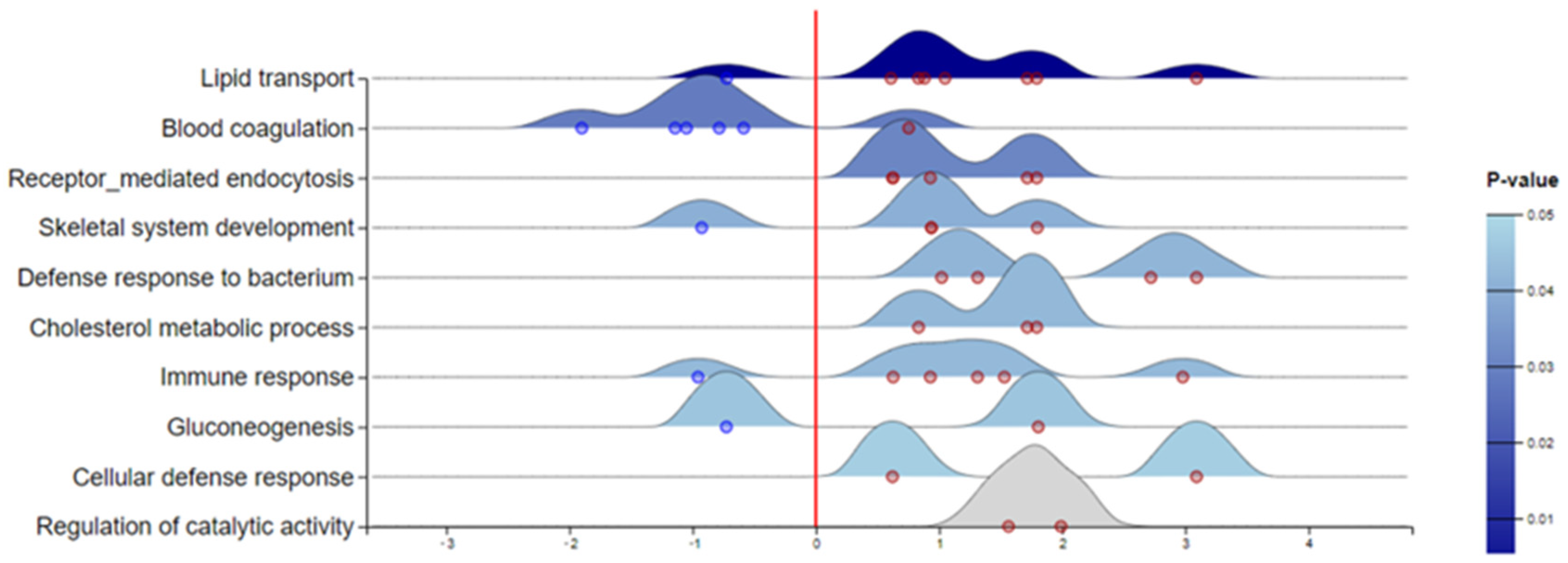
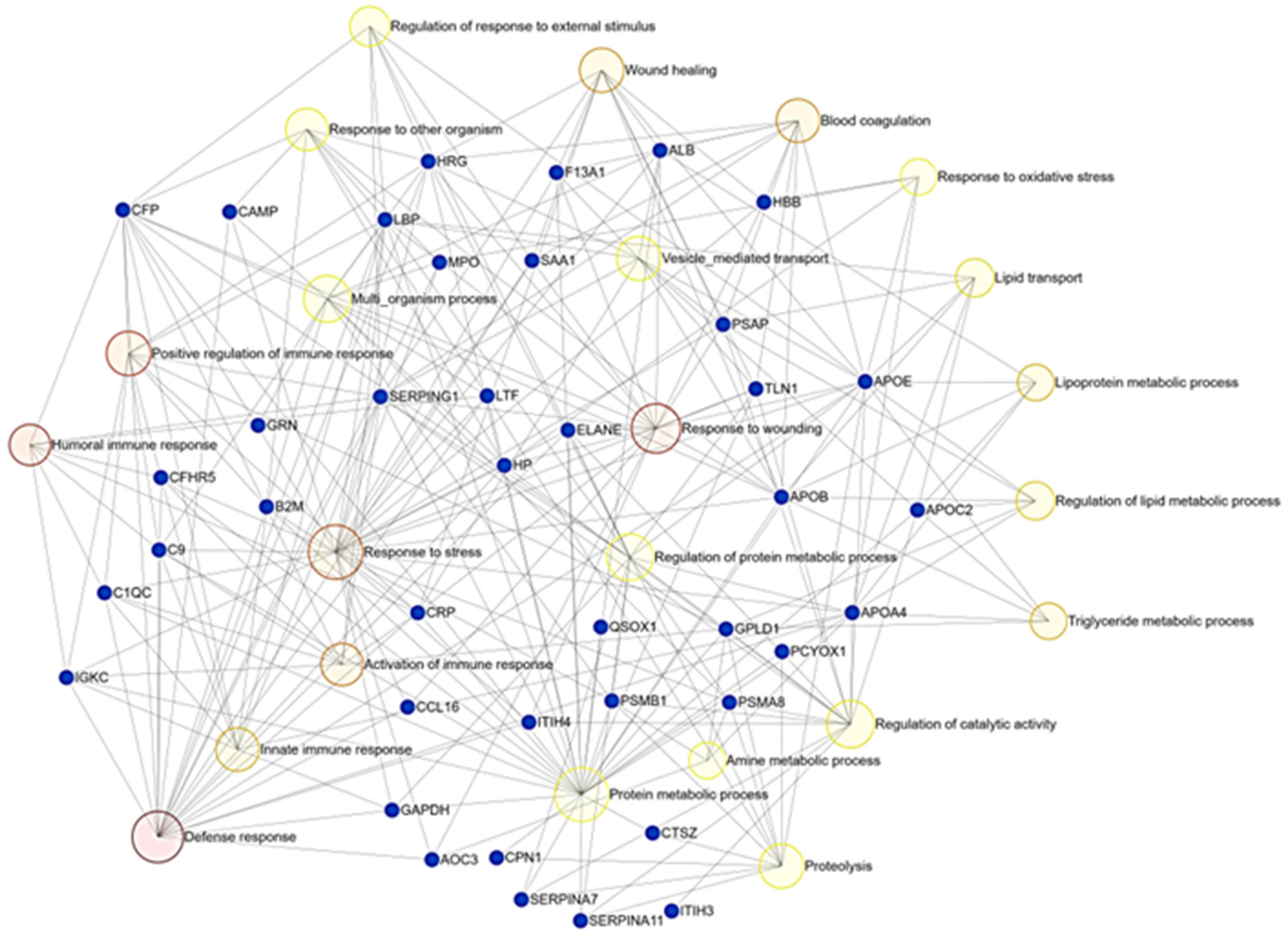
3.2. DIA-Based Proteomic Results
4. Discussion
4.1. Lipid Metabolism and Transport
4.2. Hematological Abnormalities
4.3. Immune Response
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| A2M | alpha 2 macroglobulin |
| ACN | acetonitrile |
| AGC | automatic gain control |
| Apo A4 | apolipoprotein A4 |
| Apo E | apolipoprotein E |
| APP | acute phase protein |
| ATT | alpha 1 antitrypsin |
| BCA | bicinchoninic acid |
| BP | biological process |
| CA1 | carbonic anhydrase 1 |
| CD8+ T cells | cytotoxic T lymphocytes |
| CRP | C-reactive protein, or pentraxin |
| CTSS | cathepsin S |
| DDA | data-dependent acquisition |
| DIA-MS | data-independent acquisition mass spectrometry |
| DTT | dithiothreitol |
| ELISA | enzyme-linked immunosorbent assay |
| FC | fold change |
| FDR | false discovery rate |
| GO | gene ontology |
| HB | haptoglobin |
| HCD | higher energy collisional dissociation |
| HRG | histidine-rich glycoprotein |
| IAA | iodoacetamide |
| IFAT | indirect fluorescence antibody test |
| IFN-γ | interferon gamma |
| IPC | inositol phosphorylceramide |
| iRT | indexed retention time |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| LDL | low density lipoprotein |
| Lp-PLA2 | lipoprotein-associated phospholipase A2 |
| MF | molecular function |
| MWCO | moleculat weight cut off |
| NCE | normalized collision energy |
| PAF | platelet-activating factor |
| PCA | principal component analysis |
| PSAP | prosaposin |
| rK39 | kinesin-related conserved recombinant antigen |
| SCX | strong cation exchange |
| SWATH | sequential window acquisition of all theoretical mass spectra |
| TG | triglyceride |
| TLR9 | toll-like receptor 9 |
| TMT | tandem mass tag |
| TNF-α | tumor necrosis factor alpha |
References
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Fürsch, J.; Kammer, K.-M.; Kreft, S.G.; Beck, M.; Stengel, F. Proteome-wide structural probing of low-abundant protein interactions by crosslinking mass spectrometry. Anal. Chem. 2020, 92, 4016–4022. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.; Veenstra, T.D. Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis. Separations 2022, 9, 89. [Google Scholar] [CrossRef]
- Košiček, M.; Gudelj, I.; Horvatić, A.; Jović, T.; Vučković, F.; Lauc, G.; Hećimović, S.; Kosicek, M.; Gudelj, I.; Horvatic, A.; et al. N-glycome of the lysosomal glycocalyx is altered in Niemann-Pick Type C disease model cells. Mol. Cell. Proteom. 2018, 17, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteom. 2012, 12, 1111–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Zhu, Y.; Xuan, Y.; Gao, H.; Cai, X.; Piersma, S.R.; Pham, T.V.; Schelfhorst, T.; Haas, R.R.G.D.; Bijnsdorp, I.V.; et al. DPHL: A DIA Pan-human Protein Mass Spectrometry Library for Robust Biomarker Discovery. Genom. Proteom. Bioinform. 2020, 18, 104–119. [Google Scholar] [CrossRef]
- Scarpini, S.; Dondi, A.; Totaro, C.; Biagi, C.; Melchionda, F.; Zama, D.; Pierantoni, L.; Gennari, M.; Campagna, C.; Prete, A.; et al. Visceral Leishmaniasis: Epidemiology, Diagnosis, and Treatment Regimens in Different Geographical Areas with a Focus on Pediatrics. Microorganisms 2022, 10, 1887. [Google Scholar] [CrossRef]
- Bosnić, S.; Gradoni, L.; Khoury, C.; Maroli, M. A review of leishmaniasis in Dalmatia (Croatia) and results from recent surveys on phlebotomine sandflies in three southern counties. Acta Trop. 2006, 99, 42–49. [Google Scholar] [CrossRef]
- SUNDAR, S.; SINGH, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef]
- Abass, E.; Kang, C.; Martinkovic, F.; Semião-Santos, S.J.; Sundar, S.; Walden, P.; Piarroux, R.; el Harith, A.; Lohoff, M.; Steinhoff, U. Heterogeneity of Leishmania donovani Parasites Complicates Diagnosis of Visceral Leishmaniasis: Comparison of Different Serological Tests in Three Endemic Regions. PLoS ONE 2015, 10, e0116408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvatic, A.; Guillemin, N.; Kaab, H.; McKeegan, D.; O’Reilly, E.; Bain, M.; Kules, J.; Eckersall, P.D. Quantitative proteomics using tandem mass tags in relation to the acute phase protein response in chicken challenged with Escherichia coli lipopolysaccharide endotoxin. J. Proteom. 2019, 192, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Li, L. Dynamic Range Compression with ProteoMinerTM: Principles and Examples BT. In Proteomic Profiling: Methods and Protocols; Posch, A., Ed.; Springer: New York, NY, USA, 2015; pp. 99–107. ISBN 978-1-4939-2550-6. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N. V Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef]
- Merrell, K.; Southwick, K.; Graves, S.W.; Esplin, M.S.; Lewis, N.E.; Thulin, C.D. Analysis of low-abundance, low-molecular-weight serum proteins using mass spectrometry. J. Biomol. Tech. 2004, 15, 238–248. [Google Scholar] [PubMed]
- Martinez-Subiela, S.; Horvatic, A.; Escribano, D.; Pardo-Marin, L.; Kocaturk, M.; Mrljak, V.; Burchmore, R.; Ceron, J.J.; Yilmaz, Z. Identification of novel biomarkers for treatment monitoring in canine leishmaniosis by high-resolution quantitative proteomic analysis. Vet. Immunol. Immunopathol. 2017, 191, 60–67. [Google Scholar] [CrossRef]
- Lin, L.; Zheng, J.; Yu, Q.; Chen, W.; Xing, J.; Chen, C.; Tian, R. High throughput and accurate serum proteome profiling by integrated sample preparation technology and single-run data independent mass spectrometry analysis. J. Proteom. 2018, 174, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ravuri, H.G.; Noor, Z.; Mills, P.C.; Satake, N.; Sadowski, P. Data-Independent Acquisition Enables Robust Quantification of 400 Proteins in Non-Depleted Canine Plasma. Proteomes 2022, 10, 9. [Google Scholar] [CrossRef]
- Siyal, A.A.; Chen, E.S.-W.; Chan, H.-J.; Kitata, R.B.; Yang, J.-C.; Tu, H.-L.; Chen, Y.-J. Sample Size-Comparable Spectral Library Enhances Data-Independent Acquisition-Based Proteome Coverage of Low-Input Cells. Anal. Chem. 2021, 93, 17003–17011. [Google Scholar] [CrossRef]
- Keller, M.M.; Kelly, P.H.; Pope, R.M.; Thery, F.; Wilson, M.E. Proteomic analysis of exosomes released from Leishmania infected macrophages. J. Immunol. 2020, 204, 156.19. [Google Scholar] [CrossRef]
- Martínez, C.R.; Ruiz, C.J. Alterations in Host Lipid Metabolism Produced During Visceral Leishmaniasis Infections. Curr. Trop. Med. Rep. 2019, 6, 250–255. [Google Scholar] [CrossRef]
- Bouazizi-Ben Messaoud, H.; Guichard, M.; Lawton, P.; Delton, I.; Azzouz-Maache, S. Changes in Lipid and Fatty Acid Composition During Intramacrophagic Transformation of Leishmania donovani Complex Promastigotes into Amastigotes. Lipids 2017, 52, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semini, G.; Paape, D.; Paterou, A.; Schroeder, J.; Barrios-Llerena, M.; Aebischer, T. Changes to cholesterol trafficking in macrophages by Leishmania parasites infection. Microbiologyopen 2017, 6, e00469. [Google Scholar] [CrossRef]
- Liberopoulos, E.N.; Apostolou, F.; Gazi, I.F.; Kostara, C.; Bairaktari, E.T.; Tselepis, A.D.; Elisaf, M. Visceral leishmaniasis is associated with marked changes in serum lipid profile. Eur. J. Clin. Investig. 2014, 44, 719–727. [Google Scholar] [CrossRef]
- Woods, A.G.; Sokolowska, I.; Taurines, R.; Gerlach, M.; Dudley, E.; Thome, J.; Darie, C.C. Potential biomarkers in psychiatry: Focus on the cholesterol system. J. Cell. Mol. Med. 2012, 16, 1184–1195. [Google Scholar] [CrossRef]
- Ghosh, J.; Das, S.; Guha, R.; Ghosh, D.; Naskar, K.; Das, A.; Roy, S. Hyperlipidemia offers protection against Leishmania donovani infection: Role of membrane cholesterol. J. Lipid Res. 2012, 53, 2560–2572. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, L.; Zhang, Z.; Feng, L.; Song, X.; Wu, J. Apolipoprotein A-IV involves in glucose and lipid metabolism of rat. Nutr. Metab. 2019, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- VerHague, M.A.; Cheng, D.; Weinberg, R.B.; Shelness, G.S. Apolipoprotein A-IV expression in mouse liver enhances triglyceride secretion and reduces hepatic lipid content by promoting very low density lipoprotein particle expansion. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2501–2508. [Google Scholar] [CrossRef] [Green Version]
- Tsimihodimos, V.; Kei, A.; Apostolou, F.; Elisaf, M. Diagnostic lipid changes in patients with visceral leishmaniasis. Hosp. Pract. 2018, 46, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, O.; Wilson, M.C.; Xu, W.; Hsu, F.-F.; Turk, J.; Kuhlmann, F.M.; Wang, Y.; Soong, L.; Key, P.; Beverley, S.M.; et al. Degradation of host sphingomyelin is essential for Leishmania virulence. PLoS Pathog. 2009, 5, e1000692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doering, T.; Holleran, W.M.; Potratz, A.; Vielhaber, G.; Elias, P.M.; Suzuki, K.; Sandhoff, K. Sphingolipid Activator Proteins Are Required for Epidermal Permeability Barrier Formation. J. Biol. Chem. 1999, 274, 11038–11045. [Google Scholar] [CrossRef] [Green Version]
- Kagan, T.; Stoyanova, G.; Lockshin, R.A.; Zakeri, Z. Ceramide from sphingomyelin hydrolysis induces neuronal differentiation, whereas de novo ceramide synthesis and sphingomyelin hydrolysis initiate apoptosis after NGF withdrawal in PC12 Cells. Cell Commun. Signal. 2022, 20, 15. [Google Scholar] [CrossRef]
- Ghosh, S.; Bhattacharyya, S.; Das, S.; Raha, S.; Maulik, N.; Das, D.K.; Roy, S.; Majumdar, S. Generation of ceramide in murine macrophages infected with Leishmania donovani alters macrophage signaling events and aids intracellular parasitic survival. Mol. Cell. Biochem. 2001, 223, 47–60. [Google Scholar] [CrossRef]
- Almeida, V.; Lima, I.; Fraga, D.; Carrillo, E.; Moreno, J.; Dos-Santos, W.L.C. Hematological Changes in Dogs with Visceral Leishmaniasis Are Associated with Increased IFN-γ and TNF Gene Expression Levels in the Bone Marrow. Microorganisms 2021, 9, 1618. [Google Scholar] [CrossRef]
- Menteşe, A.; Erkut, N.; Sümer, A.; Us Altay, D.; Alver, A.; Sönmez, M. Anti-carbonic anhydrase antibodies in iron deficiency anemia. Hematology 2015, 20, 363–367. [Google Scholar] [CrossRef]
- Shiferaw, E.; Murad, F.; Tigabie, M.; Abebaw, M.; Alemu, T.; Abate, S.; Mohammed, R.; Yeshanew, A.; Tajebe, F. Hematological profiles of visceral leishmaniasis patients before and after treatment of anti-leishmanial drugs at University of Gondar Hospital; Leishmania Research and Treatment Center Northwest, Ethiopia. BMC Infect. Dis. 2021, 21, 1005. [Google Scholar] [CrossRef]
- Wakabayashi Takehiko, S.K. Histidine-Rich Glycoprotein: A Possible Modulator of Coagulation and Fibrinolysis. Semin. Thromb. Hemost. 2011, 37, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Lekic, N.; Tadic, B.; Djordjevic, V.; Basaric, D.; Micev, M.; Vucelic, D.; Mitrovic, M.; Grubor, N. Splenectomy for Visceral Leishmaniasis Out of an Endemic Region: A Case Report and Literature Review. Medicina (B. Aires) 2022, 58, 184. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M. Platelet-activating factor acetylhydrolase from human plasma. In Arachidonate Related Lipid Mediators; Murphy, R.C., Fitzpatrick, F.A.B.T.-M., Eds.; Academic Press: Cambridge, MA, USA, 1990; Volume 187, pp. 344–357. ISBN 0076-6879. [Google Scholar]
- Lonardoni, M.V.; Russo, M.; Jancar, S. Essential role of platelet-activating factor in control of Leishmania (Leishmania) amazonensis infection. Infect. Immun. 2000, 68, 6355–6361. [Google Scholar] [CrossRef] [PubMed]
- Ceron, J.J.; Pardo-Marin, L.; Caldin, M.; Furlanello, T.; Solano-Gallego, L.; Tecles, F.; Bernal, L.; Baneth, G.; Martinez-Subiela, S. Use of acute phase proteins for the clinical assessment and management of canine leishmaniosis: General recommendations. BMC Vet. Res. 2018, 14, 196. [Google Scholar] [CrossRef]
- Martínez-Subiela, S.; Tecles, F.; Eckersall, P.D.; Cerón, J.J. Serum concentrations of acute phase proteins in dogs with leishmaniasis. Vet. Rec. 2002, 150, 241–244. [Google Scholar] [CrossRef]
- Brode, S.K.; Ling, S.C.; Chapman, K.R. Alpha-1 antitrypsin deficiency: A commonly overlooked cause of lung disease. C. Can. Med. Assoc. J. 2012, 184, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Kavarnos, I.; Pardali, D.; Brellou, G.D.; Papadopoulos, E.; Kritsepi-Konstantinou, M.; Adamama-Moraitou, K.K. Bronchoscopy and Lung Fine-Needle Aspiration for Antemortem Evaluation of Pulmonary Involvement in Dogs with Naturally Occurring Canine Leishmaniosis. Pathogens 2022, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.E.; Maschalidi, S.; Colisson, R.; Heslop, L.; Ghirelli, C.; Sakka, E.; Lennon-Duménil, A.-M.; Amigorena, S.; Cabanie, L.; Manoury, B. Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity 2009, 31, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Abou Fakher, F.H.; Rachinel, N.; Klimczak, M.; Louis, J.; Doyen, N. TLR9-dependent activation of dendritic cells by DNA from Leishmania major favors Th1 cell development and the resolution of lesions. J. Immunol. 2009, 182, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Rasid, O.; Mériaux, V.; Khan, E.M.; Borde, C.; Ciulean, I.S.; Fitting, C.; Manoury, B.; Cavaillon, J.-M.; Doyen, N. Cathepsin B-Deficient Mice Resolve Leishmania major Inflammation Faster in a T Cell-Dependent Manner. PLoS Negl. Trop. Dis. 2016, 10, e0004716. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, R.D.A.; Williams, R.; Scott, C.J.; Burden, R.E. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, L.; Yang, J.; Ren, A.; Sun, Z.; Yan, G.; Sun, J.; Fu, H.; Xu, W.; Hu, C.; et al. Increased serum cathepsin S in patients with atherosclerosis and diabetes. Atherosclerosis 2006, 186, 411–419. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot Accession ID | Protein Name | Gene Name | Log2FC | q-Value * |
|---|---|---|---|---|
| F1PP29 | Uncharacterized protein | ND ** | −2.60 | 0.00423 |
| A0A5F4CGT3;F1PWW0 | Filamin A | FLNA | −2.42 | 0.02513 |
| Q6JDI3 | Plasminogen (Fragment) | PLG | −1.90 | 0.00002 |
| F1PBK6 | Carbonic anhydrase | CA1 | −1.88 | 0.01058 |
| E2R5U8 | Transthyretin | TTR | −1.56 | 0.03280 |
| E2R1N7 | Prenylcysteine oxidase 1 | PCYOX1 | −1.51 | 0.00106 |
| F1PCG4 | Peroxiredoxin 2 | PRDX2 | −1.49 | 0.00102 |
| E2RT38;F1PAQ3 | Maltase-glucoamylase | MGAM | −1.47 | 0.00048 |
| A0A5F4D5S2;J9PAD1 | C4a anaphylatoxin | LOC481722 | −1.41 | 0.00000 |
| F1PZC6 | Histidine rich glycoprotein | HRG | −1.14 | 0.00139 |
| A0A5F4C5S3;F1PNR5 | Coagulation factor XI | F11 | −1.05 | 0.00722 |
| E2RA67 | C-C motif chemokine | CCL14 | −0.96 | 0.00516 |
| F1PD34 | Secreted phosphoprotein 2 | SPP2 | −0.93 | 0.00371 |
| A1ILJ0 | Alpha 1 antitrypsin | SERPINA1 | −0.92 | 0.01290 |
| A0A5F4CEP4 | Fetuin B | FETUB | −0.90 | 0.00197 |
| A0A5F4D9Z3;F1PB85 | Serpin family A member 7 | SERPINA7 | −0.88 | 0.03724 |
| D2K841;J9P346 | C-X-C motif chemokine; | PPBP | −0.87 | 0.00888 |
| E2RPW3 | Paraoxonase | PON3 | −0.85 | 0.00029 |
| A0A5F4BY85;A0A5F4CHL2; E2RK02 | Glycosyl-phosphatidylinositol-specific phospholipase D | GPLD1 | −0.84 | 0.00129 |
| A0A5F4CZV0;F2Z4P4;J9P9I7 | Elongation factor 1-alpha | LOC102155289 | −0.83 | 0.02608 |
| A0A5F4DCK5;F1PKX3 | Coagulation factor XIII A chain | F13A1 | −0.78 | 0.04243 |
| Q28275-1 | Isoform 1 of Fibronectin | FN1 | −0.77 | 0.00018 |
| A0A5F4CT11;E2RE97 | Numb-like protein | COQ8B | −0.77 | 0.00044 |
| A0A5F4CD78;A0A5F4DFV6;E2R3V1 | Fc receptor like 4 | FCRL4 | −0.74 | 0.03556 |
| A0A5F4CED7;F1Q4D9 | Plasma retinol-binding protein | RBP4 | −0.73 | 0.01947 |
| F1P8Z5 | Apolipoprotein B | APOB | 1.72 | 0.00011 |
| F1PNY2 | Immunoglobulin kappa | IGKC | 1.73 | 0.00066 |
| A0A5F4D392;A0A5F4D5Q6;J9NX46 | Clathrin light chain A | CLTA | 1.73 | 0.01765 |
| Q2EG92 | Lubricin (Fragment) | PRG4 | 1.79 | 0.00138 |
| A0A5S7EUL7;P18649 | Apolipoprotein E | APOE | 1.79 | 0.00194 |
| A0A5F4BSV4;A0A5F4C284;A0A5F4C8H6;A0A5F4CPB8;F1PIA3 | Fibrillin 1 | FBN1 | 1.80 | 0.02898 |
| F1PBT3;J9P7A6 | Fructose-bisphosphate aldolase | ALDOA | 1.81 | 0.00606 |
| A0A5F4CYW8;E2R6Q7 | Cathepsin B | CTSB | 1.99 | 0.00071 |
| A0A5F4C3X0;A0A5F4CAZ5;E2QY07 | Actinin alpha 1 | ACTN1 | 2.05 | 0.04209 |
| F1PKL6;L7N0K1 | Ig-like domain-containing protein | ND | 2.44 | 0.00506 |
| E2R5J0;T2KEN6 | Pentaxin | CRP | 2.45 | 0.00301 |
| J9NT12 | Fibrinogen like 1 | FGL1 | 2.47 | 0.01586 |
| A0A5F4D8N3;A0A5F4DDP8 | Lymphocyte cytosolic protein 1 | LCP1 | 2.50 | 0.00036 |
| E2RJY0 | Potassium channel tetramerization domain containing 12 | KCTD12 | 2.67 | 0.01294 |
| F1PQ52 | Myeloperoxidase | MPO | 2.72 | 0.01008 |
| E2RHN1 | Granulin precursor | GRN | 2.84 | 0.00137 |
| A0A5F4C236;A0A5F4CUH8;A0A5F4DFI4;E2RLA5 | Golgi membrane protein 1 | GOLM1 | 2.92 | 0.00622 |
| F1PAK0;Q8HY81 | Cathepsin S | CTSS | 2.98 | 0.00043 |
| E2QW61 | Lipopolysaccharide-binding protein | LBP | 3.09 | 0.00238 |
| E2R8C5 | Immunoglobulin lambda variable 2-33 | IGLV2-33 | 4.04 | 0.00158 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinković, F.; Popović, M.; Smolec, O.; Mrljak, V.; Eckersall, P.D.; Horvatić, A. Data Independent Acquisition Reveals In-Depth Serum Proteome Changes in Canine Leishmaniosis. Metabolites 2023, 13, 365. https://doi.org/10.3390/metabo13030365
Martinković F, Popović M, Smolec O, Mrljak V, Eckersall PD, Horvatić A. Data Independent Acquisition Reveals In-Depth Serum Proteome Changes in Canine Leishmaniosis. Metabolites. 2023; 13(3):365. https://doi.org/10.3390/metabo13030365
Chicago/Turabian StyleMartinković, Franjo, Marin Popović, Ozren Smolec, Vladimir Mrljak, Peter David Eckersall, and Anita Horvatić. 2023. "Data Independent Acquisition Reveals In-Depth Serum Proteome Changes in Canine Leishmaniosis" Metabolites 13, no. 3: 365. https://doi.org/10.3390/metabo13030365