
Tetraenone A: A New β-Ionone Derivative from Tetraena aegyptia

 , , , , ,
, , , , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
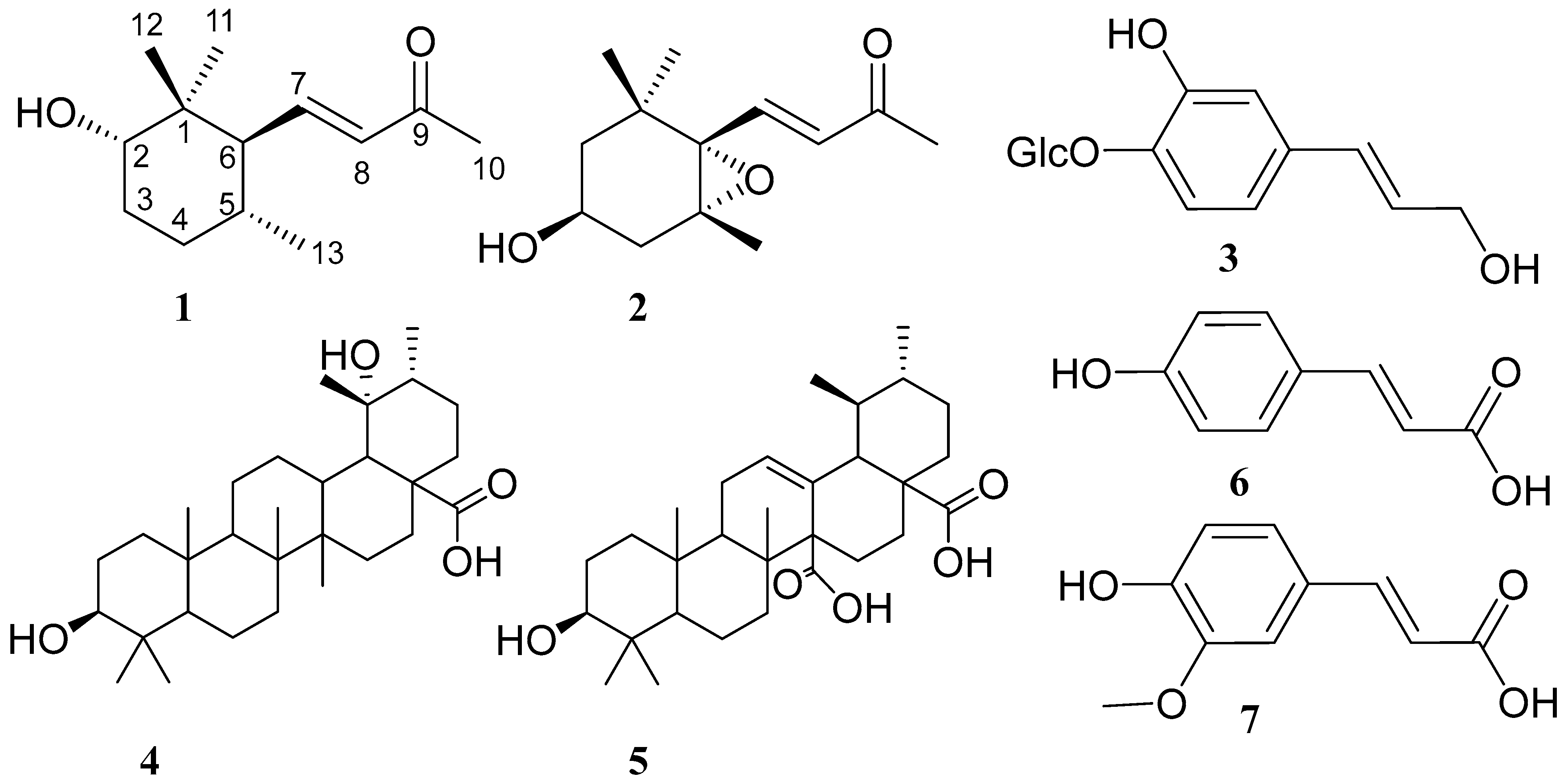
2.1. Structural Characterization of Compounds 1 and 2
2.2. Conformational Analysis of Compounds 1 and 2
2.2.1. Conformational Analysis of Compound 1
2.2.2. Conformational Analysis of Compound 2
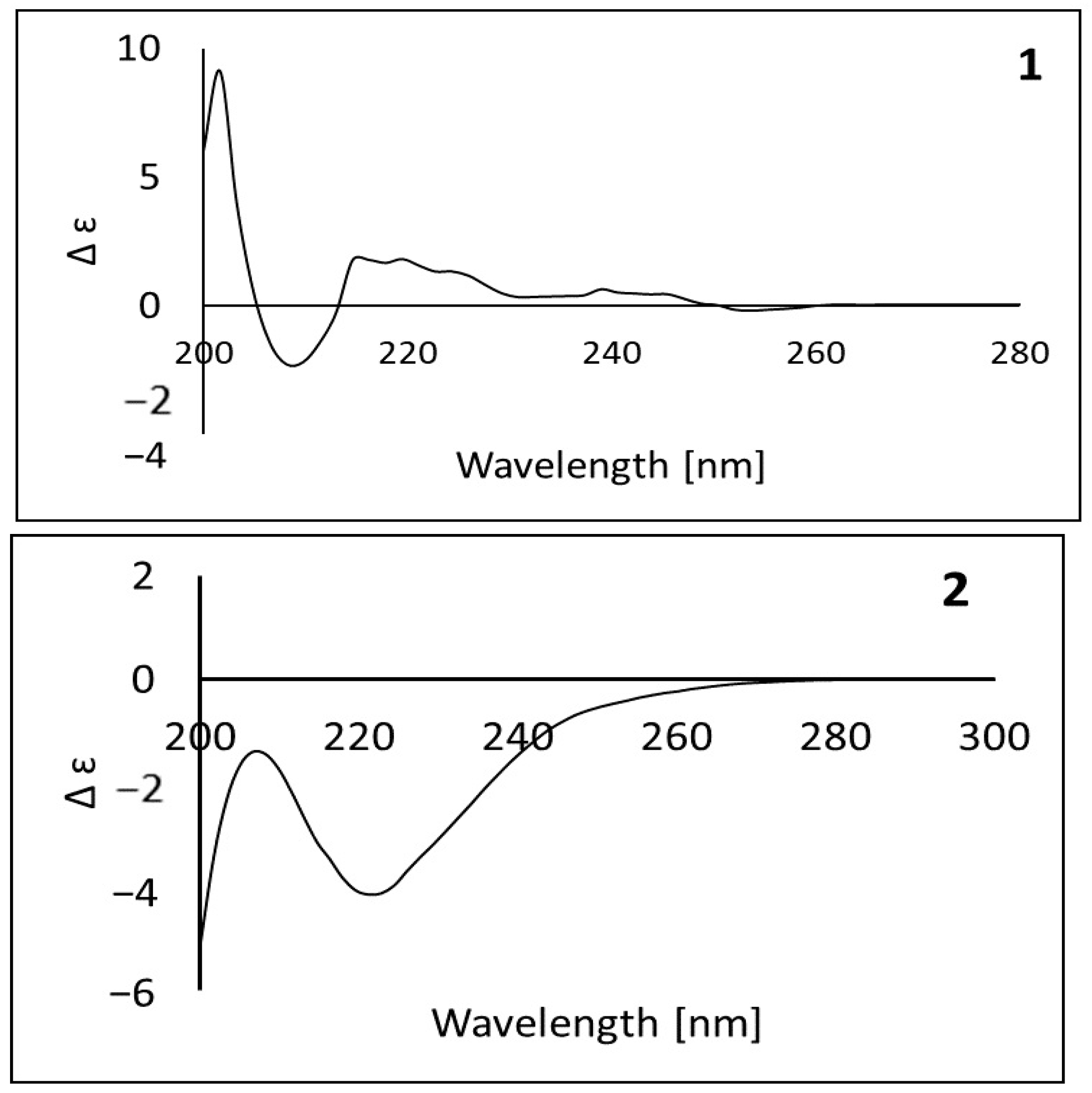
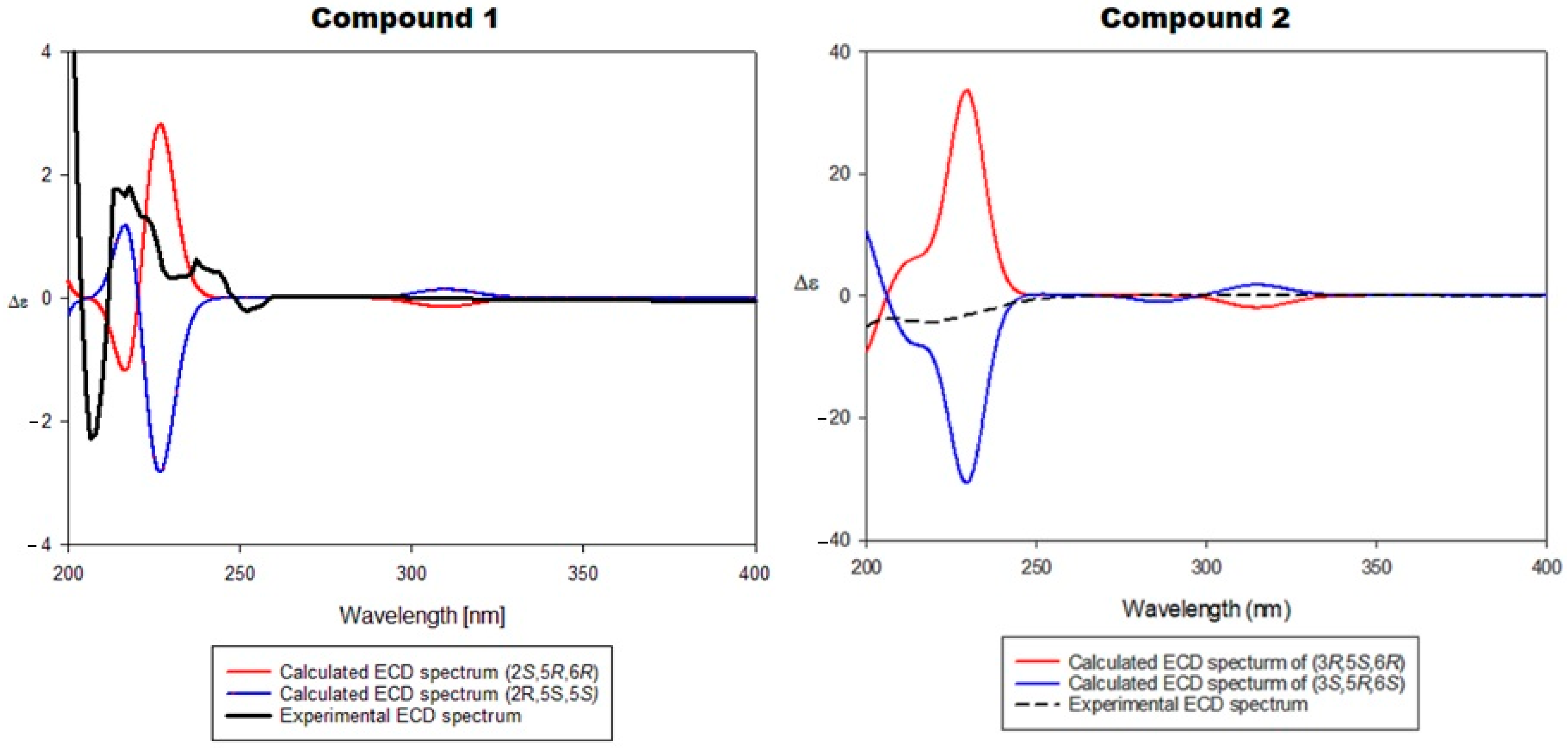
2.2.3. Assignment of the Absolute Stereochemical Configuration of Compounds 1 and 2 (ECD Spectra)
2.3. In Silico Evaluation of Bioactivity of Compounds 1 and 2
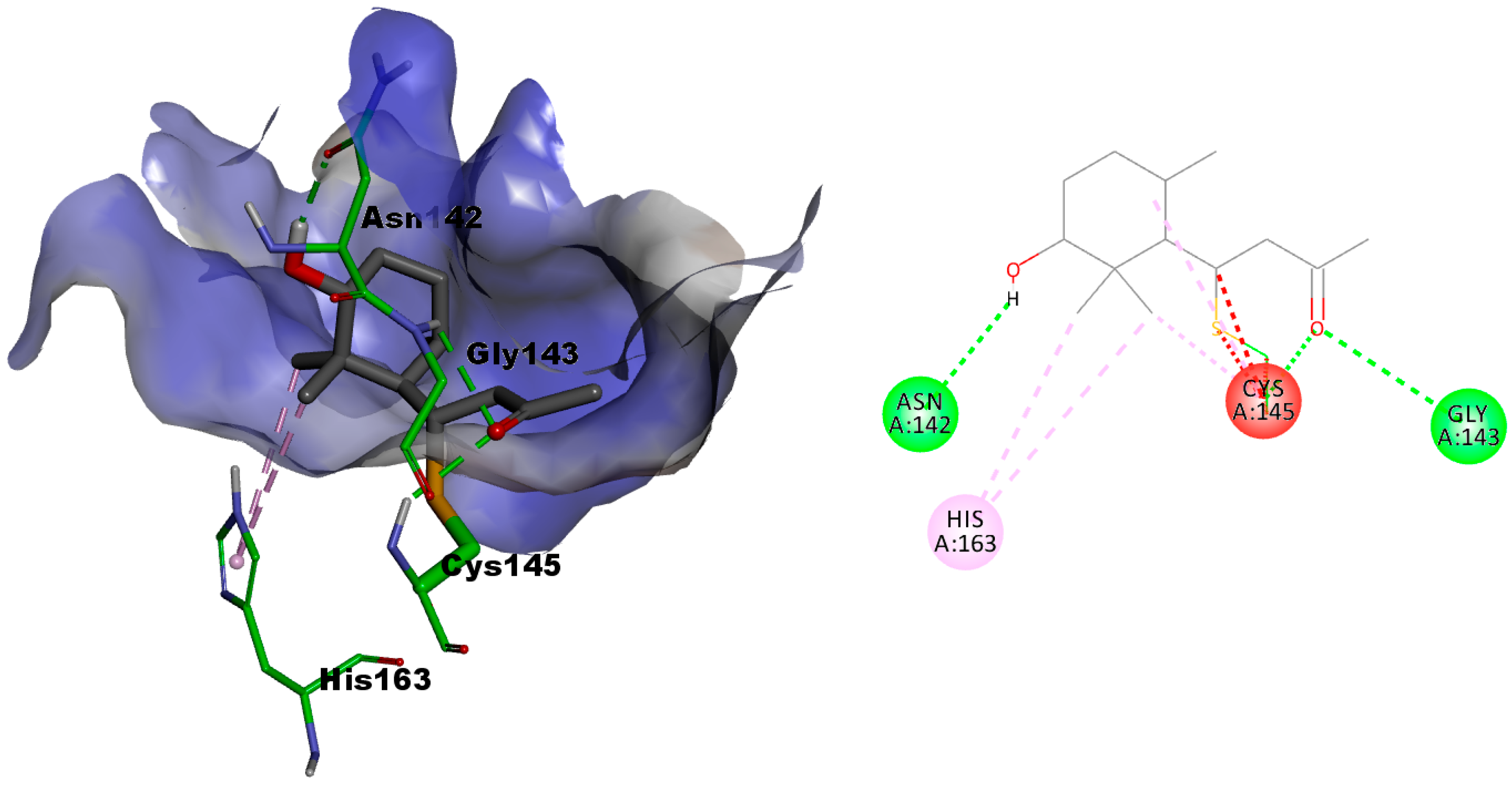
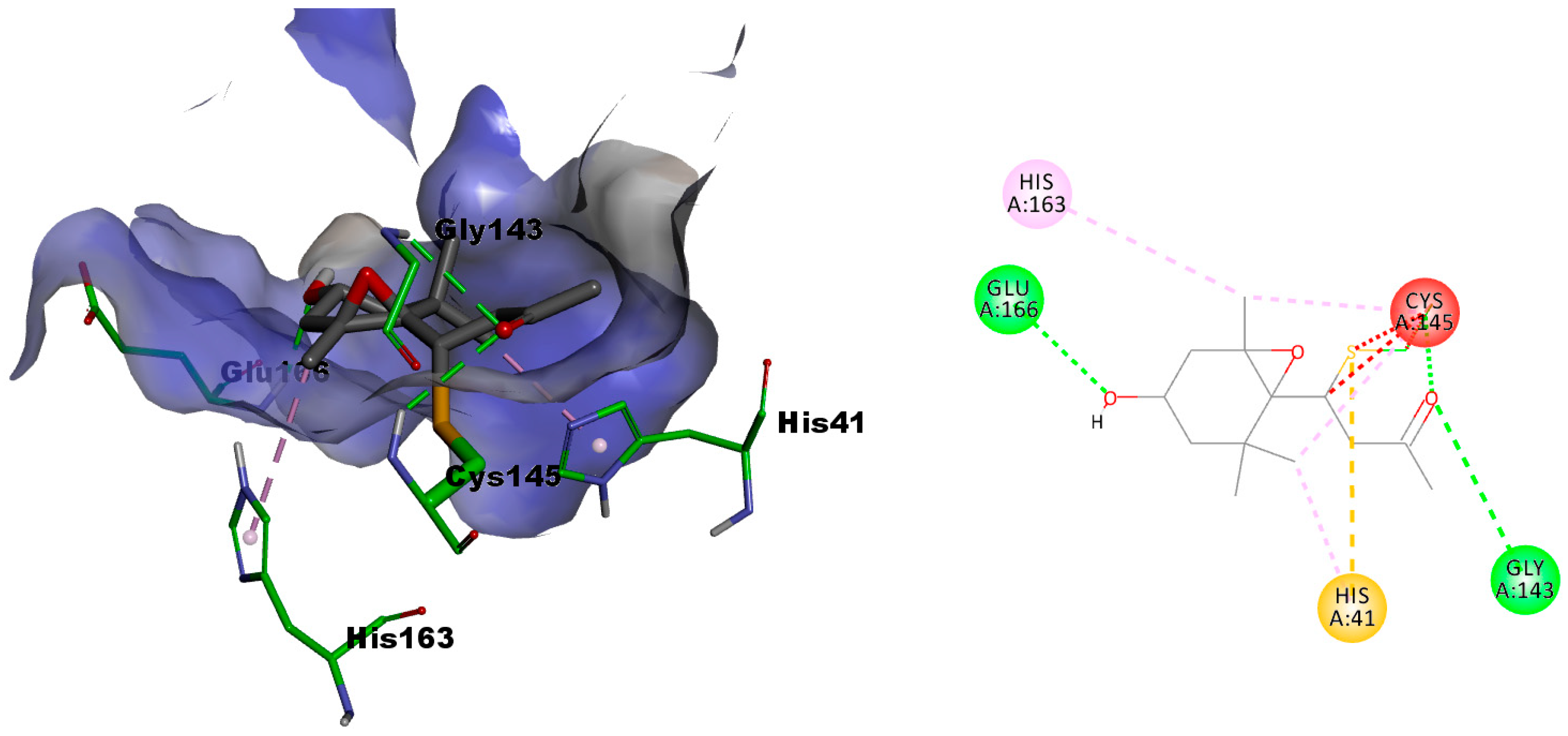
2.3.1. In Silico Evaluation of Compounds 1 and 2 against SARS-CoV-2 Main Protease
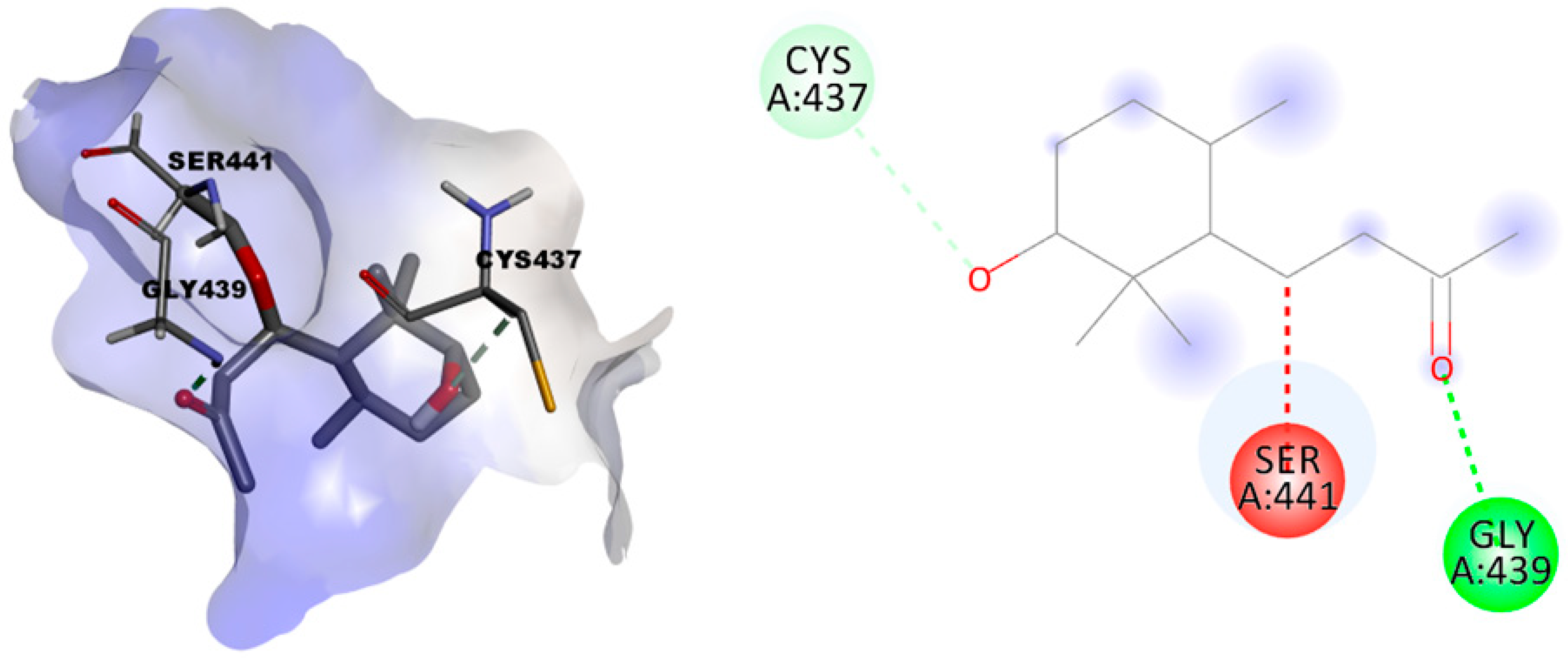
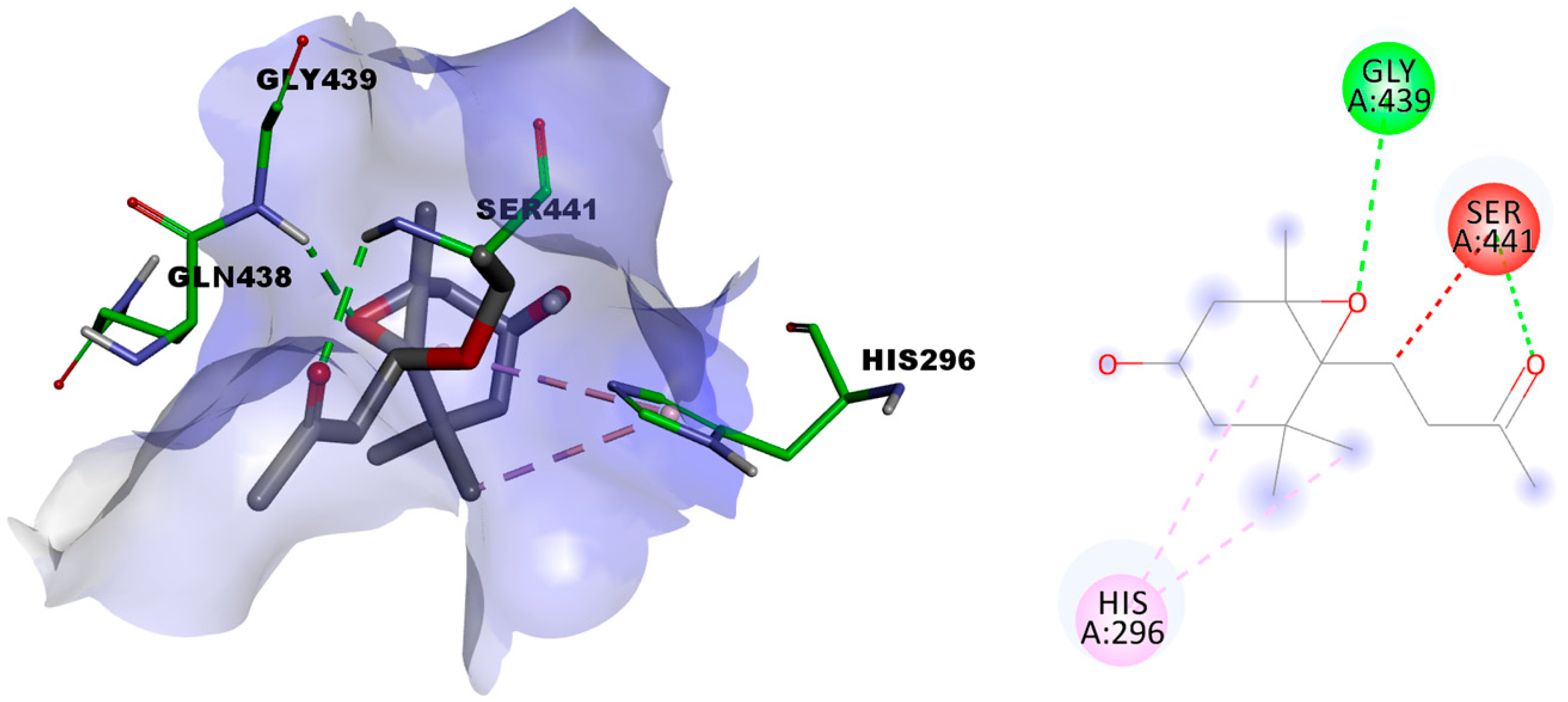
2.3.2. In Silico Evaluation of Compounds 1 and 2 against Transmembrane Serine Protease 2 (TMPRSS2)
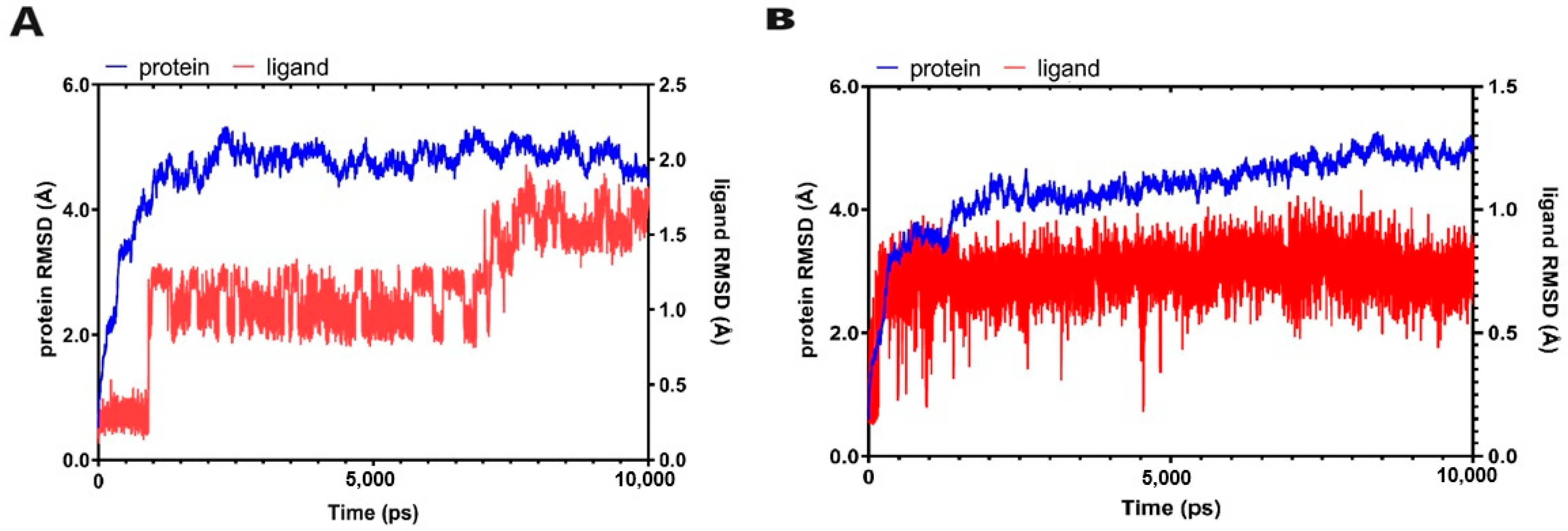
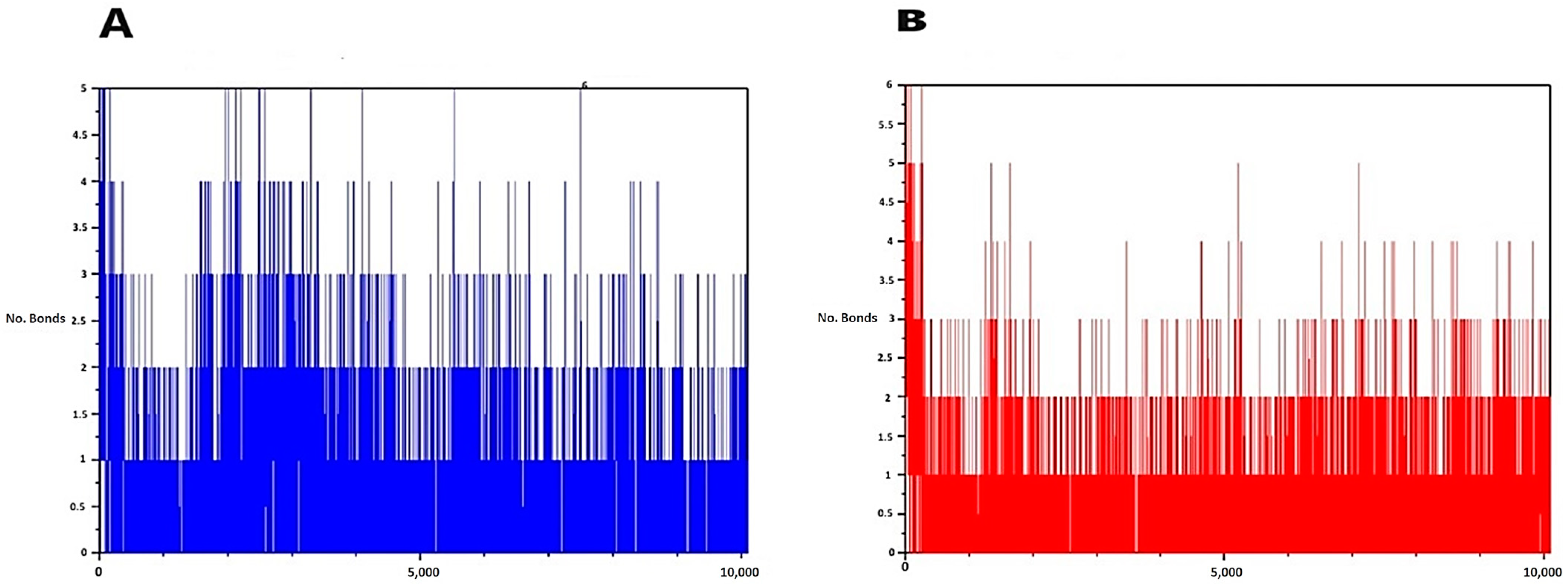
2.3.3. Molecular Dynamic Simulations
3. Materials and Methods
3.1. Plant Material
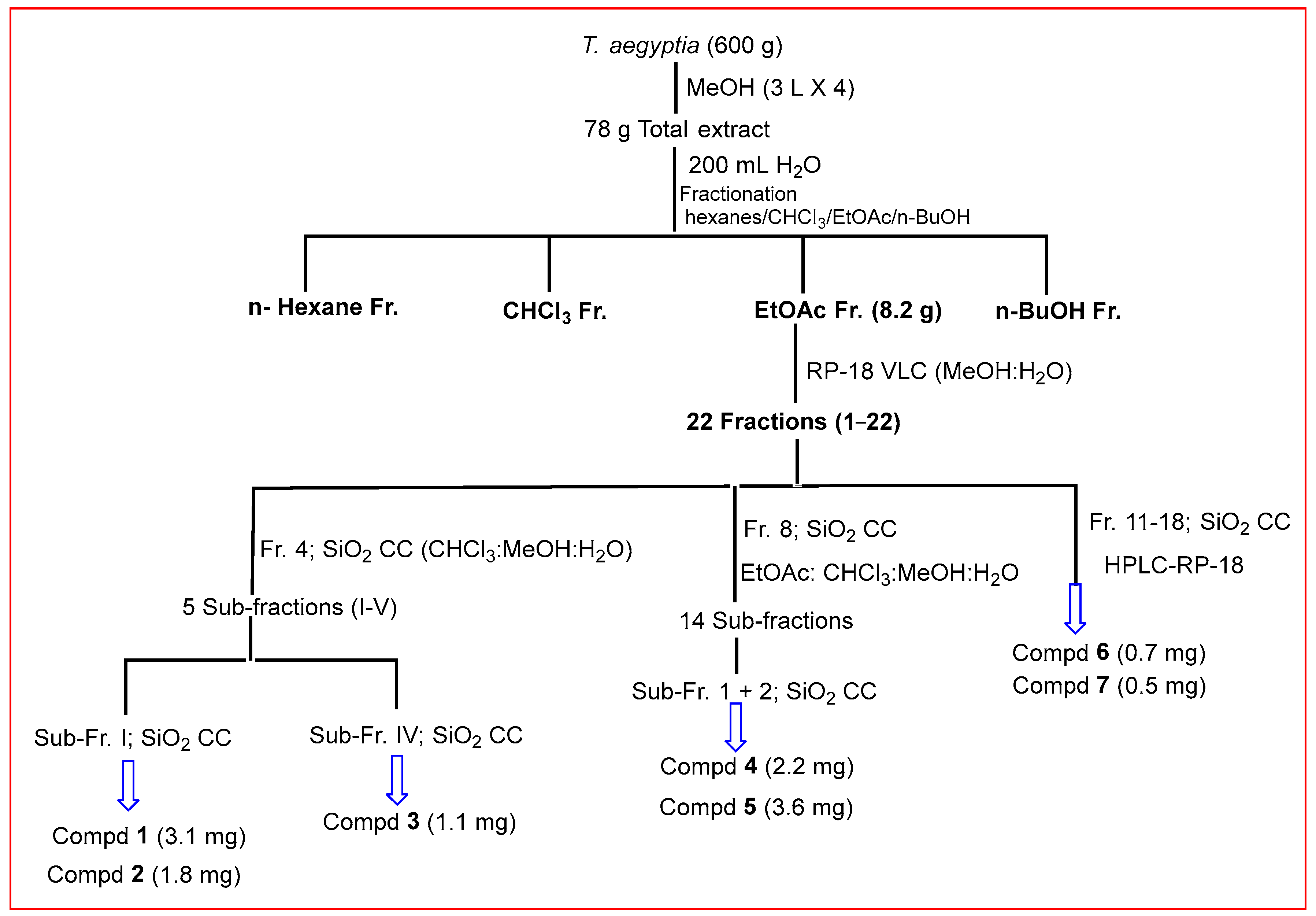
3.2. Extraction and Isolation
3.3. Conformational Analysis and Electronic Circular Dichroism (ECD) Spectra
3.4. In Silico Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shawky, E.M.; Gabr, N.M.; El-Gindi, M.R.; Mekky, R.H. A Comprehensive Review on Genus Zygophyllum. J. Adv. Pharm. Res. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Sheahan, M.; Cutler, D. Contribution of vegetative anatomy to the systematics of the Zygophyllaceae R.Br. Bot. J. Linn. Soc. 1993, 113, 227–262. [Google Scholar] [CrossRef]
- Belguidoum, M.; Dendougui, H.; Kendour, Z.; Belfar, A.; Bensaci, C.; Hadjadj, M. Antioxidant activities, phenolic, flavonoid and tannin contents of endemic Zygophyllum Cornutum Coss. From Algerian Sahara. Der Pharma Chem. 2015, 7, 312–317. [Google Scholar]
- Guenzet, A.; Krouf, D.; Zennaki, S.; Berzou, S. Zygophyllum gaetulum Aqueous Extract Protects against Diabetic Dyslipidemia and Attenuates Liver and Kidney Oxidative Damage in Streptozotocin Induced-Diabetic Rats. Int. J. Pharm. Sci. Res. 2014, 5, 4709. [Google Scholar]
- Kchaou, M.; Ben Salah, H.B.; Mhiri, R.; Allouche, N. Anti-oxidant and anti-acetylcholinesterase activities of Zygophyllum album. Bangladesh J. Pharmacol. 2016, 11, 54–62. [Google Scholar] [CrossRef]
- Pöllmann, K.; Gagel, S.; Elgamal, M.A.; Shaker, K.H.; Seifert, K. Triterpenoid saponins from the roots of Zygophyllum species. Phytochemistry 1997, 44, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.A.; Ali, Z.; El-Amier, Y.A.; Khan, I.A. A new lignan from Zygophyllum aegyptium. Magn. Reson. Chem. 2016, 54, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, C.; Gruner, M.; Tunsag, J.; Batsuren, D.; Ganpurev, B.; Chuluunnyam, L.; Sodbayar, B.; Schmidt, A.W.; Knölker, H.-J. Chemical constituents isolated from Zygophyllum melongena Bunge growing in Mongolia. Nat. Prod. Res. 2016, 30, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Ferreira, D.; Ding, Y. Determination of Absolute Configuration of Natural Products: Theoretical Calculation of Electronic Circular Dichroism as a Tool. Curr. Org. Chem. 2010, 14, 1678–1697. [Google Scholar] [CrossRef]
- Sasaki, H.; Morota, T.; Nishimura, H.; Ogino, T.; Katsuhara, T.; Sugama, K.; Chin, M.; Mitsuhashi, H. Norcarotenoids of Rehmannia glutinosa var. Hueichingensis. Phytochemistry 1991, 30, 1997–2001. [Google Scholar] [CrossRef]
- Machida, K.; Kikuchi, M. Norisoprenoids from Viburnum dilatatum. Phytochemistry 1996, 41, 1333–1336. [Google Scholar] [CrossRef]
- Weyerstahl, P.; Marschall, H.; Bork, W.-R.; Rilk, R. Megastigmanes and other constituents of the absolute of Boronia megastigma from Tasmania. Eur. J. Org. Chem. 1994, 1994, 1043–1047. [Google Scholar] [CrossRef]
- Feng, W.-S.; Li, M.; Zheng, X.-K.; Zhang, N.; Song, K.; Wang, J.-C.; Kuang, H.-X. Two new ionone glycosides from the roots of Rehmannia glutinosa Libosch. Nat. Prod. Res. 2015, 29, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhan, Y.-C.; Sha, Y.; Pei, Y.-H. Norisoprenoids from Ulva lactuca. J. Asian Nat. Prod. Res. 2007, 9, 321–325. [Google Scholar] [CrossRef]
- Lv, X.-J.; Li, Y.; Ma, S.-G.; Qu, J.; Liu, Y.-B.; Li, Y.-H.; Zhang, D.; Li, L.; Yu, S.-S. Bioactive megastigmane glucosides and monoterpenes from Lyonia ovalifolia. J. Asian Nat. Prod. Res. 2018, 21, 559–572. [Google Scholar] [CrossRef]
- Mori, K. Syntheses of optically active grasshopper ketone and dehydrovomifoliol as a synthetic support for the revised absolute configuration of (+)-abscisic acid. Tetrahedron 1974, 30, 1065–1072. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Ji, Y.-P.; Zhao, Y.; Wang, C.-G.; Hu, J.-F. Gynostemosides A–E, megastigmane glycosides from Gynostemma pentaphyllum. Phytochemistry 2010, 71, 693–700. [Google Scholar] [CrossRef]
- Wolfram, K.; Schmidt, J.; Wray, V.; Milkowski, C.; Schliemann, W.; Strack, D. Profiling of phenylpropanoids in transgenic low-sinapine oilseed rape (Brassica napus). Phytochemistry 2010, 71, 1076–1084. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Z.; Cao, P.; Tan, X.; Ding, L. Chemical constituents of Idesia polycarpa var. vestita. Nat. Prod. Res. Dev. 2003, 15, 13–14. [Google Scholar]
- Misra, L.; Laatsch, H. Triterpenoids, essential oil and photo-oxidative 28 → 13-lactonization of oleanolic acid from Lantana camara. Phytochemistry 2000, 54, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Miana, G.A.; Al-Hazimi, H.M. Assignment of the 13C NMR spectrum of quinovic acid. Phytochemistry 1986, 26, 225–227. [Google Scholar] [CrossRef]
- Soliman, G. 359. Quinovic acid from Zygophyllum coccineum, L. J. Chem. Soc. 1939, 1760–1761. [Google Scholar] [CrossRef]
- Schneider, B.; Gershenzon, J.; Graser, G.; Hölscher, D.; Schmitt, B. One-dimensional 13C NMR and HPLC-1H NMR techniques for observing carbon-13 and deuterium labelling in biosynthetic studies. Phytochem. Rev. 2003, 2, 31–43. [Google Scholar] [CrossRef]
- Pawar, D.M.; Noe, E.A. Conformational Study of Cyclohexene Oxide by Dynamic NMR Spectroscopy and Ab Initio Molecular Orbital Calculations. J. Am. Chem. Soc. 1998, 120, 1485–1488. [Google Scholar] [CrossRef]
- Todeschini, R.; Pitea, D.; Favini, G. Conformation of bicyclo[n.1.0] derivatives: Part 2. Norcarane and cyclohexene epoxide. J. Mol. Struct. 1981, 71, 279–286. [Google Scholar] [CrossRef]
- Liu, J.; Wang, R. Classification of Current Scoring Functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conf. No. | Relative Energy (kJ/mol) | Boltzmann Weights | Cumulative Boltzmann Weights | Compound 1 (α-OH) | Compound 1 (β-OH) |
|---|---|---|---|---|---|
 |  | ||||
| 1 | 0.00 | 0.406 | 0.406 |  |  |
| 2 | 0.75 | 0.300 | 0.705 |  |  |
| 3 | 3.07 | 0.117 | 0.823 |  |  |
| 4 | 3.16 | 0.114 | 0.936 |  |  |
| 5 | 7.17 | 0.023 | 0.959 |  |  |
| Conf. No. | Relative Energy (kJ/mol) | Boltzmann Weights | Cumulative Boltzmann Weights | Compound 2 (α-OH) | Compound 2 (β-OH) |
|---|---|---|---|---|---|
 |  | ||||
| 1 | 0.00 | 0.319 | 0.319 |  |  |
| 2 | 1.15 | 0.201 | 0.520 |  |  |
| 3 | 3.19 | 0.088 | 0.608 |  |  |
| 4 | 3.87 | 0.067 | 0.675 |  |  |
| 5 | 3.91 | 0.066 | 0.740 |  |  |
| 6 | 4.69 | 0.048 | 0.788 |  |  |
| 7 | 4.78 | 0.046 | 0.835 |  |  |
| 8 | 5.08 | 0.041 | 0.876 |  |  |
| 9 | 6.15 | 0.027 | 0.902 |  |  |
| 10 | 6.18 | 0.026 | 0.929 |  |  |
| 11 | 7.09 | 0.018 | 0.947 |  |  |
| 12 | 7.50 | 0.015 | 0.963 |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashour, A.; Sherif, A.E.; El-Sayed, S.M.; Kim, J.-Y.; Jang, D.S.; Anvari, A.; Farahat, A.A.; Ibrahim, S.R.M.; Mohamed, G.A.; Ainousah, B.E.; et al. Tetraenone A: A New β-Ionone Derivative from Tetraena aegyptia. Metabolites 2023, 13, 1202. https://doi.org/10.3390/metabo13121202
Ashour A, Sherif AE, El-Sayed SM, Kim J-Y, Jang DS, Anvari A, Farahat AA, Ibrahim SRM, Mohamed GA, Ainousah BE, et al. Tetraenone A: A New β-Ionone Derivative from Tetraena aegyptia. Metabolites. 2023; 13(12):1202. https://doi.org/10.3390/metabo13121202
Chicago/Turabian StyleAshour, Ahmed, Asmaa E. Sherif, Selwan M. El-Sayed, Ji-Young Kim, Dae Sik Jang, Abtin Anvari, Abdelbasset A. Farahat, Sabrin R. M. Ibrahim, Gamal A. Mohamed, Bayan E. Ainousah, and et al. 2023. "Tetraenone A: A New β-Ionone Derivative from Tetraena aegyptia" Metabolites 13, no. 12: 1202. https://doi.org/10.3390/metabo13121202