

In Vitro, In Silico and Network Pharmacology Mechanistic Approach to Investigate the α-Glucosidase Inhibitors Identified by Q-ToF-LCMS from Phaleria macrocarpa Fruit Subcritical CO2 Extract
 , , and
, , and
Abstract
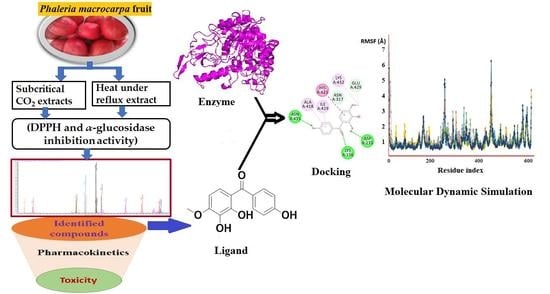
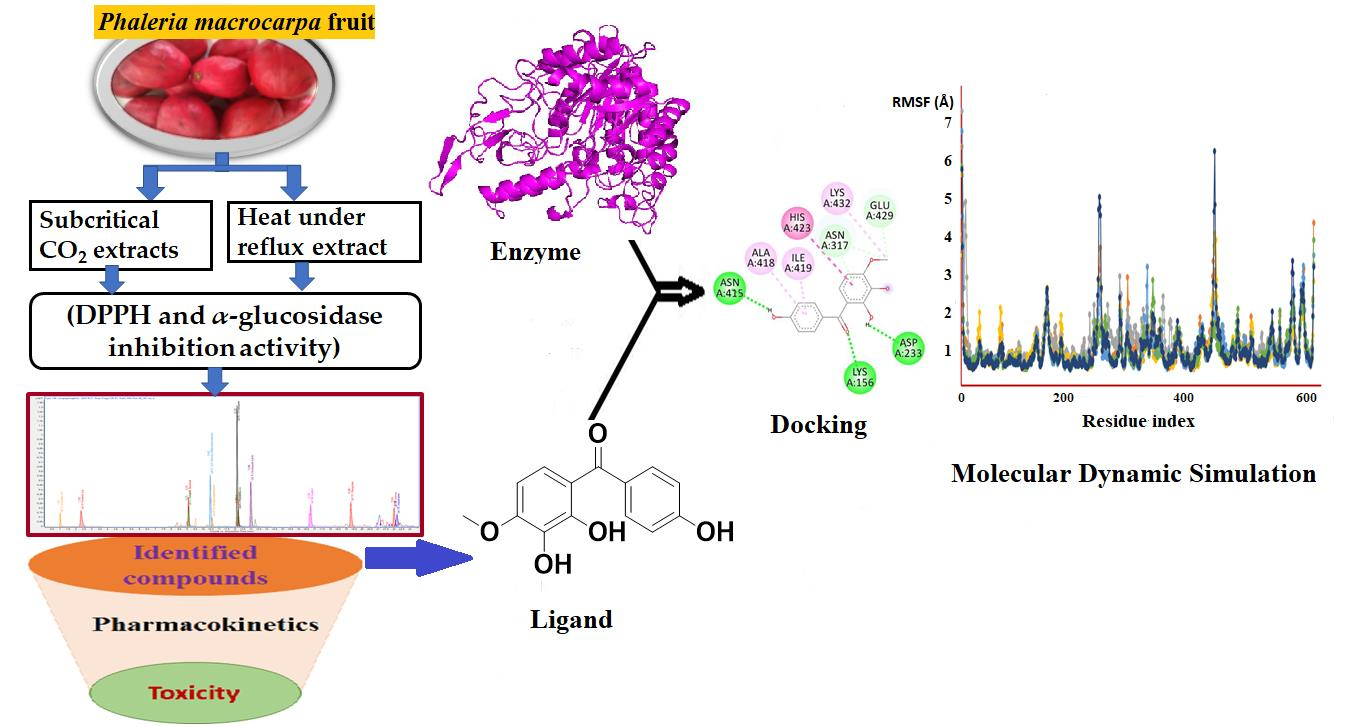
:
1. Introduction
2. Materials and Methods
2.1. Plant Material Collection and Identification
2.2. Extraction Process
2.2.1. Subcritical Carbon Dioxide Extract (SCE) Preparation
2.2.2. Heating under Reflux Extract (HRE) Preparation
2.3. Bioactivity Measurement
2.3.1. DPPH Free Radical Scavenging Activity
2.3.2. α-Glucosidase Inhibitory Activity
2.4. Detection of Bioactive Compounds of SCE-2 by Quadrupole Time-of-Flight Liquid Chromatography Mass Spectrometry (Q-ToF-LCMS)
2.5. Pharmacokinetic and Toxicity Studies
2.6. In Silico Molecular Docking Analysis
2.7. Molecular Dynamic Simulation (MDS) Approach
2.8. Compounds Target Pathway and Data Set Enrichment Analysis
2.9. Statical Analysis
3. Results
3.1. DPPH Free Radical Scavenging Activity
3.2. Enzyme Inhibition Assay (α-Glucosidase)
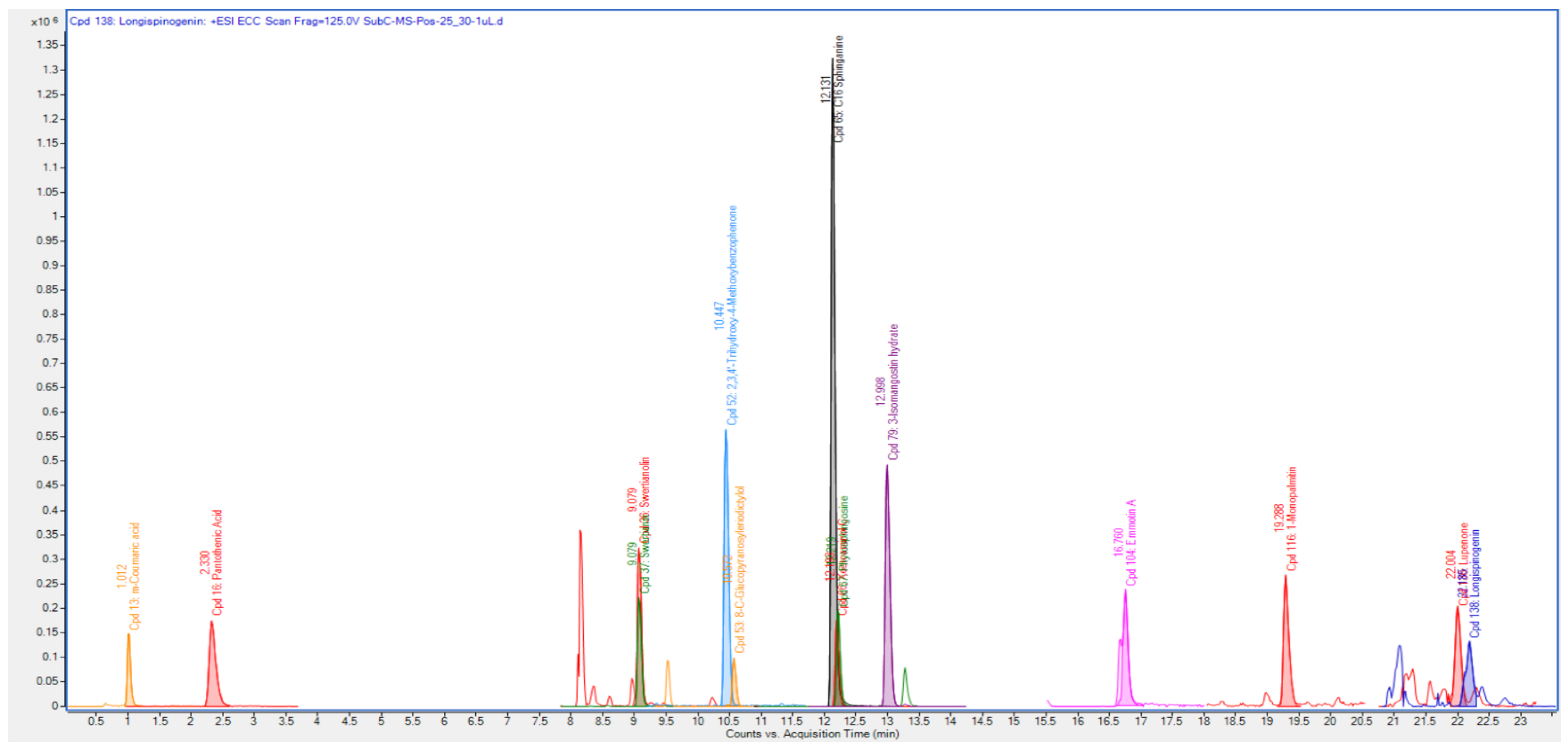
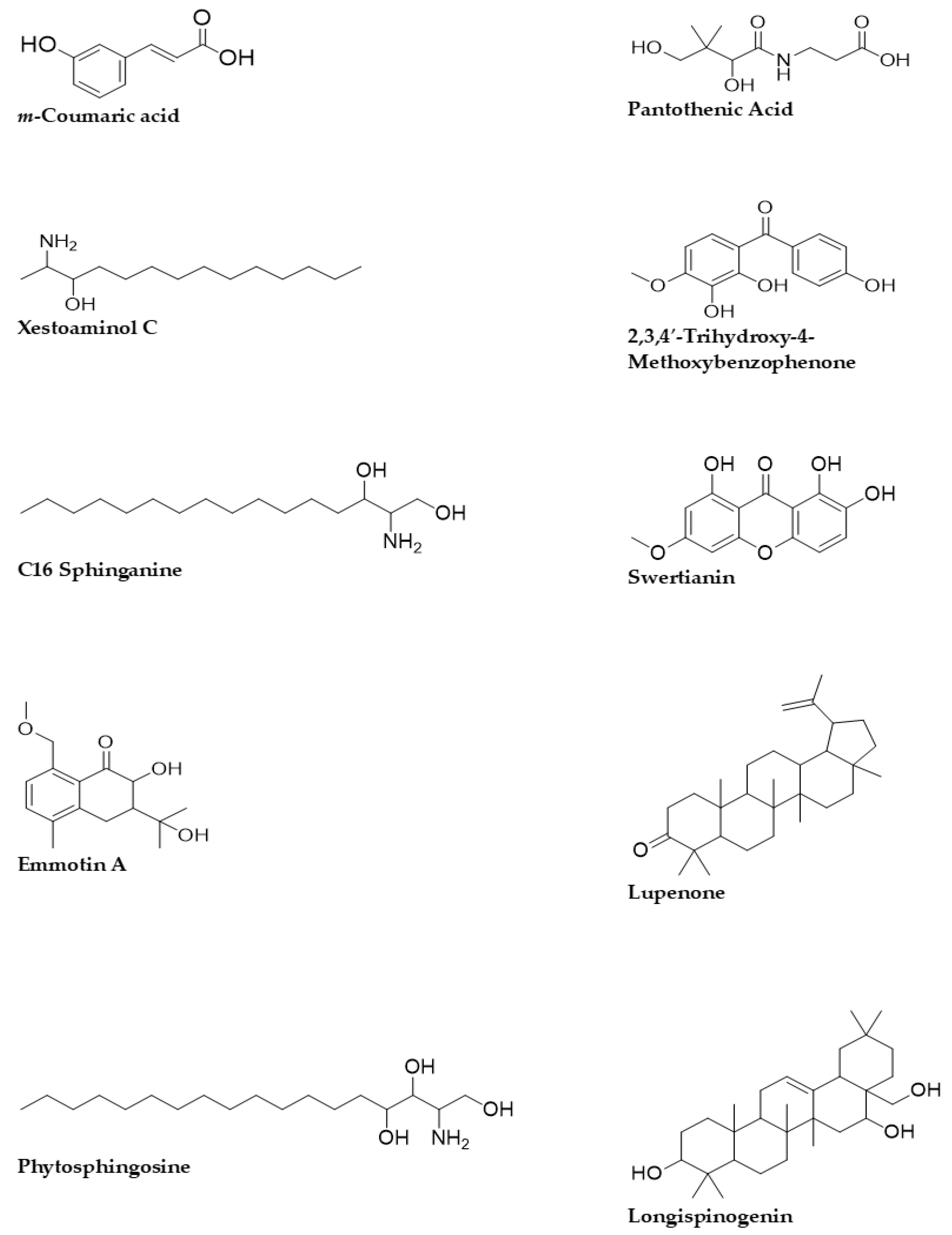
3.3. Quadrupole Time-of-Flight Liquid Chromatography Mass Spectrometry (Q-ToF-LCMS) Analysis
3.4. Physiochemical, Pharmacokinetic and Toxicity Studies
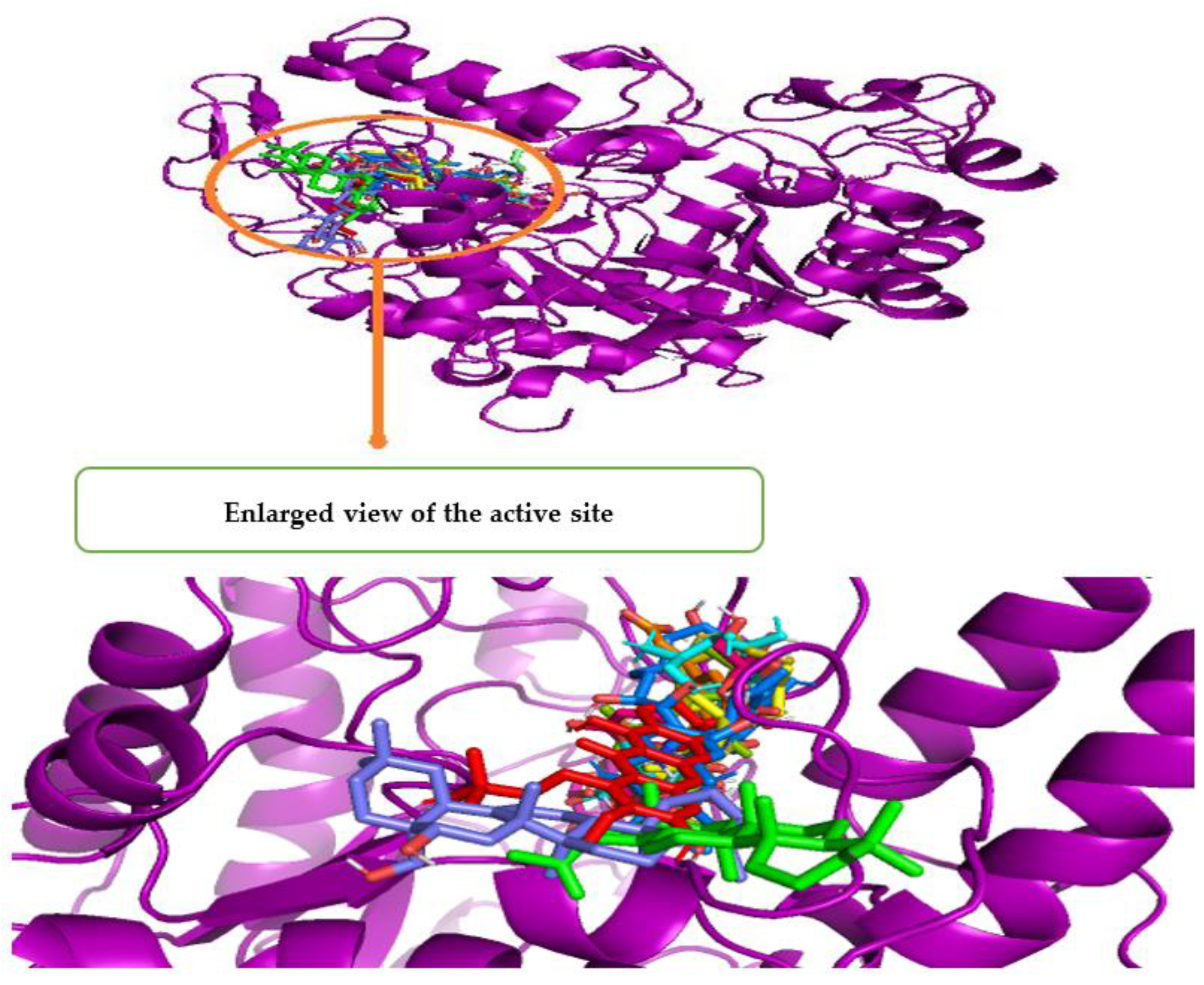
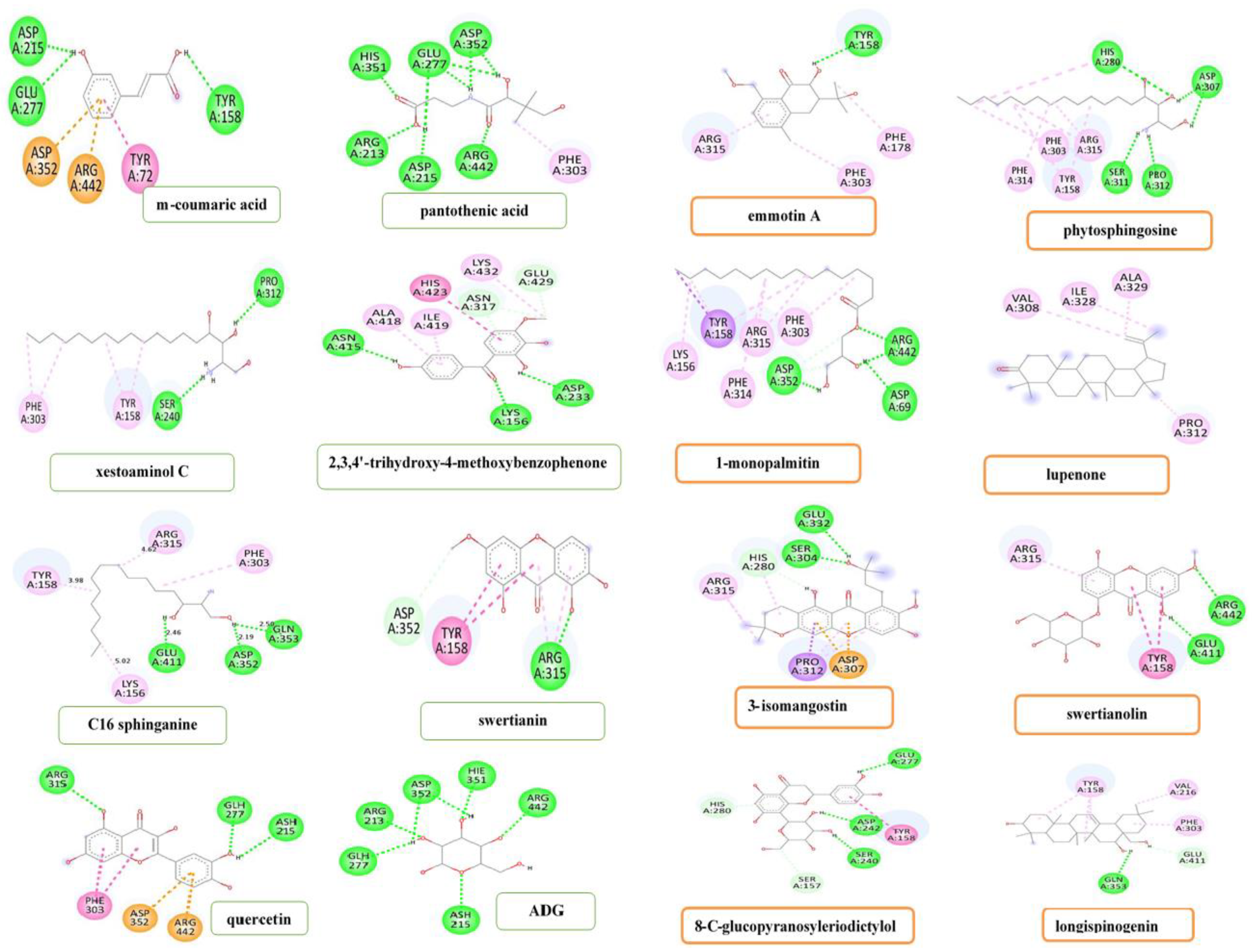
3.5. In Silico Study of Compounds Identified by Q-ToF-LCMS Analysis
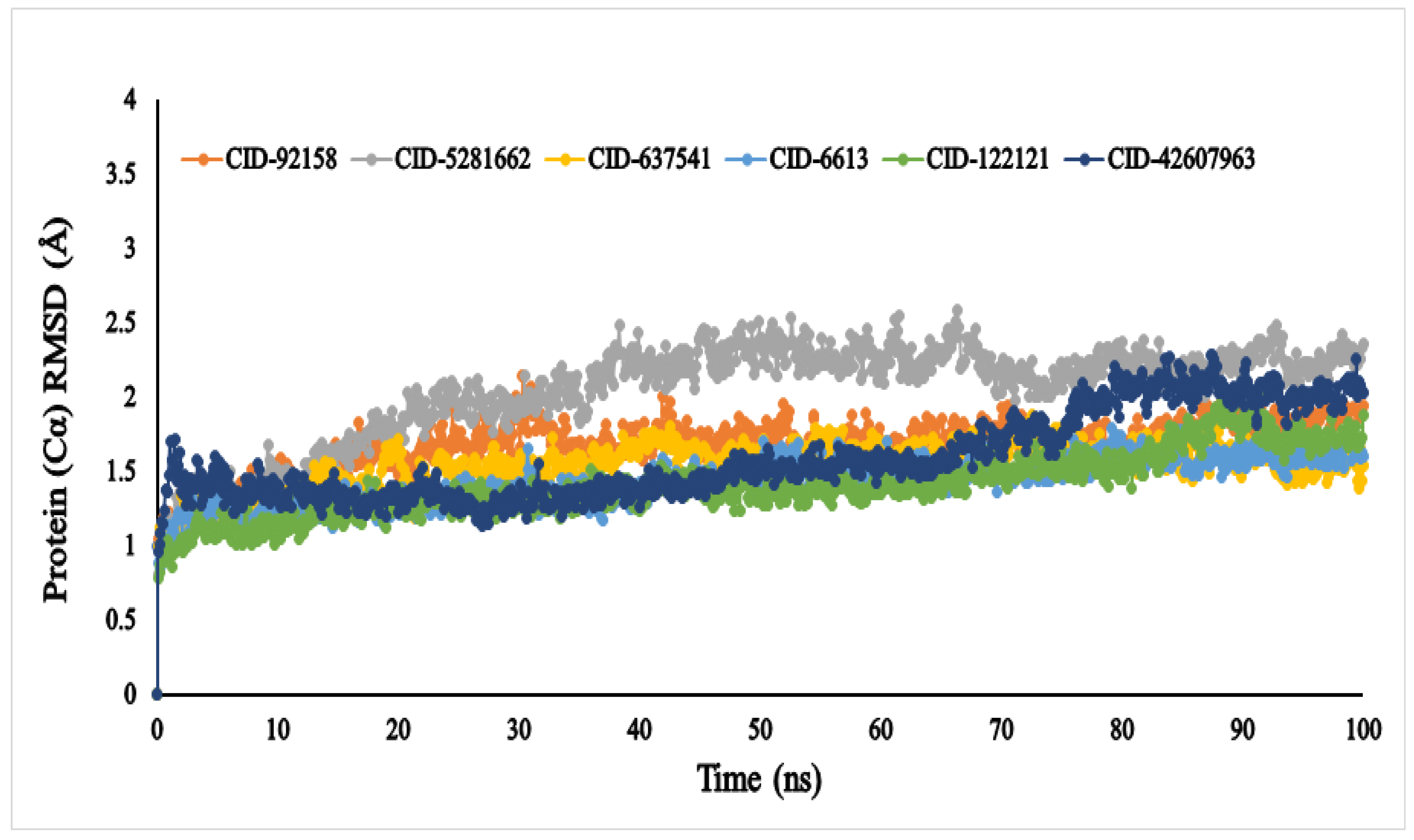
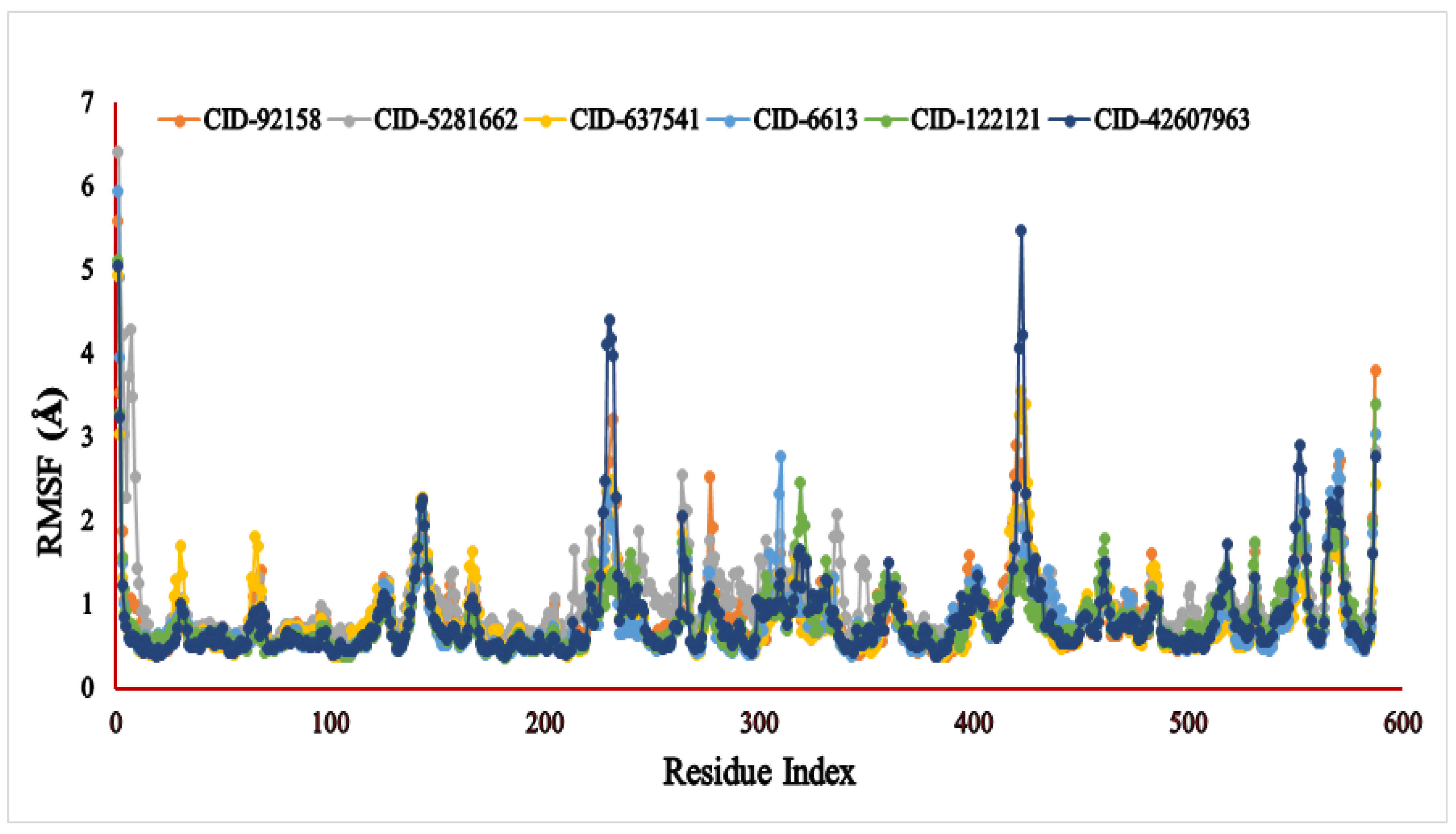
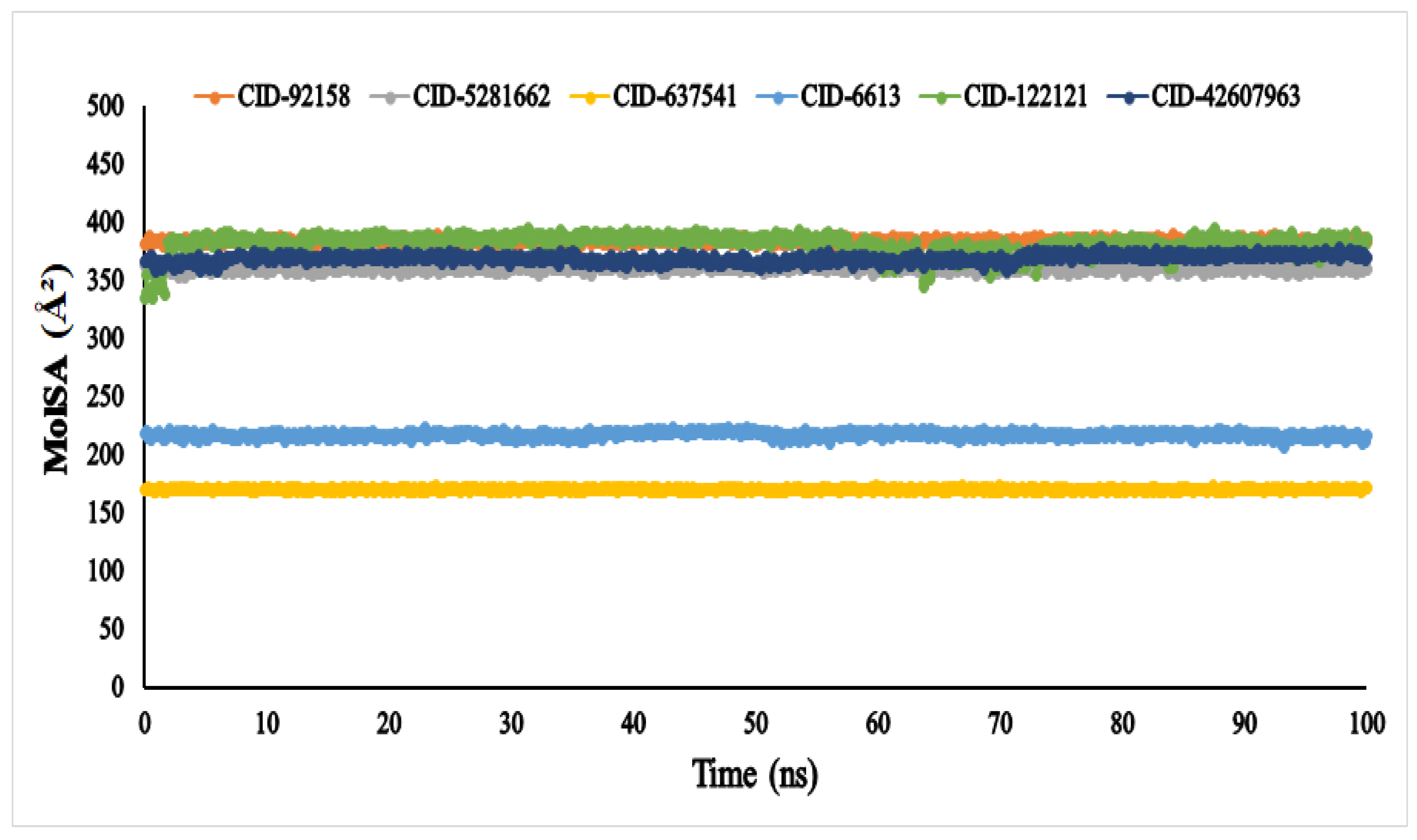
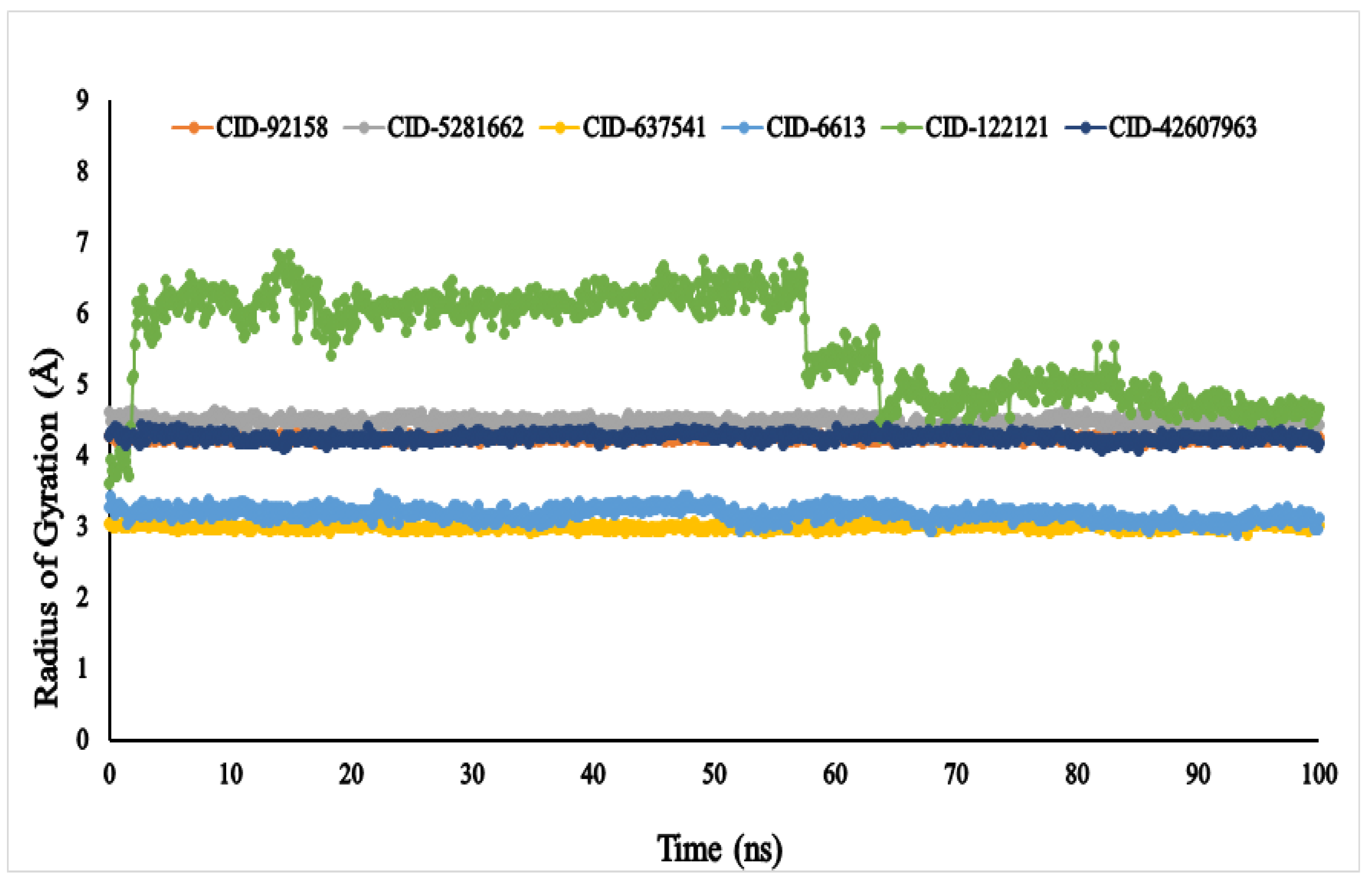
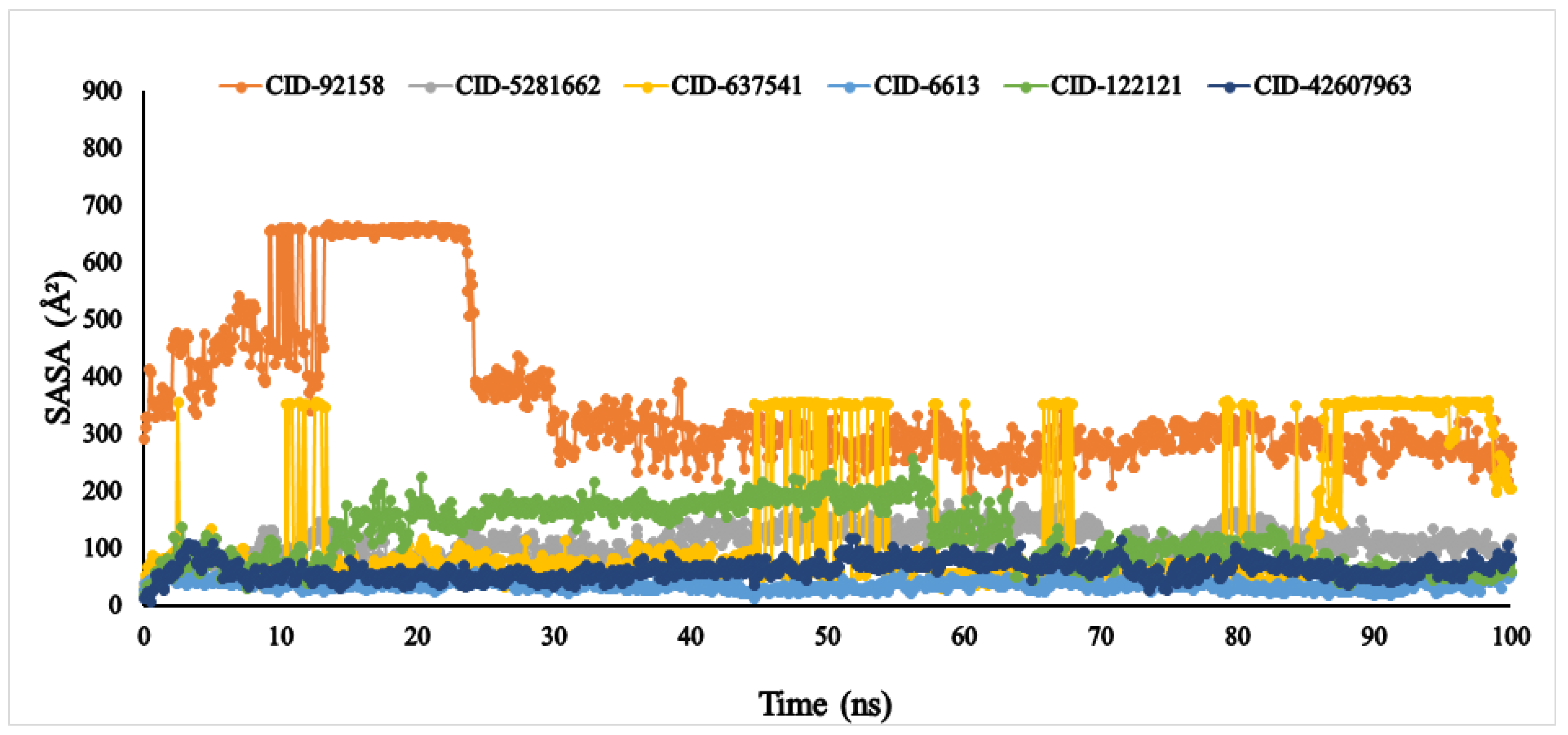
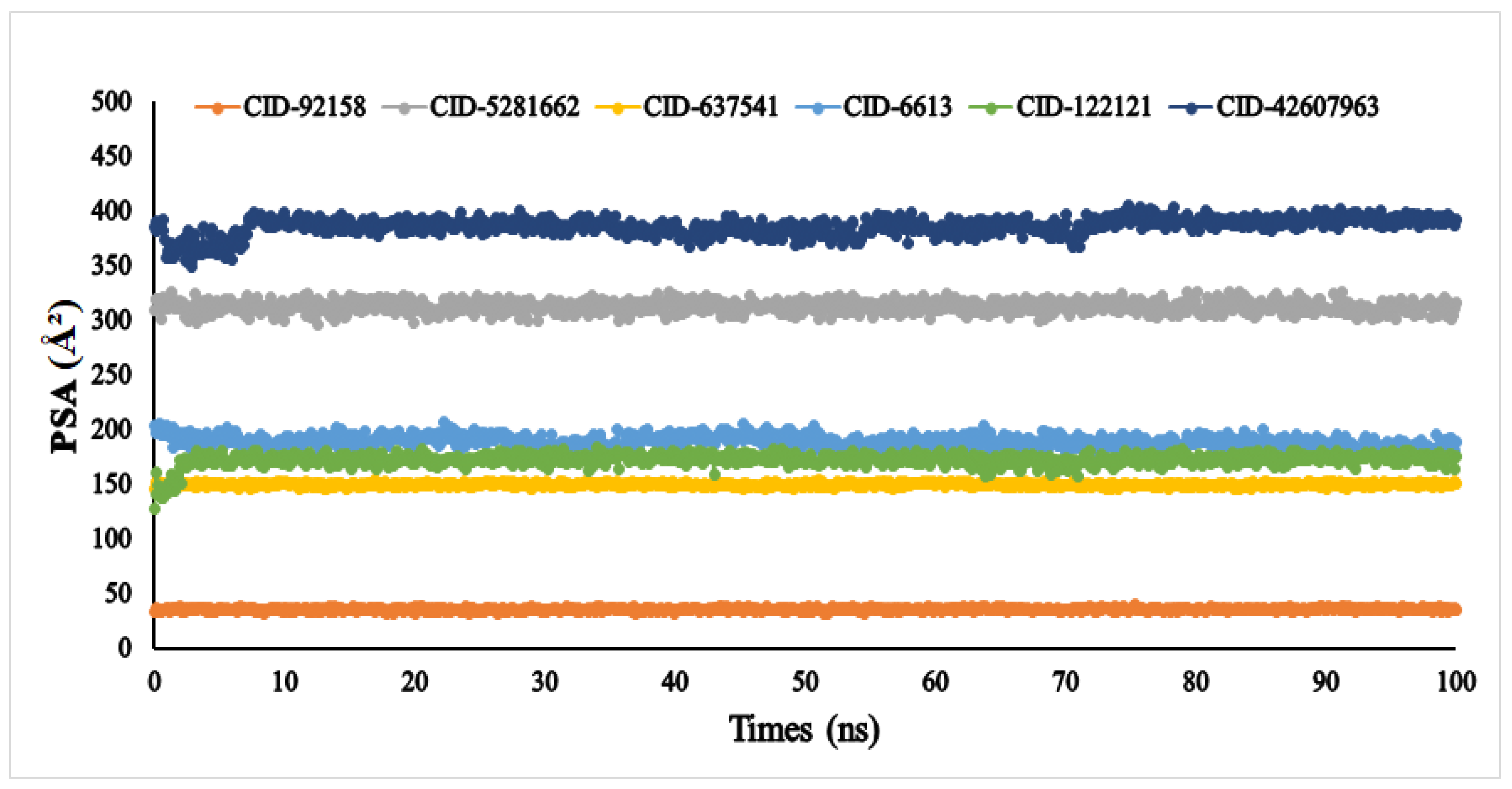
3.6. Molecular Dynamic Simulation (MDS)
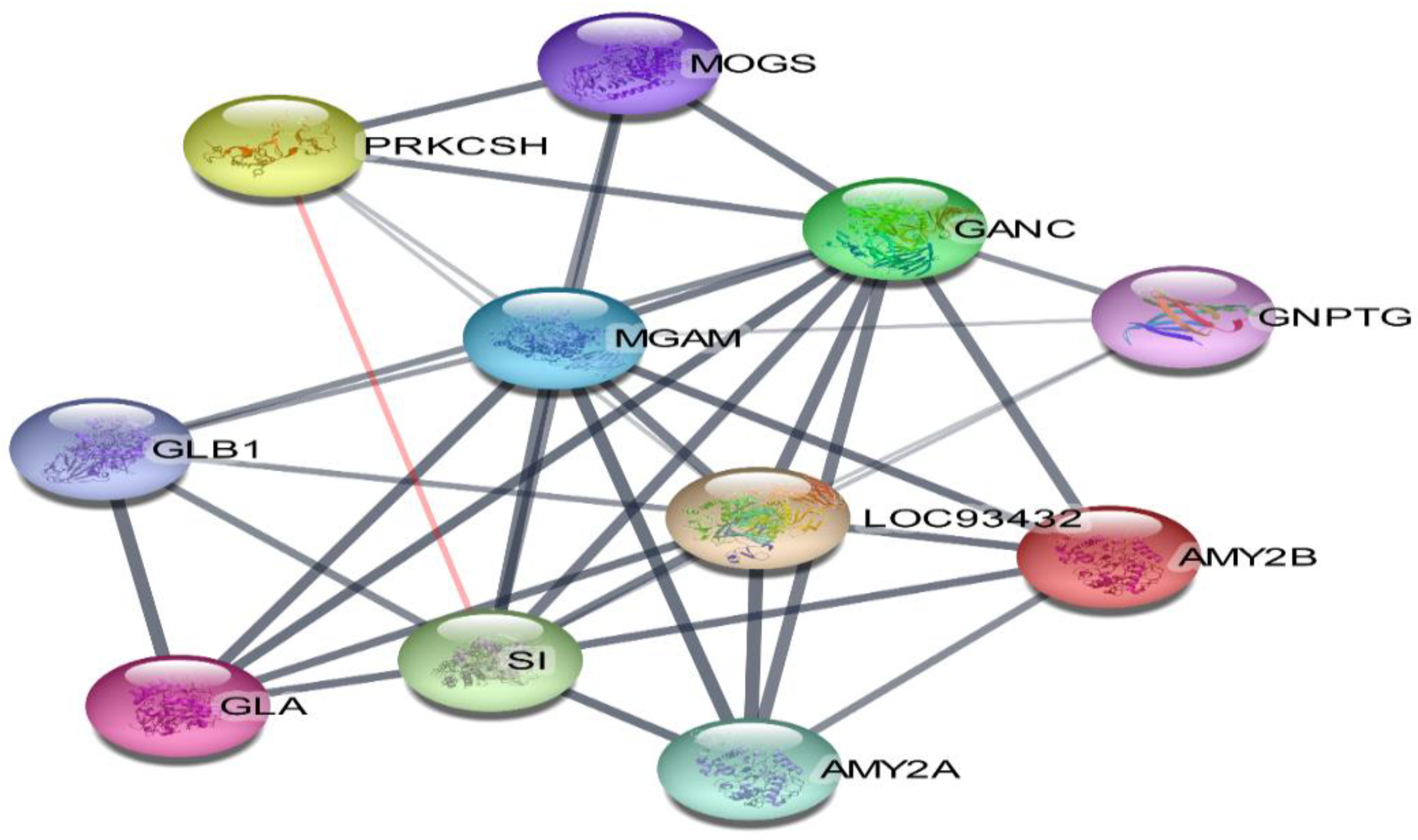
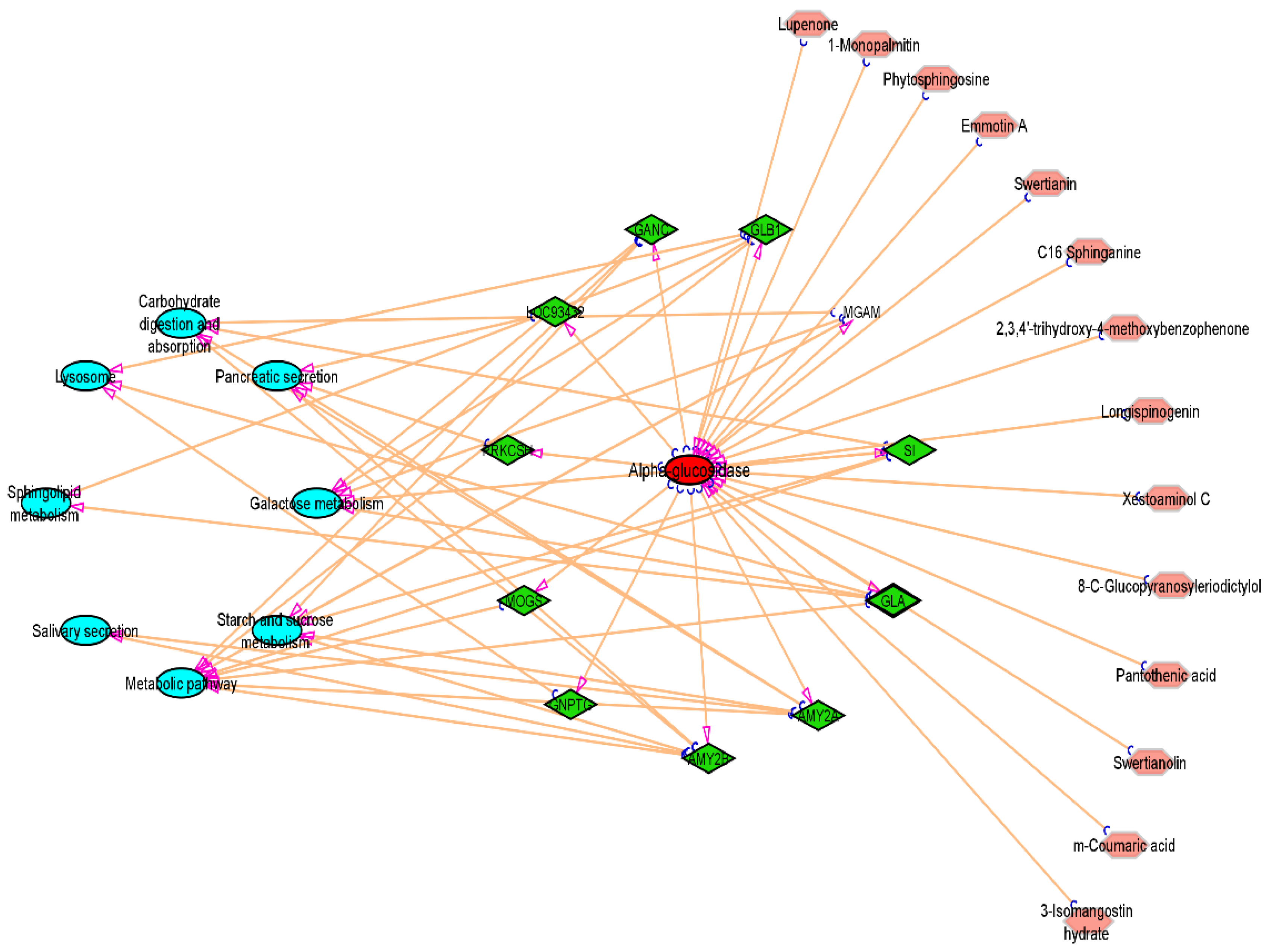
3.7. Compound-Target-Pathway Network
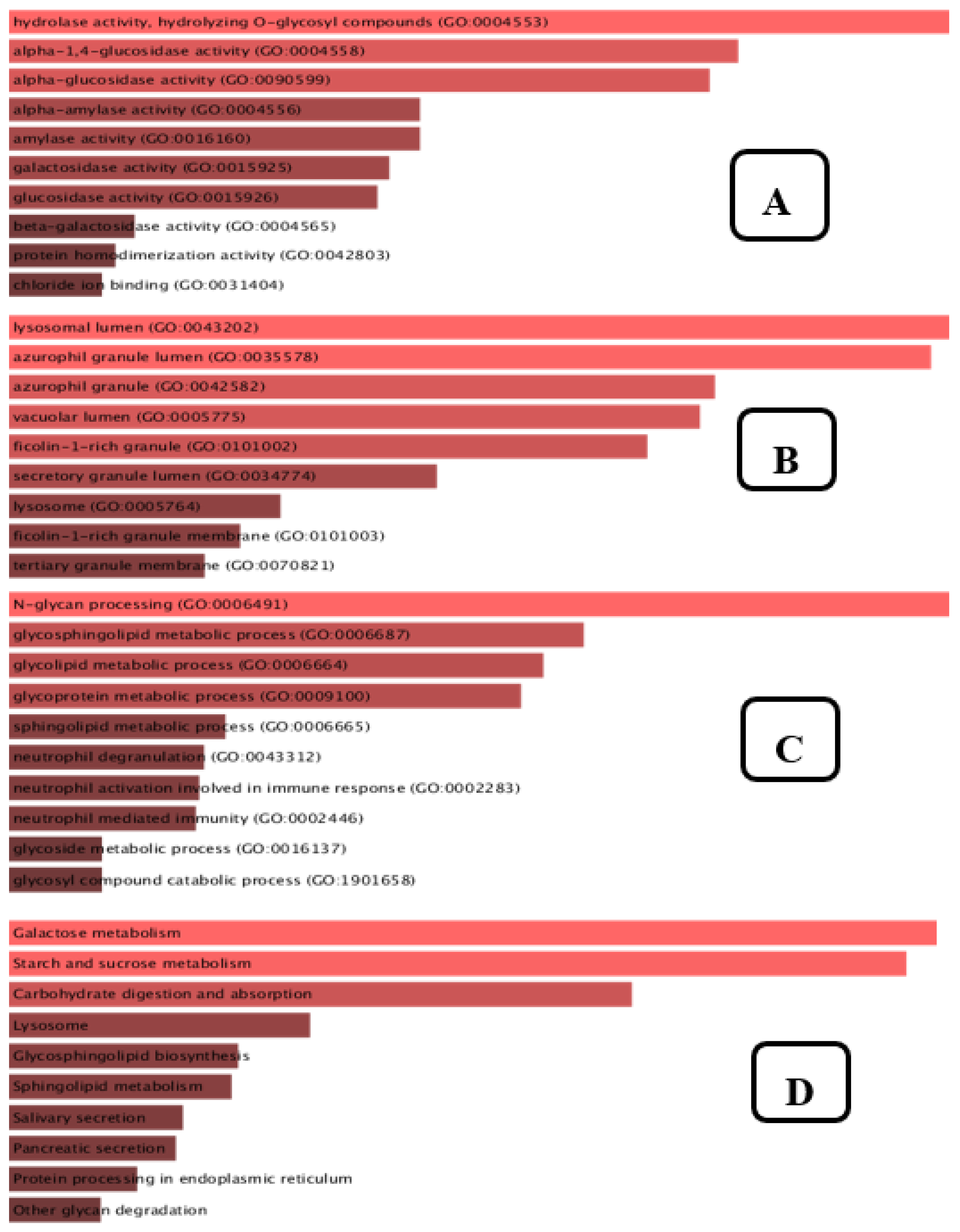
3.8. Gene Set Enrichment Analysis
4. Discussion
4.1. In Vitro Bioactivity Assay
4.2. Qualitative Tandem Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry (Q-ToF-LCMS) Analysis
4.3. Pharmacokinetics and Physiochemical Analysis
4.4. Molecular Docking of Compounds Identified by Q-ToF-LCMS Analysis
4.5. Molecular Dynamic Simulation
4.6. Compound Target Pathway and Gene Set Enrichment Analysis
4.7. Limitation and Future Directions of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hossain, U.; Das, A.K.; Ghosh, S.; Sil, P.C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. [Google Scholar] [CrossRef] [PubMed]
- Kumar Thakur, A.; Kumar, Y.; Goyal, R.K. Pharmacotherapeutics of miglitol: An α-glucosidase inhibitor. J. Anal. Pharm. Res. 2018, 7, 617–619. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Mirza, W. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, I.A.; Gill, S.; Wilcox, M.D.; Pearson, J.P.; Chater, P.I. Starch digestion in the upper gastrointestinal tract of humans. Starch-Starke 2018, 70, 1700111. [Google Scholar] [CrossRef]
- He, J.H.; Chen, L.X.; Li, H. Progress in the discovery of naturally occurring antidiabetic drugs and in the identification of their molecular targets. Fitoterapia 2019, 134, 270–289. [Google Scholar] [CrossRef]
- Rajendiran, D.; Packirisamy, I.; Gunasekaran, K.Y. A review on role of antioxidants in diabetes. Asian J. Pharm. Clin. Res. 2018, 11, 48–53. [Google Scholar] [CrossRef]
- Mia, M.A.R.; Ahmed, Q.U.; Helaluddin, A.B.M.; Ferdosh, S.; Siddique, M.M.; Azmi, S.N.H.; Sarker, M.Z.I. Acute and subacute toxicity assessment of liquid CO2 extract of Phaleria macrocarpa fruits flesh in mice model. J. King Saud Univ. Sci. 2022, 34, 101912. [Google Scholar] [CrossRef]
- Altaf, R.; Asmawi, M.Z.; Dewa, A.; Sadikun, A.; Umar, M.I. Phytochemistry and medicinal properties of Phaleria macrocarpa (Scheff.) Boerl. extracts. Phcog Rev. 2013, 7, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Ali, R.B.; Atangwho, I.J.; Kaur, N.; Abraika, O.S.; Ahmad, M.; Mahmud, R.; Asmawi, M.Z. Bioassay-guided antidiabetic study of Phaleria macrocarpa fruit extract. Molecules 2012, 17, 4986–5002. [Google Scholar] [CrossRef]
- Mia, M.A.; Ferdosh, S.; Ahmed, Q.U.; Helaluddin, A.B.M.; Sarker, M.; Islam, Z. Bridging Indigenous Knowledge and Scientific Evidence for Pharmacological Studies of Phaleria macrocarpa: A Systematic Review. Nat. Prod. J. 2022, 12, 29–45. [Google Scholar] [CrossRef]
- Hendra, R.; Haryani, Y. Phaleria macrocarpa (Boerl.) Scheff fruit: A potential source of natural antioxidant. Pharmcol. Clin. Pharm. Res. 2018, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Nirmala, C.; Bisht, M.S.; Bajwa, H.K.; Santosh, O. Bamboo: A rich source of natural antioxidants and its applications in the food and pharmaceutical industry. Trends Food Sci. Technol. 2018, 77, 91–99. [Google Scholar] [CrossRef]
- Liang, N.J.; Kitts, D.D. Antioxidant property of coffee components: Assessment of methods that define mechanisms of action. Molecules 2014, 19, 19180–19208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibi-Sadeer, N.; Montesano, D.; Albrizio, S.; Zengin, G.; Mahomoodally, M.F. The versatility of antioxidant assays in food science and safety Chemistry, applications, strengths, and limitations. Antioxidants 2020, 9, 709. [Google Scholar] [CrossRef]
- Sadeer, N.B.; Llorent-Martínez, E.J.; Bene, K.; Mahomoodally, M.F.; Mollica, A.; Sinan, K.I.; Zengin, G. Chemical profiling, antioxidant, enzyme inhibitory and molecular modelling studies on the leaves and stem bark extracts of three African medicinal plants. J. Pharm. Biomed. Anal. 2019, 174, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Lazim, R.; Suh, D.; Choi, S. Advances in molecular dynamics simulations and enhanced sampling methods for the study of protein systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Easmin, S.; Sarker, M.Z.I.; Ghafoor, K.; Ferdosh, S.; Jaffri, J.M.; Akanda, M.J.H.; Khatib, A. Extraction of α-glucosidase inhibitory compounds from Phaleria macrocarpa fruit flesh using solvent, sonication, and subcritical carbon dioxide soxhlet methods. J. Food Biochem. 2017, 41, e12399. [Google Scholar] [CrossRef]
- Pudziuvelyte, L.; Jakštas, V.; Ivanauskas, L.; Laukevičienė, A.; Ibe, C.F.D.; Kursvietiene, L.; Bernatoniene, J. Different extraction methods for phenolic and volatile compounds recovery from Elsholtzia ciliata fresh and dried herbal materials. Ind. Crops Prod. 2018, 1, 286–294. [Google Scholar] [CrossRef]
- Aryal, S.; Baniya, M.K.; Danekhu, K.; Kunwar, P.; Gurung, R.; Koirala, N. Total phenolic content, flavonoid content and antioxidant potential of wild vegetables from Western Nepal. Plants 2019, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Murugesu, S.; Ahmed, Q.U.; Uzir, B.F.; Yusoff, N.I.N.; Perumal, V.; Ibrahim, Z.; Khatib, A. Rapid investigation of α-glucosidase inhibitory activity of Clinacanthus nutans leaf using infrared fingerprinting. Vib. Spectrosc. 2019, 100, 22–29. [Google Scholar] [CrossRef]
- Saleem, H.; Htar, T.T.; Naidu, R.; Nawawi, N.S.; Ahmad, I.; Ashraf, M.; Ahemad, N. Biological, chemical and toxicological perspectives on aerial and roots of Filago germanica (L.) huds: Functional approaches for novel phyto-pharmaceuticals. Food Chem. Toxicol. 2019, 123, 363–373. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lestari, W.; Dewi, R.T.; Kardono, L.B.S.; Yanuar, A. Docking sulochrin and its derivative as α-glucosidase inhibitors of Saccharomyces cerevisiae. Indones. J. Chem. 2017, 17, 144–150. [Google Scholar] [CrossRef]
- Leong, S.W.; Awin, T.; Faudzi, S.M.M.; Maulidiani, M.; Shaari, K.; Abas, F. Synthesis and biological evaluation of asymmetrical diarylpentanoids as antiinflammatory, anti-α-glucosidase, and antioxidant agents. Med. Chem. Res. 2019, 28, 2002–2009. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Margolis, K.G. Building community in the gut: A role for mucosal serotonin. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 6–8. [Google Scholar] [CrossRef]
- Wang, J.; Chu, H.; Li, H.; Yang, W.; Zhao, Y.; Shen, T.; Zhang, L. A network pharmacology approach to investigate the mechanism of erjing prescription in type 2 diabetes. Evid.-Based Complement. Altern. Med. 2021, 2021, 9933236. [Google Scholar] [CrossRef]
- Onyebuchi, C.; Kavaz, D. Effect of extraction temperature and solvent type on the bioactive potential of Ocimum gratissimum L. extracts. Sci. Rep. 2020, 10, 21760. [Google Scholar] [CrossRef]
- Reverchon, E.; De Marco, I. Supercritical fluid extraction and fractionation of natural matter. J. Supercrit. Fluids. 2006, 38, 146–166. [Google Scholar] [CrossRef]
- Soeksmanto, A.; Hapsari, Y.; Simanjuntak, P. Antioxidant content of parts of Mahkota dewa, Phaleria macrocarpa [Scheff] Boerl. (Thymelaceae). Biodiversitas 2007, 8, 92–95. [Google Scholar] [CrossRef]
- Adil, I.H.; Cetin, H.I.; Yener, M.E.; Bayındırlı, A. Subcritical (carbon dioxide+ ethanol) extraction of polyphenols from apple and peach pomaces, and determination of the antioxidant activities of the extracts. J. Supercrit. Fluids 2007, 43, 55–63. [Google Scholar] [CrossRef]
- Lopez, A.; Rico, M.; Rivero, A.; Suárez de Tangil, M. The effects of solvents on the phenolic contents and antioxidant activity of Stypocaulon scoparium algae extracts. Food Chem. 2011, 125, 1104–1109. [Google Scholar] [CrossRef]
- Ali, R.B.; Atangwho, I.J.; Kuar, N.; Ahmad, M.; Mahmud, R.; Asmawi, M.Z. In vitro and in vivo effects of standardized extract and fractions of Phaleria macrocarpa fruits pericarp on lead carbohydrate digesting enzymes. BMC Complement. Altern. Med. 2013, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che Sulaiman, I.S.; Basri, M.; Fard Masoumi, H.R.; Chee, W.J.; Ashari, S.E.; Ismail, M. Effects of temperature, time, and solvent ratio on the extraction of phenolic compounds and the anti-radical activity of Clinacanthus nutans Lindau leaves by response surface methodology. Chem. Cent. J. 2017, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhassan, A.M.; Ahmed, Q.U.; Latip, J.; Shah, S.A.A. A new sulphated flavone and other phytoconstituents from the leaves of Tetracera indica Merr. and their α-glucosidase inhibitory activity. Nat. Prod. Res. 2019, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moselhy, S.S.; Razvi, S.S.; ALshibili, F.A.; Kuerban, A.; Hasan, M.N.; Balamash, K.S.; Ismail, I.M. m-coumaric acid attenuates non-catalytic protein glycosylation in the retinas of diabetic rats. J. Pestic Sci. 2018, 43, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.A.; Subhan, N.; Hossain, H.; Hossain, M.; Reza, H.M.; Rahman, M.M.; Ullah, M.O. Hydroxycinnamic acid derivatives: A potential class of natural compounds for the management of lipid metabolism and obesity. Nutr. Metab. 2016, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Hazelwood, R.L.; Bennett, L.L.; Nelson, A.M.M. Effect of pantothenic acid deficiency on insulin sensitivity and response to ACTH of intact and diabetic rats. Endocrinology 1956, 58, 427–434. [Google Scholar] [CrossRef]
- Adam, S.H.; Giribabu, N.; Bakar, N.M.A.; Salleh, N. Marantodes pumilum (Kacip fatimah) enhances in-vitro glucose uptake in 3T3-L1 adipocyte cells and reduces pancreatic complications in streptozotocin-nicotinamide induced male diabetic rats. Biomed. Pharmacother. 2017, 96, 716–726. [Google Scholar] [CrossRef]
- Susilawati, S.; Matsjeh, S.; Pranowo, H.D.; Anwar, C. Antioxidant activity of 2, 6, 4’-trihydroxy-4-methoxy benzophenone from ethyl acetate extract of leaves of mahkota dewa (Phaleria macrocarpa (Scheff.) Boerl.). Indones. J. Chem. 2011, 11, 180–185. [Google Scholar] [CrossRef]
- Andriani, Y.; Tengku-Muhammad, T.S.; Mohamad, H.; Saidin, J.; Syamsumir, D.F.; Chew, G.S.; Abdul Wahid, M.E. Phaleria macrocarpa Boerl. (Thymelaeaceae) leaves increase SR-BI expression and reduce cholesterol levels in rats fed a high cholesterol diet. Molecules 2015, 20, 4410–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anyanwu, G.O.; Iqbal, J.; Khan, S.U.; Zaib, S.; Rauf, K.; Onyeneke, C.E.; Ojo, O.O. Antidiabetic activities of chloroform fraction of Anthocleista vogelii planch root bark in rats with diet-and alloxan-induced obesity-diabetes. J. Ethnopharmacol. 2019, 229, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.S.A.; Giribabu, N.; Rao, P.V.; Salleh, N. Pancreato protective effects of Geniotrigona thoracica stingless bee honey in streptozotocin-nicotinamide-induced male diabetic rats. Biomed. Pharmacother. 2017, 89, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Ambika; Chauhan, S.M.S. Activity-guided isolation of antioxidant xanthones from Swertia chirayita (Roxb.) H. Karsten (Gentianaceae). Nat. Prod. Res. 2012, 26, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.S.; Chen, C.P.; Xiao, Z.Q.; Wang, Y.L.; Min, Q.X.; Yue, Y.; Chen, J. In vitro and in vivo antidiabetic activity of Swertia kouitchensis extract. J. Ethnopharmacol. 2013, 147, 622–630. [Google Scholar] [CrossRef]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Le Stunff, H. Sphingosine-1-Phosphate metabolism in the regulation of obesity/type 2 diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef]
- Snel, M.; Sleddering, M.A.; Pijl, H.; Nieuwenhuizen, W.F.; Frolich, M.; Havekes, L.M.; Jazet, I.M. The effect of dietary phytosphingosine on cholesterol levels and insulin sensitivity in subjects with the metabolic syndrome. Eur. J. Clin. Nutr. 2010, 64, 419–423. [Google Scholar] [CrossRef] [Green Version]
- Murakami, I.; Mitsutake, S.; Kobayashi, N.; Matsuda, J.; Suzuki, A.; Shigyo, T.; Igarashi, Y. Improved high-fat diet-induced glucose intolerance by an oral administration of phytosphingosine. Biosci. Biotechnol. Biochem. 2013, 77, 120644. [Google Scholar] [CrossRef]
- Zeng, M.; Che, Z.; Liang, Y.; Wang, B.; Chen, X.; Li, H.; Zhou, Z. GC–MS based plasma metabolic profiling of type 2 diabetes mellitus. Chromatographia 2009, 69, 941–948. [Google Scholar] [CrossRef]
- Su, K.; Yi, B.; Yao, B.Q.; Xia, T.; Yang, Y.F.; Zhang, Z.H.; Chen, C. Liraglutide attenuates renal tubular ectopic lipid deposition in rats with diabetic nephropathy by inhibiting lipid synthesis and promoting lipolysis. Pharmacol. Res. 2020, 156, 104778. [Google Scholar] [CrossRef]
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.U.; Nik Yusoff, N.I.; Uzir, B.F.; Perumal, V.; Khatib, A. Characterization of α-glucosidase inhibitors from Clinacanthus nutans Lindau leaves by gas chromatography-mass spectrometry-based metabolomics and molecular docking simulation. Molecules 2018, 23, 2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wu, H.; Wang, X.; Yang, Y.; Wang, Y.; Qian, H.; Zhang, Y. RP-HPLC characterization of lupenone and β-sitosterol in Rhizoma musae and evaluation of the antidiabetic activity of lupenone in diabetic Sprague-Dawley rats. Molecules 2014, 19, 14114–14127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, E.K.; Oh, J.S. Lupenone isolated from Adenophora triphylla var. japonica extract inhibits adipogenic differentiation through the downregulation of PPARγ in 3T3-L1 cells. Phytother. Res. 2013, 27, 761–766. [Google Scholar] [CrossRef]
- Zarena, A.S.; Sachindra, N.M.; Sankar, K.U. Optimisation of ethanol modified supercritical carbon dioxide on the extract yield and antioxidant activity from Garcinia mangostana L. Food Chem. 2012, 130, 203–208. [Google Scholar] [CrossRef]
- Ali, J.S.; Saleem, H.; Mannan, A.; Zengin, G.; Mahomoodally, M.F.; Locatelli, M.; Zia, M. Metabolic fingerprinting, antioxidant characterization, and enzyme-inhibitory response of Monotheca buxifolia (Falc.) A. DC. extracts. BMC Complement. Med. Ther. 2020, 20, 313. [Google Scholar] [CrossRef]
- Akinyede, K.A.; Oyewusi, H.A.; Hughes, G.D.; Ekpo, O.E.; Oguntibeju, O.O. In vitro evaluation of the anti-diabetic potential of aqueous acetone Helichrysum petiolare Extract (AAHPE) with molecular docking relevance in diabetes mellitus. Molecules 2021, 27, 155. [Google Scholar] [CrossRef]
- Ali, M.Y.; Kim, D.H.; Seong, S.H.; Kim, H.-R.; Jung, H.A.; Choi, J.S. α-glucosidase and protein tyrosine phosphatase 1B inhibitory activity of plastoquinones from marine brown alga Sargassum serratifolium. Mar. Drugs 2017, 15, 368. [Google Scholar] [CrossRef] [Green Version]
- Duke, J.A.; Cseke, L.J.; Warber, S.; Kirakosyan, A.; Brielmann, H.L.; Kaufman, P.B. Natural Products from Plants, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef]
- Guarnera, E.; Berezovsky, I.N. Allosteric sites: Remote control in regulation of protein activity. Curr. Opin. Struct. Biol. 2016, 37, 1–8. [Google Scholar] [CrossRef]
- Nussinov, R. Introduction to protein ensembles and allostery. Chem Rev. 2016, 116, 6263–6266. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef] [PubMed]
- Nokhala, A.; Siddiqui, M.J.; Ahmed, Q.U.; Ahamad Bustamam, M.S.; Zakaria, Z.A. Investigation of α-glucosidase inhibitory metabolites from Tetracera scandens leaves by GC–MS metabolite profiling and docking studies. Biomolecules 2020, 10, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargsyan, K.; Grauffel, C.; Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Libera, J.L.; Duran-Verdugo, F.; Valdés-Jimenez, A.; Nunez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Bibi, N.; Gul, S.; Ali, J.; Kamal, M.A. Bioinformatics approach to explore the inhibitory mechanism of existing drugs repurposed to fight against COVID-19. Eur. J. Pharmacol. 2020, 885, 173496. [Google Scholar] [CrossRef]
- Beura, S.; Chetti, P. In silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3747–3759. [Google Scholar] [CrossRef]
- Baildya, N.; Khan, A.A.; Ghosh, N.N.; Dutta, T.; Chattopadhyay, A.P. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: An insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1227, 129390. [Google Scholar] [CrossRef]
- Elebeedy, D.; Elkhatib, W.F.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; Al-Karmalawy, A.A. Anti-SARS-CoV-2 activities of tanshinone IIA, carnosic acid, rosmarinic acid, salvianolic acid, baicalein, and glycyrrhetinic acid between computational and in vitro insights. RSC Adv. 2021, 11, 29267–29286. [Google Scholar] [CrossRef]
- Nipun, T.S.; Ema, T.I.; Mia, M.A.R.; Hossen, M.S.; Arshe, F.A.; Ahmed, S.Z.; Dey, D. Active site-specific quantum tunneling of hACE2 receptor to assess its complexing poses with selective bioactive compounds in co-suppressing SARS-CoV-2 influx and subsequent cardiac injury. J. Adv. Vet. Anim. Res. 2021, 8, 540. [Google Scholar] [CrossRef]
- Tekin, H.O.; ALMisned, G.; Issa, S.A.; Kasikci, E.S.; Arooj, M.; Ene, A.; Zakaly, H.M. Molecular polar surface area, total solvent accessible surface area (SASA), heat of formation, and gamma-ray attenuation properties of some flavonoids. Front. Phys. 2022, 10, 838725. [Google Scholar] [CrossRef]
- Hasan, M.M.; Ahmed, Q.U.; Soad, S.Z.M.; Tunna, T.S. Animal models and natural products to investigate in vivo and in vitro antidiabetic activity. Biomed. Pharmacother. 2018, 101, 833–841. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration µg/mL | SCE-1 (%) Mean ± SEM | SCE-2 (%) Mean ± SEM | HRE (%) Mean ± SEM | Ascorbic Acid (%) Mean ± SEM |
|---|---|---|---|---|
| 400 | 61.49 ± 0.25 | 75.36 ± 0.82 | 63.63 ± 0.64 | 98.00 ± 0.46 |
| 200 | 51.25 ± 0.57 | 61.34 ± 0.45 | 53.84 ± 1.73 | 92.83 ± 0.85 |
| 100 | 42.28 ± 1.39 | 44.43 ± 1.04 | 43.26 ± 0.95 | 87.49 ± 0.43 |
| 50 | 37.84 ± 0.19 | 36.56 ± 0.71 | 39.16 ± 0.95 | 70.99 ± 1.18 |
| 25 | 29.60 ± 0.70 | 26.58 ± 0.87 | 31.53 ± 1.61 | 54.06 ± 1.16 |
| 12.5 | 27.87 ± 2.00 | 23.75 ± 0.39 | 26.40 ± 2.21 | 46.21 ± 0.66 |
| Concentration (μg/mL) | SCE-1 (%) Mean ± SEM | SCE-2 (%) Mean ± SEM | HRE (%) Mean ± SEM | Quercetin (%) Mean ± SEM |
|---|---|---|---|---|
| 80.0 | 73.21 ± 0.31 | 81.79 ± 0.82 | 67.57 ± 0.68 | 91.53 ± 0.24 |
| 40.0 | 65.47 ± 0.62 | 68.16 ± 0.16 | 59.24 ± 0.49 | 79.48 ± 0.52 |
| 20.0 | 54.70 ± 0.51 | 60.97 ± 0.49 | 52.08 ± 0.21 | 64.43 ± 0.15 |
| 10.0 | 43.46 ± 0.71 | 53.47 ± 0.15 | 40.35 ± 0.28 | 61.09 ± 0.28 |
| 5.0 | 31.16 ± 0.41 | 48.39 ± 0.18 | 26.33 ± 0.20 | 54.21 ± 0.30 |
| 2.5 | 20.22 ± 0.98 | 37.28 ± 0.92 | 22.56 ± 0.18 | 43.70 ± 0.36 |
| 1.25 | 8.911 ± 0.31 | 32.07 ± 0.45 | 8.68 ± 0.28 | 35.62 ± 0.43 |
| Identified Compounds | Formula | M/Z | Mass | Retention Time | Score |
|---|---|---|---|---|---|
| m-Coumaric acid | C9H8O3 | 182.080 | 164.046 | 1.012 | 98.35 |
| Pantothenic acid | C9H17NO5 | 220.118 | 219.110 | 2.33 | 99.41 |
| Xestoaminol C | C14 H31NO | 230.248 | 229.241 | 12.193 | 97.9 |
| 2,3,4′-Trihydroxy-4-Methoxybenzophenone | C14H12O5 | 261.076 | 260.069 | 10.447 | 98.66 |
| C16 Sphinganine | C16H35NO2 | 274.274 | 273.267 | 12.131 | 98.49 |
| Swertianin | C14 H10 O6 | 275.054 | 274.047 | 9.079 | 99.39 |
| Emmotin A | C16 H22 O4 | 279.160 | 278.152 | 16.76 | 95.7 |
| Phytosphingosine | C18H39NO3 | 318.300 | 317.292 | 12.219 | 99.64 |
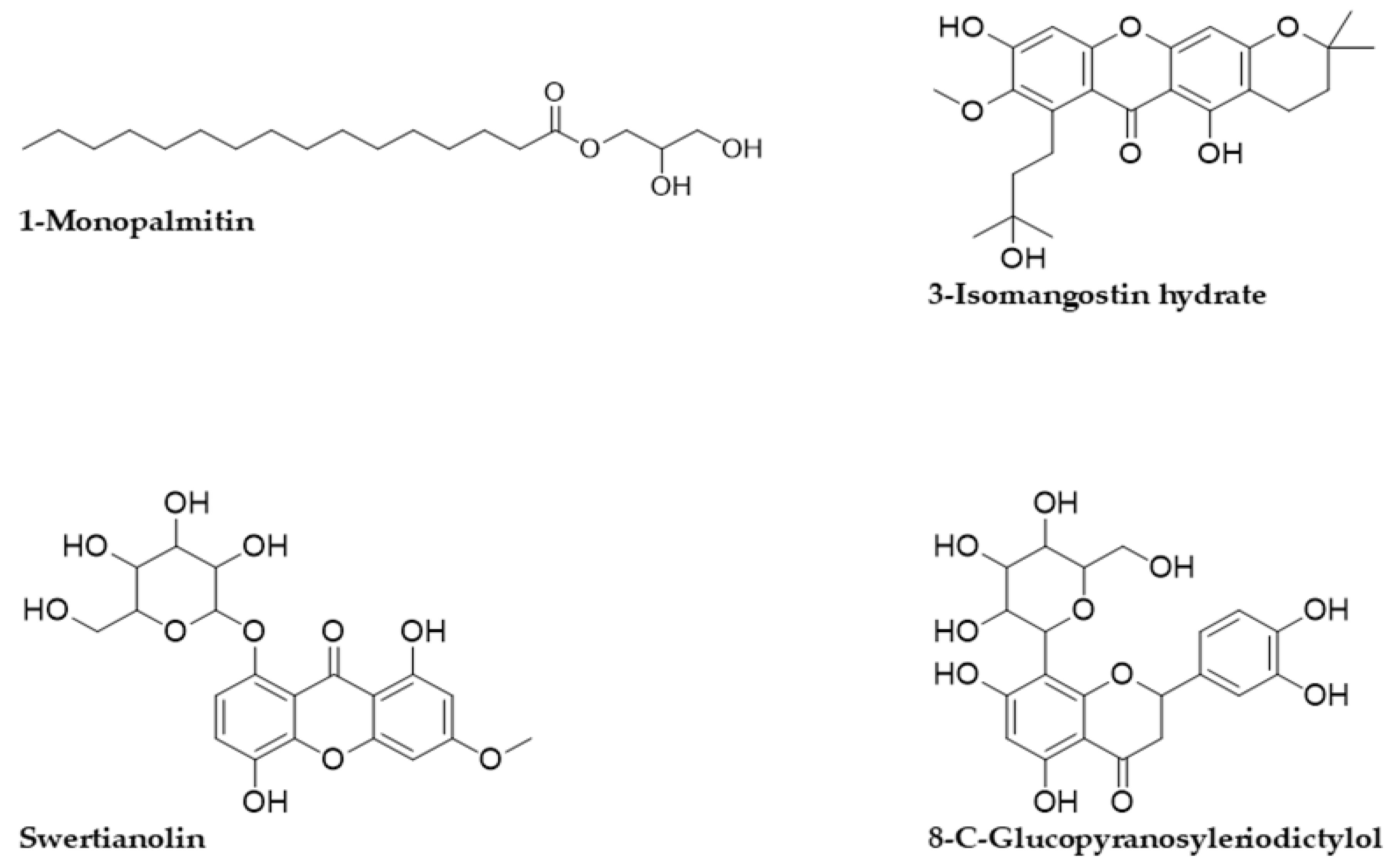
| 1-Monopalmitin | C19 H38O4 | 331.283 | 330.276 | 19.288 | 95.23 |
| Lupenone | C30H48O | 425.377 | 424.370 | 22.004 | 99.61 |
| 3-Isomangostin hydrate | C24H28O7 | 429.191 | 428.183 | 12.998 | 99.35 |
| Swertianolin | C20H20O11 | 437.108 | 436.100 | 9.079 | 99.68 |
| 8-C-Glucopyranosyleriodictylol | C21H22O11 | 451.123 | 450.116 | 10.572 | 99.32 |
| Longispinogenin | C30H50O3 | 476.409 | 458.376 | 22.185 | 99.2 |
| Physiochemical Properties | Pharmacokinetic Criteria | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Com. | MW | LogP | HA | HD | NRB | MR | SA | NL | DL | IA | BBA | AT | LD50 | Class | HT |
| 1 | 164.16 | 1.49 | 2 | 2 | 2 | 45.13 | 69.59 | 0 | Yes | 92.86 | Yes | No | 2980 | 5 | No |
| 2 | 219.24 | −1.04 | 4 | 4 | 6 | 52.21 | 87.91 | 0 | Yes | 30.44 | No | No | 10000 | 6 | No |
| 3 | 229.41 | 3.62 | 2 | 2 | 11 | 73.28 | 101.62 | 0 | Yes | 90.37 | Yes | No | 3500 | 5 | No |
| 4 | 260.24 | 2.04 | 5 | 3 | 3 | 68.88 | 108.88 | 0 | Yes | 93.67 | No | No | 2000 | 4 | No |
| 5 | 273.46 | 3.37 | 3 | 3 | 14 | 84.06 | 119.14 | 0 | Yes | 91.79 | Yes | No | 3500 | 5 | No |
| 6 | 274.23 | 2.07 | 6 | 3 | 1 | 72.55 | 111.62 | 0 | Yes | 77.18 | No | Yes | 4000 | 5 | No |
| 7 | 278.34 | 1.63 | 4 | 2 | 3 | 76.90 | 118.94 | 0 | Yes | 95.02 | Yes | Yes | 2500 | 5 | No |
| 8 | 317.51 | 3.12 | 4 | 4 | 16 | 94.83 | 136.67 | 0 | Yes | 94.24 | No | No | 3500 | 5 | No |
| 9 | 330.50 | 4.36 | 4 | 2 | 17 | 97.06 | 142.17 | 0 | Yes | 90.92 | Yes | No | 5000 | 5 | No |
| 10 | 424.71 | 4.54 | 1 | 0 | 1 | 129.18 | 191.77 | 0 | Yes | 98.47 | No | No | 5000 | 5 | No |
| 11 | 428.48 | 4.17 | 7 | 3 | 4 | 119.72 | 179.38 | 0 | Yes | 92.34 | No | Yes | 550 | 4 | No |
| 12 | 436.37 | −0.46 | 11 | 6 | 4 | 104.67 | 173.42 | 2 | No | 49.54 | No | No | 5000 | 5 | No |
| 13 | 450.40 | −0.27 | 11 | 8 | 3 | 106.21 | 180.50 | 2 | No | 33.39 | No | No | 2000 | 4 | No |
| 14 | 458.73 | 6.11 | 3 | 3 | 1 | 137.21 | 201.99 | 1 | Yes | 90.18 | No | No | 4300 | 5 | No |
| Compounds | Binding Affinity (kcal/mol) |
|---|---|
| Control ligand (ADG) | −6.0 |
| Quercetin | −8.4 |
| m-Coumaric acid | −7.0 |
| Pantothenic acid | −6.6 |
| Xestoaminol C | −5.5 |
| 2,3,4′-Trihydroxy-4-Methoxybenzophenone | −7.8 |
| C16 Sphinganine | −6.0 |
| Swertianin | −7.9 |
| Emmotin A | −7.2 |
| Phytosphingosine | −5.8 |
| 1-Monopalmitin | −6.2 |
| Lupenone | −9.5 |
| 3-Isomangostin hydrate | −9.0 |
| Swertianolin | −9.2 |
| 8-C-Glucopyranosyleriodictylol | −9.6 |
| Longispinogenin | −8.0 |
| Compounds | Interacting Amino Acid Residues | Bond Type | Bond Distance (Å) |
|---|---|---|---|
| m-Coumaric acid | TYR158 | Hydrogen bond | 2.54 |
| ASP215 | Hydrogen bond | 1.96 | |
| GLU277 | Hydrogen bond | 2.66 | |
| ARG442 | Pi-cation | 3.69 | |
| ASP352 | Pi-anion | 4.54 | |
| TYR72 | Pi-Pi-T shaped | 5.47 | |
| Pantothenic acid | ARG442 | Hydrogen bond | 2.66 |
| ASP215 | Hydrogen bond | 1.99 | |
| ARG213 | Hydrogen bond | 2.18 | |
| HIS351 | Hydrogen bond | 1.94 | |
| GLU277 | Hydrogen bond | 2.66, 2.66, 2.19 | |
| ASP352 | Hydrogen bond | 2.16, 2.99 | |
| PHE303 | Pi-alkyl interaction | 4.87 | |
| Xestoaminol C | PRO312 | Hydrogen bond | 2.70 |
| SER240 | Hydrogen bond | 2.45 | |
| TYR158 | Pi-alkyl interaction | 5.0, 4.97 | |
| PHE303 | Pi-alkyl interaction | 4.84, 5.10 | |
| 2,3,4′-Trihydroxy-4-Methoxybenzophenone | ASP233 | Hydrogen bond | 2.68 |
| LYS156 | Hydrogen bond | 2.85 | |
| ASN415 | Hydrogen bond | 2.53 | |
| GLU429 | Carbon-hydrogen bond | 3.62 | |
| ASN317 | Carbon-hydrogen bond | 3.06, 3.58 | |
| HIS423 | Pi-Pi-T-shaped interaction | 5.16 | |
| LYS432 | Alkyl interaction | 3.99 | |
| ALA418 | Pi-alkyl interaction | 4.87 | |
| ILE419 | Pi-alkyl interaction | 4.19 | |
| C16 Sphinganine | GLN353 | Hydrogen bond | 2.50 |
| ASP352 | Hydrogen bond | 2.19 | |
| GLU411 | Hydrogen bond | 2.46 | |
| LYS156 | Alkyl interaction | 5.02 | |
| ARG315 | Alkyl interaction | 4.62 | |
| PHE303 | Pi-alkyl interaction | 5.43 | |
| TYR158 | Pi-alkyl interaction | 3.98 | |
| Swertianin | ASP352 | Carbon-hydrogen bond | 3.59 |
| ARG315 | Hydrogen bond | 2.05 | |
| ARG315 | Pi-alkyl interaction | 4.52, 4.89 | |
| TYR158 | Pi-Pi-T-shaped interaction | 4.95, 5.52 | |
| Emmotin A | TYR158 | Hydrogen bond | 2.40 |
| PHE178 | Pi-alkyl interaction | 5.16 | |
| PHE303 | Pi-alkyl interaction | 4.45 | |
| ARG315 | Pi-alkyl interaction | 4.38 | |
| Phytosphingosine | HIS280 | Hydrogen bond | 2.72 |
| ASP307 | Hydrogen bond | 2.19, 2.45 | |
| PRO312 | Hydrogen bond | 2.20 | |
| SER311 | Hydrogen bond | 2.46 | |
| ARG315 | Alkyl interaction | 4.81, 4.84 | |
| TYR158 | Pi-alkyl interaction | 4.78, 5.41 | |
| PHE303 | Pi-alkyl interaction | 5.03 | |
| PHE314 | Pi-alkyl interaction | 5.48 | |
| HIS280 | Pi-alkyl interaction | 5.47 | |
| 1-Monopalmitin | ARG442 | Hydrogen bond | 2.38, 5.98 |
| ASP69 | Hydrogen bond | 2.50 | |
| ASP352 | Hydrogen bond | 2.52 | |
| ASP352 | Carbon-hydrogen bond | 3.55, 3.61 | |
| ASP303 | Pi-alkyl interaction | 5.08 | |
| PHE314 | Pi-alkyl interaction | 5.46 | |
| ARG315 | Alkyl interaction | 4.11, 5.06 | |
| LYS156 | Alkyl interaction | 4.20 | |
| TYR158 | Pi-sigma interaction | 3.60 | |
| TYR158 | Pi-alkyl interaction | 4.58, 5.18 | |
| Lupenone | VAL308 | Alkyl interaction | 5.29 |
| ILE328 | Alkyl interaction | 5.29 | |
| ALA329 | Alkyl interaction | 3.59 | |
| PRO312 | Alkyl interaction | 4.55 | |
| 3-Isomangostin hydrate | GLU332 | Hydrogen bond | 2.54 |
| SER304 | Hydrogen bond | 2.16 | |
| HIS280 | Pi-donor hydrogen bond | 2.66 | |
| HIS280 | Pi-alkyl interaction | 5.29 | |
| ARG315 | Alkyl interaction | 4.33, 4.69 | |
| PRO312 | Pi-sigma interaction | 3.69 | |
| ASP307 | Pi-anion interaction | 3.79, 3.84 | |
| Swertianolin | ARG442 | Hydrogen bond | 2.92 |
| GLU411 | Hydrogen bond | 2.35 | |
| TYR158 | Pi-Pi-T-shaped interaction | 5.50, 5.53 | |
| ARG315 | Pi-alkyl interaction | 4.74 | |
| 8-C-Glucopyranosyleriodictylol | GLU277 | Hydrogen bond | 2.78 |
| ASP242 | Hydrogen bond | 1.89 | |
| SER240 | Hydrogen bond | 2.36 | |
| SER157 | Carbon-hydrogen bond | 3.38 | |
| HIS280 | Pi-donor hydrogen bond | 3.21 | |
| TYR158 | Pi-Pi-T-shaped interaction | 4.99 | |
| Longispinogenin | GLN353 | Hydrogen bond | 2.91 |
| GLU411 | Carbon hydrogen bond | 3.55 | |
| VAL216 | Alkyl interaction | 4.76 | |
| PHE303 | Pi-alkyl interaction | 5.36 | |
| TYR158 | Pi-alkyl interaction | 5.41, 4.59, 4.64 | |
| Quercetin | ARG315 | Hydrogen bond | 2.73 |
| GLH277 | Hydrogen bond | 2.01 | |
| ASH215 | Hydrogen bond | 2.46 | |
| PHE303 | Pi-Pi T-shaped | 4.96, 5.13 | |
| ARG442 | Pi-cation | 3.75 | |
| ASP352 | Pi-anion | 4.28 | |
| Control ligand (ADG) | ARG213 | Hydrogen bond | 1.80 |
| GLH277 | Hydrogen bond | 2.13 | |
| ASH215 | Hydrogen bond | 2.44 | |
| ASP352 | Hydrogen bond | 2.18, 2.37 | |
| HIE351 | Hydrogen bond | 1.96 | |
| ARG442 | Hydrogen bond | 1.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mia, M.A.R.; Ahmed, Q.U.; Ferdosh, S.; Helaluddin, A.B.M.; Awal, M.S.; Sarian, M.N.; Sarker, M.Z.I.; Zakaria, Z.A. In Vitro, In Silico and Network Pharmacology Mechanistic Approach to Investigate the α-Glucosidase Inhibitors Identified by Q-ToF-LCMS from Phaleria macrocarpa Fruit Subcritical CO2 Extract. Metabolites 2022, 12, 1267. https://doi.org/10.3390/metabo12121267
Mia MAR, Ahmed QU, Ferdosh S, Helaluddin ABM, Awal MS, Sarian MN, Sarker MZI, Zakaria ZA. In Vitro, In Silico and Network Pharmacology Mechanistic Approach to Investigate the α-Glucosidase Inhibitors Identified by Q-ToF-LCMS from Phaleria macrocarpa Fruit Subcritical CO2 Extract. Metabolites. 2022; 12(12):1267. https://doi.org/10.3390/metabo12121267
Chicago/Turabian StyleMia, Md. Abdur Rashid, Qamar Uddin Ahmed, Sahena Ferdosh, Abul Bashar Mohammed Helaluddin, Md. Shihabul Awal, Murni Nazira Sarian, Md. Zaidul Islam Sarker, and Zainul Amiruddin Zakaria. 2022. "In Vitro, In Silico and Network Pharmacology Mechanistic Approach to Investigate the α-Glucosidase Inhibitors Identified by Q-ToF-LCMS from Phaleria macrocarpa Fruit Subcritical CO2 Extract" Metabolites 12, no. 12: 1267. https://doi.org/10.3390/metabo12121267