3.1. Model Architecture
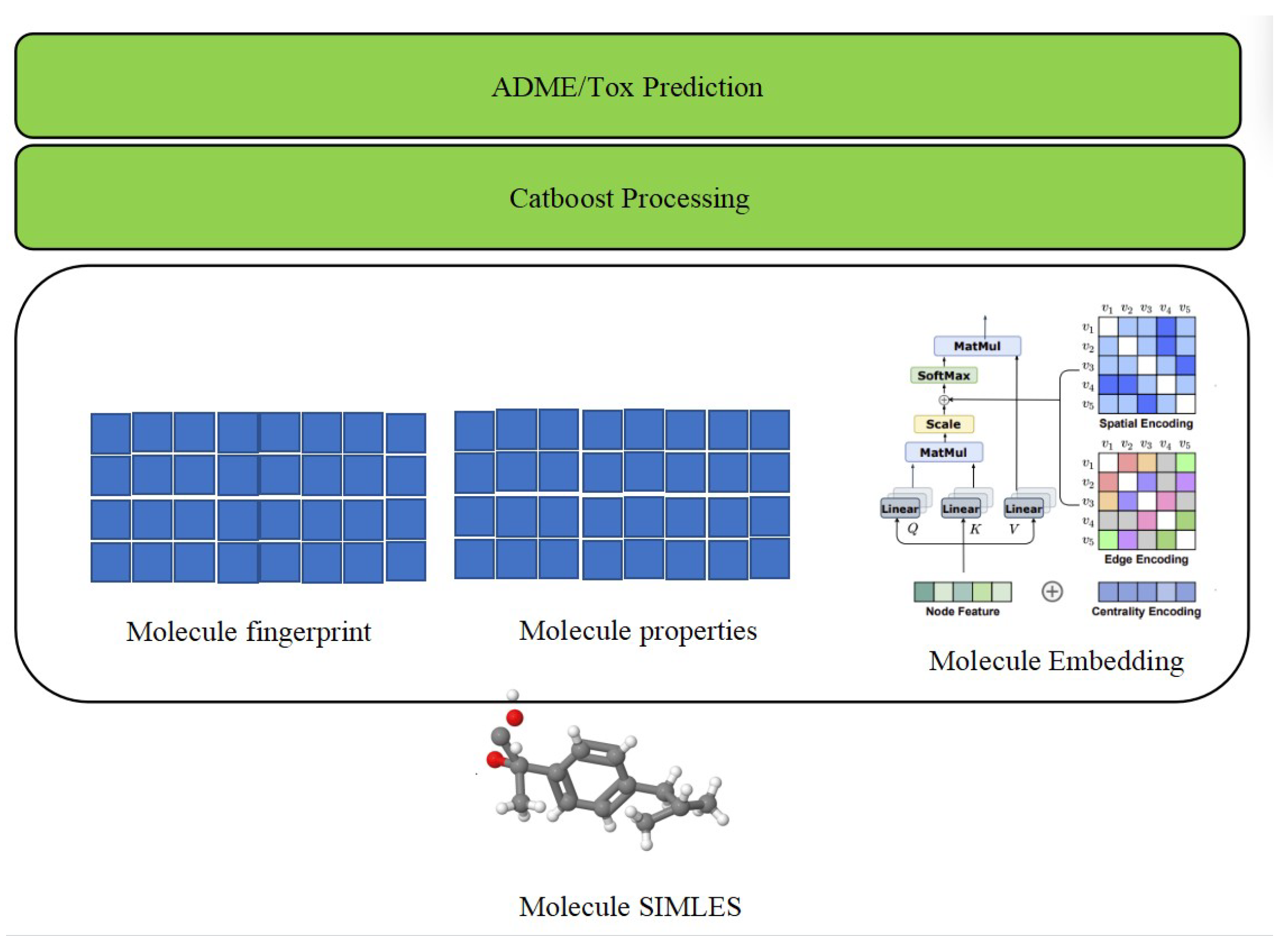
This study builds upon the foundational architecture of the traditional sequence transformer model, extending its capabilities to incorporate graph-oriented features. This augmentation enhances the model’s capacity to effectively discern spatial topology information and connectivity details within molecular structures, shown by
Figure 1. In cognizance of the evolving requirements for ADME/Tox prediction, our methodology acknowledges specific limitations within the traditional model. Notably, it lacks the inclusion of molecular fingerprint-like features, recognized for their historical efficacy in traditional computational chemistry research. These features, often grounded in expert knowledge, along with the consideration of key physicochemical properties, are deemed indispensable for a comprehensive understanding of molecular behavior. Our proposed methodology, rooted in the transformer architecture, systematically addresses these limitations through a meticulously designed training regimen. This involves pre-training—initial model training to capture foundational patterns in molecular data; fine-tuning—iterative refinement of the model using task-specific data, adapting it to the nuances of ADME/Tox prediction; and end-to-end training—a comprehensive training phase incorporating both pre-training and fine-tuning, culminating in the generation of specialized embedding for small molecules.
To augment predictive performance, our methodology introduces a secondary training phase utilizing a tree model, specifically CatBoost. This strategic addition is aimed at comprehensively integrating diverse information types, encompassing molecularity. At last, model evaluation is conducted using rigorous performance metrics tailored for ADME/Tox prediction tasks. The key metrics include Symmetric Mean Absolute Percentage Error, which is used to calculate an accuracy measure based on percentage (or relative) errors and is defined as the following:
where
is the target value, and (
) is the predictions. The Pearson Correlation Coefficient (PCC) is measuring the linear relationship between predicted and actual values. It is defined as the following:
where
and
are the mean value
x and sample value
y, respectively. These metrics provide a quantitative assessment of the model’s predictive capabilities, ensuring a robust evaluation of its performance.
(R-Square) is a statistical measure in a regression model that determines the proportion of variance in the dependent variable that can be explained by the independent variable. It is defined as the following:
In the initial phase of our model development, we fine-tune the Graphormer model based on the PCQM4Mv2 dataset, originally curated under the PubChemQC project. This dataset, rooted in quantum chemistry, enables the transfer of 2D/3D molecular graphs represented in SMILES format into embeddings:
Subsequently, we integrate molecular fingerprinting and molecular properties based on the embeddings generated by the Graphormer model in the preceding step. This integration encompasses three types of information. First, the Molecule Embedding is computed, incorporating Centrality Encoding to describe the importance levels of nodes in a graph, Spatial Encoding to represent the spatial positions of nodes, and Edge Encoding in Graphormer to encode relationships between nodes. Notably, this differs from traditional graph neural networks by not relying solely on the Euclidean distance between node embeddings of connected nodes:
Centrality Encoding refers to describing the importance level of nodes in a graph. Spatial Encoding refers to the process of representing the spatial position of nodes in a graph as part of the input to the Graphormer model. Edge Encoding in Graphormer refers to the process of encoding the relationships between nodes in a graph. This is unlike traditional graph neural networks, which are generated by the Euclidean distance between the node embeddings of two connected nodes. Moreover, the prediction model not only considers the embedding feature, which represents position and connection information, but also considers the molecular fingerprint, as well as the physicochemicals (connectivity, estate, kappa, burden, charge, property, etc.) that have been proved effective in traditional computational drug research. Here, we define the COMBINE operation.
Furthermore, our prediction model extends beyond considering embedding features that capture position and connection information. It incorporates molecular fingerprinting and various physicochemical properties (connectivity, estate, kappa, burden, charge, property, etc.) that have demonstrated effectiveness in traditional computational drug research:
In this paper, we employ CatBoost as the final processing step on the last COMBINE result, the detailed procedure shown by following algorithm. CatBoost is chosen for its robustness in handling heterogeneous data, as it consistently outperforms the majority of boosting algorithms in the initial run. This selection is grounded in its ability to effectively navigate diverse data types and optimize predictive performance, ensuring a reliable and comprehensive analysis of the combined molecular features obtained from the preceding steps.
| Algorithm 1: Generative tree. |
![Electronics 13 00624 i001]() |
3.2. Benchmark
The properties of drug molecules typically encompass fundamental attributes, ADME-related pharmacokinetic properties, and toxicity properties. Given the multitude of relevant properties and to ensure comparability with commercial software and web platforms, we selected five benchmark properties: solubility (logS), clearance (CL), permeability (PAPP), plasma protein binding (PPB), and median lethal dose (LD50). These chosen properties serve as key benchmarks, providing a focused and standardized set of criteria for evaluating and comparing the performance of our model against established commercial tools and web-based platforms in the field of drug discovery and development.
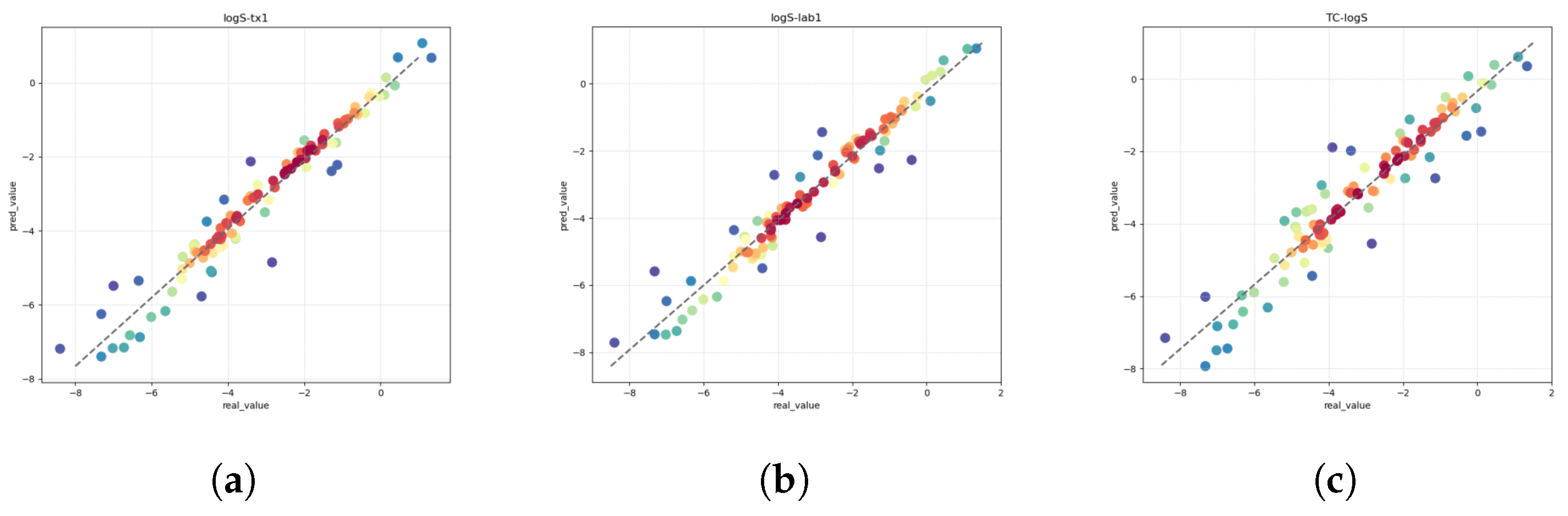
Solubility (logS) constitutes a pivotal parameter in the comprehensive assessment of drug molecules, bearing direct implications for their absorption and distribution within the ADMET framework. This parameter holds particular significance, as it profoundly influences the oral bioavailability of a drug, emerging as a critical pharmacokinetic property necessitating meticulous optimization in the drug discovery process. The quantification of solubility, denoted by the 10-based logarithm (logS), assumes paramount importance in shaping formulations and determining the subsequent therapeutic efficacy of a drug [
35,
36,
37].
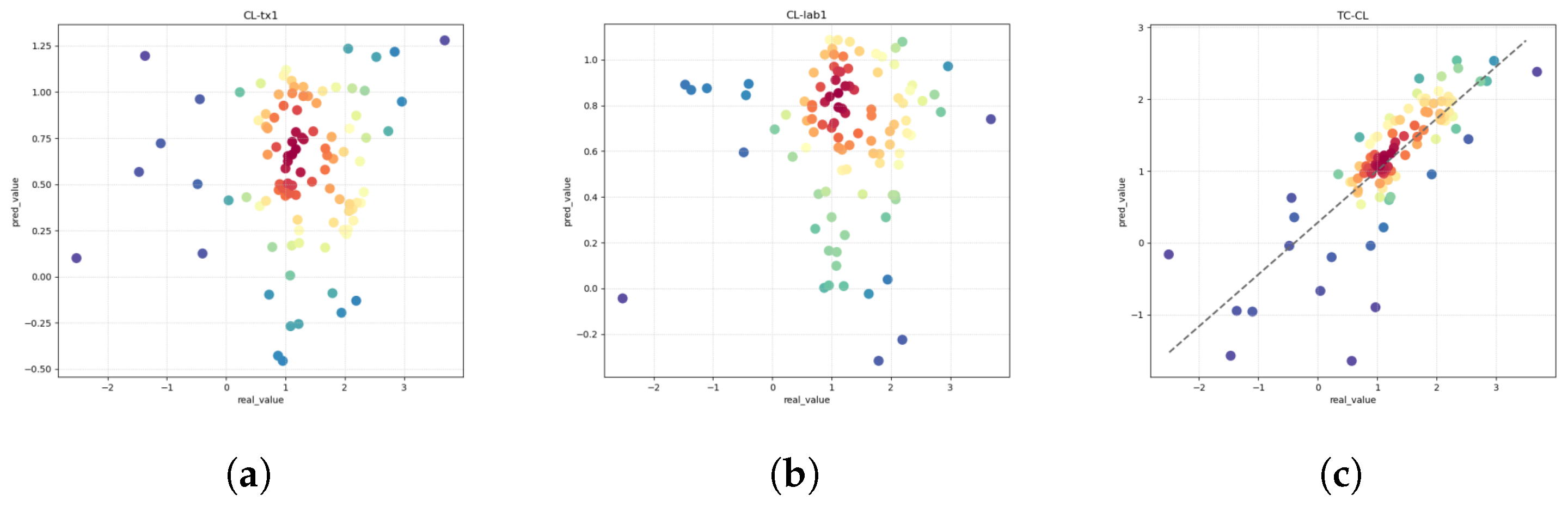
Clearance (CL) is a pivotal pharmacokinetic parameter denoting the volume of plasma from which a drug is systematically removed per unit time. This parameter serves as a critical indicator for evaluating drug metabolism, reflecting the rate at which a drug undergoes elimination from the body. Key organs involved in drug elimination encompass the liver, kidneys, lungs, and intestines, each contributing to diverse drug-specific elimination pathways. Within the intricate landscape of drug metabolism, the clearance rate, represented by the CL value, is subject to multifaceted influences. These encompass intrinsic factors, such as the physicochemical properties of the drug, the prevailing physiological state of the organism, and the activity of the metabolic enzyme system. The resultant CL value becomes a vital quantitative measure, offering insights into the dynamic interplay between a drug and the biological milieu. In academic and clinical contexts, the discernment of drug clearance intricacies stands as an essential foundation, contributing profoundly to the formulation of evidence-based pharmaceutical strategies and clinical decision making [
38,
39,
40].
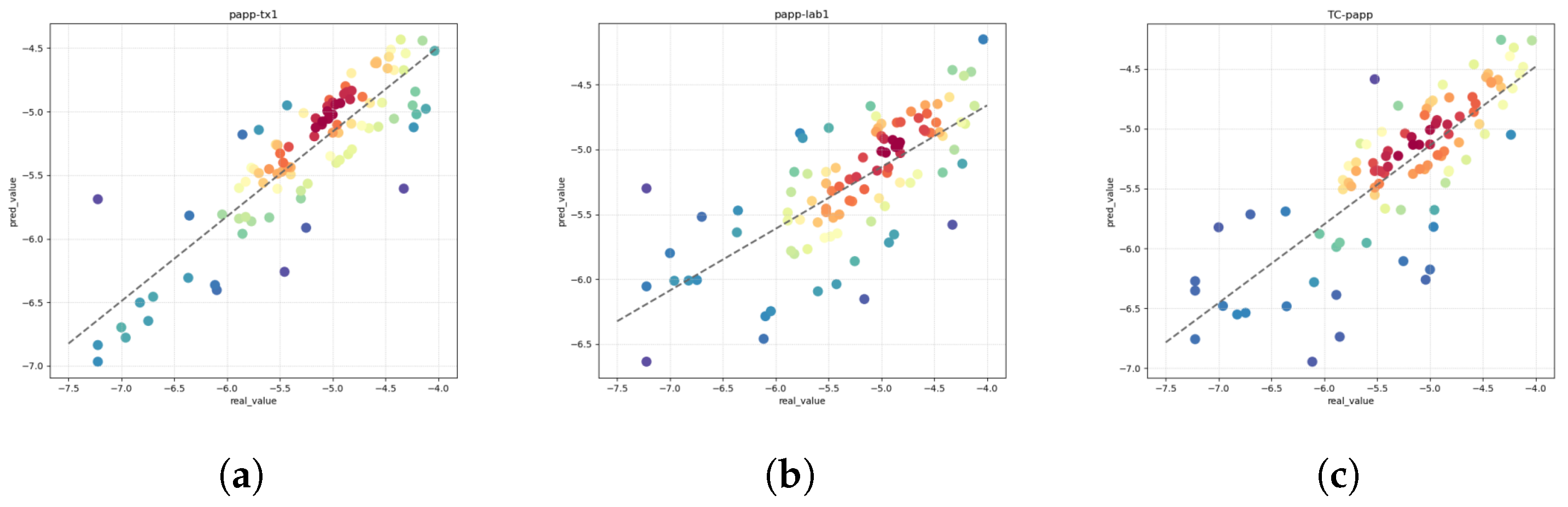
Permeability (PAPP) stands as a pivotal index in assessing a drug’s capability to traverse cell membranes and reach its intended site of action. Widely employed in the realm of drug discovery and development, PAPP furnishes indispensable insights into the pharmacokinetic attributes of potential drug candidates. The discernment gleaned from PAPP assessments serves as a cornerstone for optimizing drug design, enhancing the likelihood of success in subsequent clinical trials. The adoption of PAPP as a metric underscores its significance in shaping the trajectory of drug development endeavors, propelling advancements in the rational design of pharmacotherapeutics [
41,
42,
43,
44].
Plasma protein (PPB) denotes the extent to which a drug molecule associates with proteins in the plasma, predominantly with albumin. Upon introduction into the bloodstream, a drug may manifest in two distinct states: as free or unbound drug molecules and as drug molecules bound to plasma proteins. Only the unbound drug molecules retain pharmacological activity, enabling interaction with target sites in the body to elicit the desired therapeutic effects. The intricate binding of drugs to plasma proteins significantly governs their distribution, metabolism, and elimination within the biological milieu. Elevated plasma protein binding has the potential to constrain the availability of free, active drug molecules, thereby exerting a pivotal influence on the drug’s therapeutic efficacy. This underscores the imperative for the meticulous consideration of plasma protein binding dynamics in the determination of dosage regimens [
45,
46].
The median lethal dose (LD50) stands as a critical measure in the assessment of substance toxicity. This standardized metric involves the systematic administration of progressively increasing doses of the substance to discrete groups of test animals until 50% of them succumb. The ensuing dose–response data are then utilized to derive the LD50 value, representing the quantity of the substance expected to induce mortality in 50% of a population upon exposure. Embraced extensively in regulatory toxicology, the LD50 test retains its fundamental role in evaluating acute toxicity and establishing safety thresholds for substances that carry potential risks to human health. Conventionally expressed in milligrams of the substance per kilogram of the body weight of the test animal, LD50 data are pivotal for informed risk assessment and the development of safety guidelines [
47,
48].
In our comprehensive investigation of bioactive molecules and their drug-like properties, we meticulously compiled a valuable dataset by amalgamating information from two distinct sources. The primary contributor to our dataset is the Chembl database, a meticulously curated repository of bioactive molecules renowned for possessing drug-like characteristics. This database seamlessly integrates chemical, bioactivity, and genomic data, providing an invaluable resource to facilitate the translation of genomic information into the development of effective new drugs. Specifically, forecast data related to CL (clearance), caco-2 permeability, PPB (plasma protein binding), and LD50 were extracted from the Chembl database. These parameters play pivotal roles in understanding the pharmacokinetics and toxicity of bioactive compounds, critical aspects in drug development. Following a meticulous filtering process, our local dataset now comprises a total of 1128 entries for logS (logarithm of aqueous solubility), 2999 entries for CL, 1209 entries for caco-2 permeability, 2081 entries for PPB, and 2633 entries for LD50. Each entry in this curated dataset encapsulates essential information contributing to a holistic understanding of the properties and behavior of the bioactive molecules under investigation. To enhance the robustness of our dataset and ensure its applicability across diverse scenarios, we incorporated additional test data points for logS, CL, PPB, and LD50. These test data points were randomly generated to provide a representative sample for further validation and assessment of the predictive models. For a more detailed breakdown of the dataset, please refer to
Table 1, which encapsulates a comprehensive overview of the entries and properties included in our exploration of bioactive molecules.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}