
Dynamics of Medical Screening: A Simulation Model of PSA Screening for Early Detection of Prostate Cancer


Abstract
:1. Introduction and Motivation
2. Methodology and Background of Systems Models for Cancer
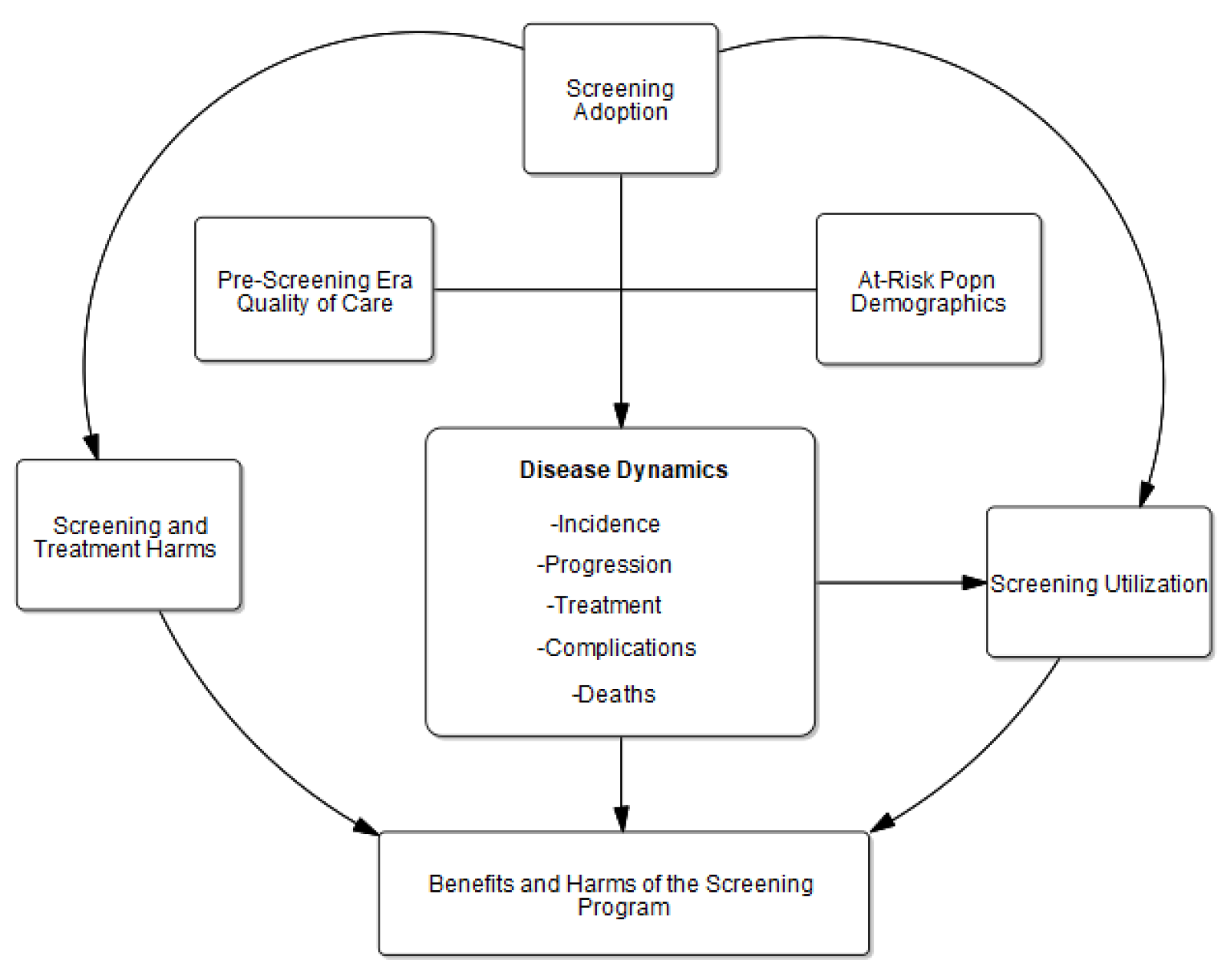
3. Overview of the PSA Screening Model
3.1. Data Types and Inputs
3.2. Population and Natural History of Disease
3.2.1. Population Increase and Aging
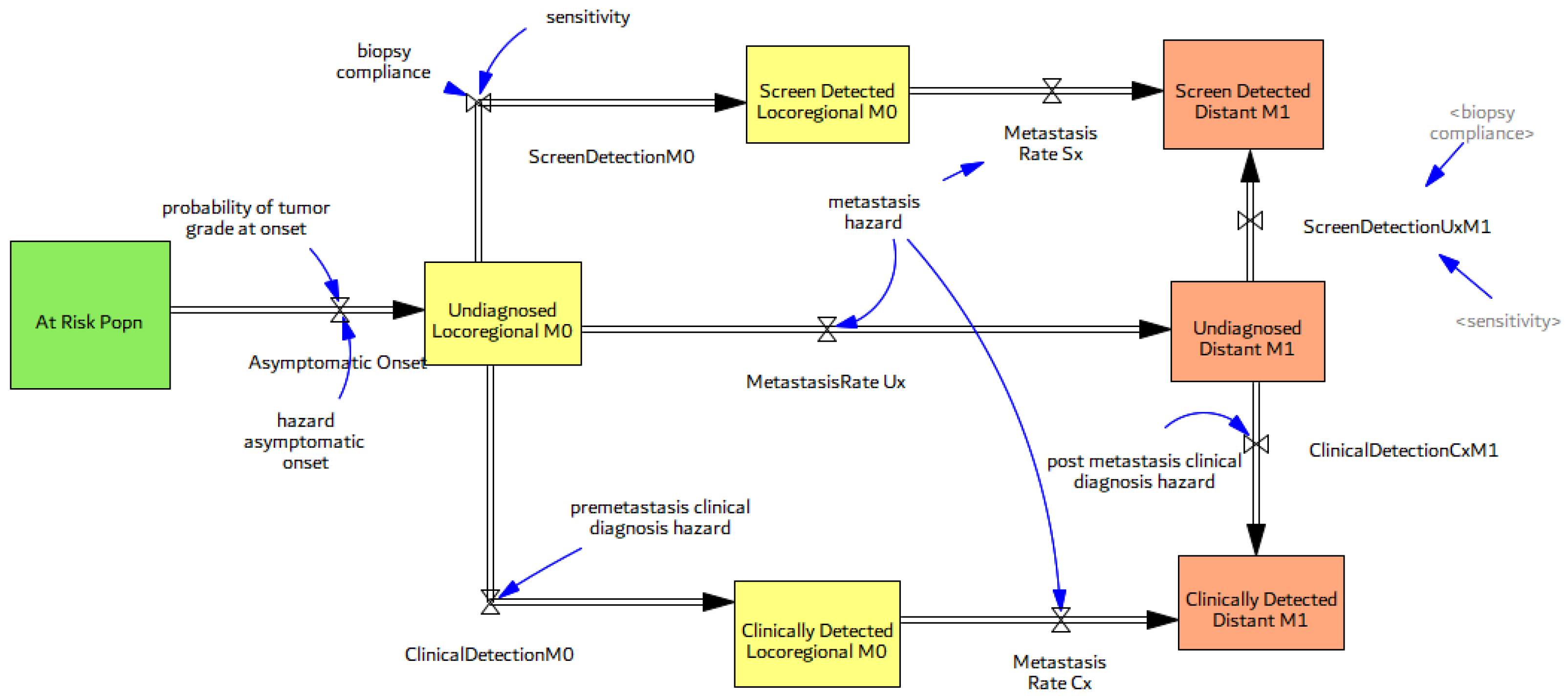
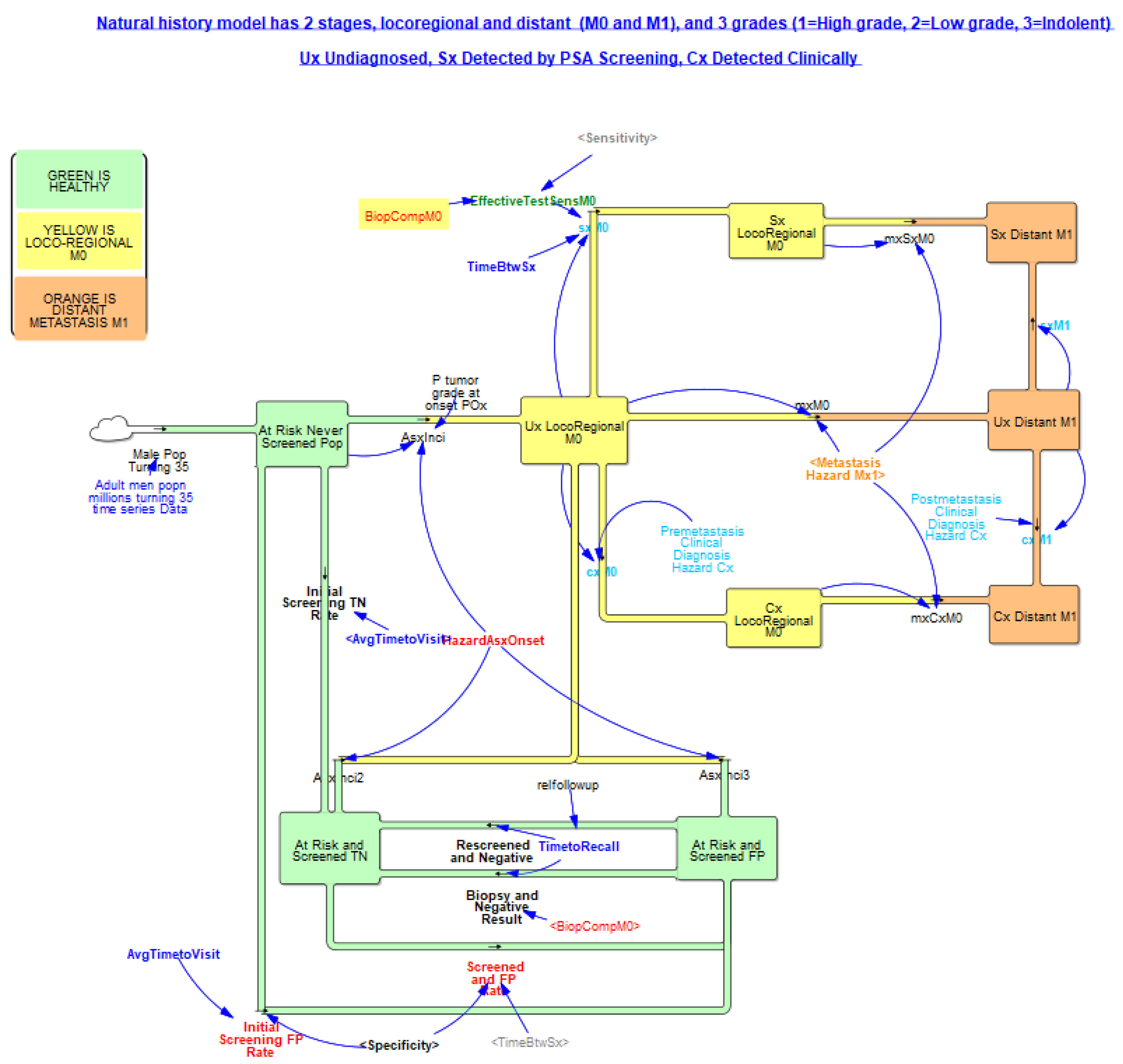
3.2.2. Natural History of Disease
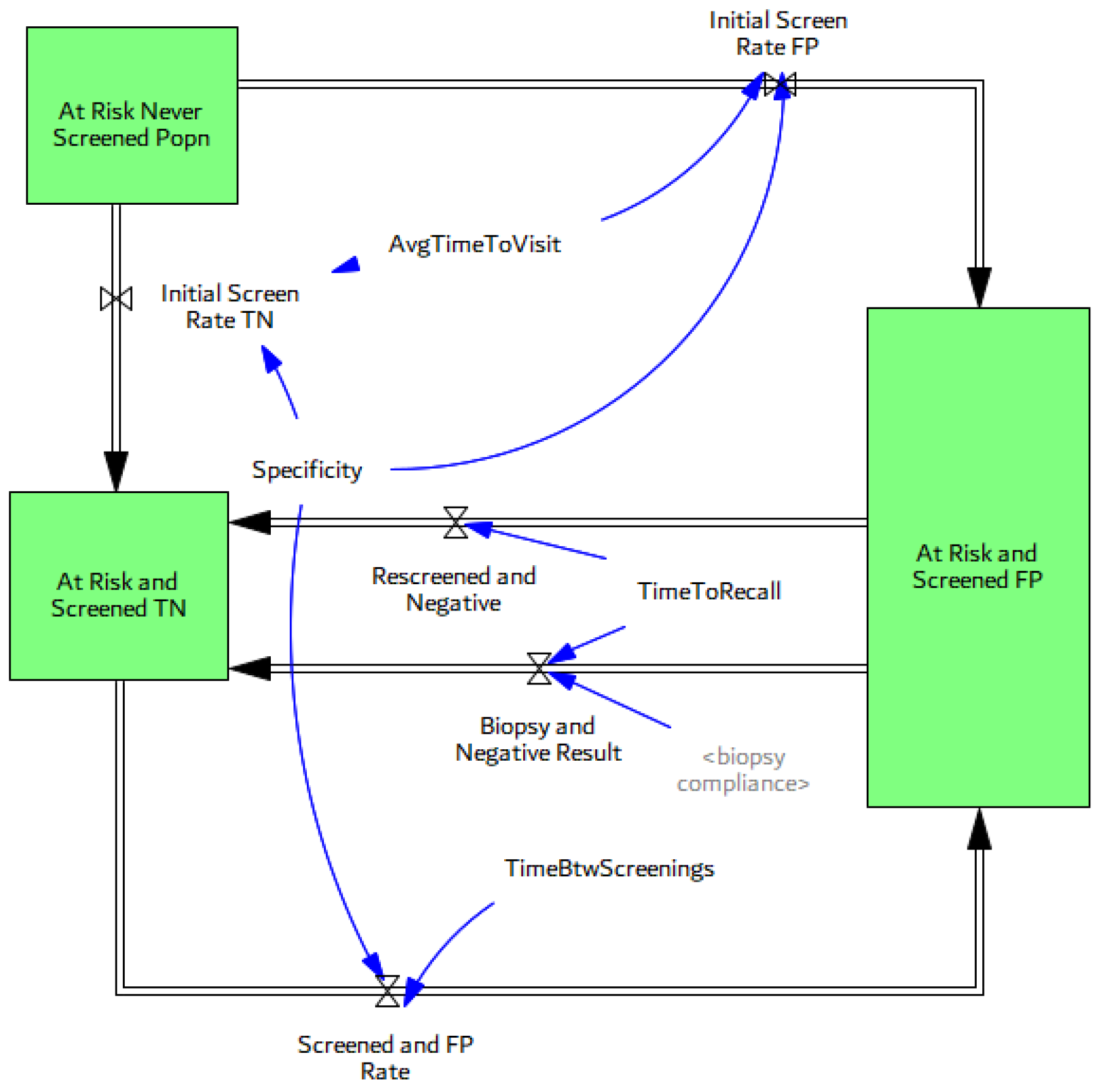
3.3. Screening, Clinical Detection, Dissemination of Screening
3.3.1. Screening Structure and Test Specifics
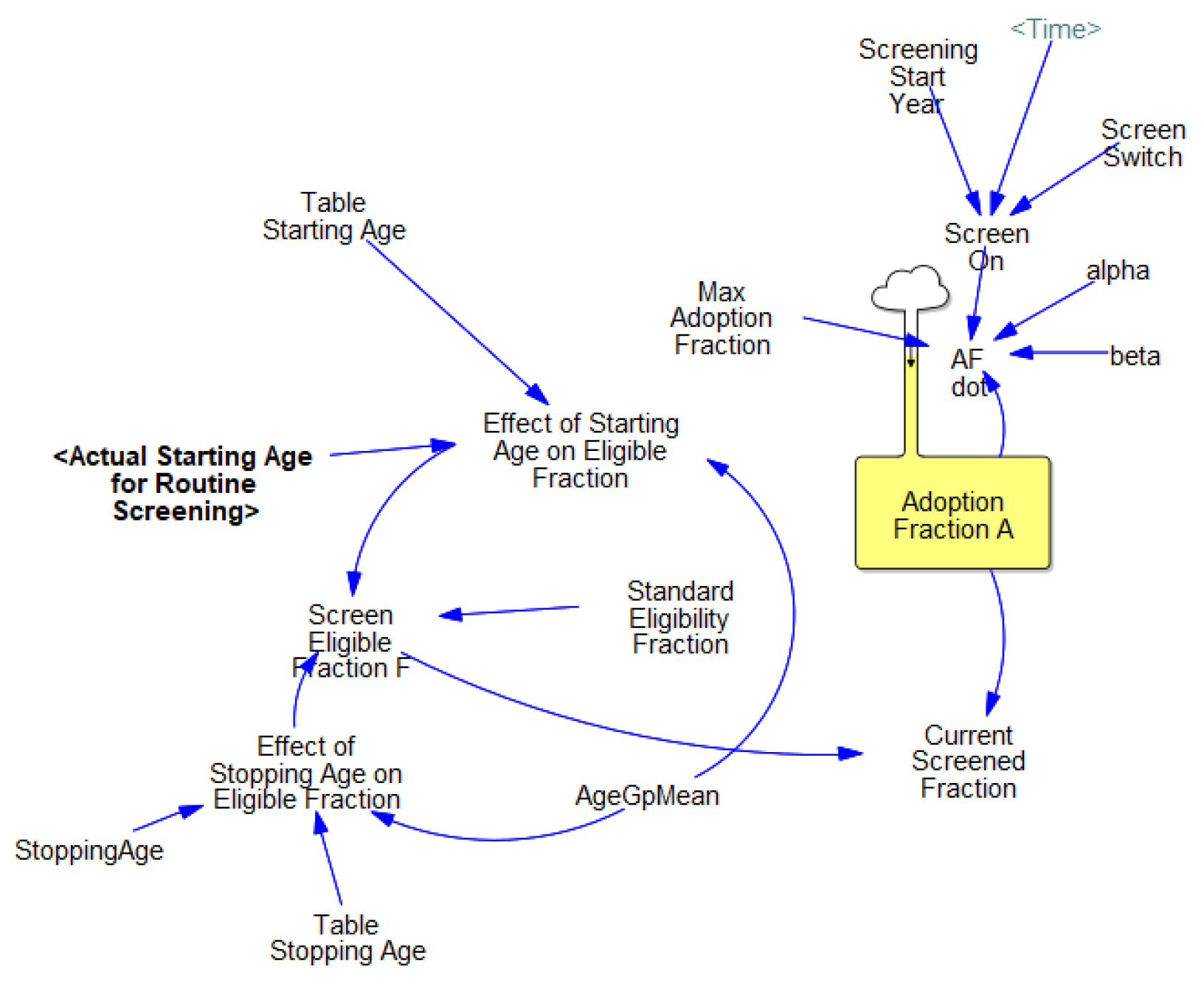
3.3.2. Screening Dissemination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Parameter [Unit] | Sensitivity Range | Source(s) for Base Case |
|---|---|---|---|
| Probability of indolent tumor at onset | pOx [dmnl] | 0.2–0.6 | Expert judgement |
| Hazard Asymptomatic Onset (by age group) PCa specific mortality fraction (by grade) | Oxi [1/year] dfM0, dfM1 [1/year] | 0.0–0.05 0.07–0.37 | [36,41,42] SEER survival curves by stage, [37,47,48] |
| Pre-metastasis clinical diagnosis hazard (by age, grade) | Cx1, Cx2 [1/year] | 0–0.03 | [37,39] |
| Multiplier for Hazard of Clinical Diagnosis (by age group) | MCx [dmnl] | 15–25 | [39] |
| Time between screenings | TimeBtwSx [year] | 1.5–2.5 | [39] |
| Biopsy compliance (by stage) | BiopCompM0/M1 [dmnl] | 0.3–0.7; 0.9 | [39,45] |
| Time to act | τ [year] | 0.25–0.5 | [49] |
| alpha | ⍺ [1/year] | 0.015–0.03 | Based on PSA curve [46] |
| beta | β [1/year] | 0.45–0.65 | Based on PSA curve [46] |
| Max adoption fraction | Amax [dmnl] | 0.25–0.9 | Expert judgement |
| Stopping age to screen | Agestop [years] | 70–85 | Expert judgement |
| HBR Translation Delay | λt [year] | 2–10 | Expert judgement |
4. Simulation Results
4.1. Basic Dynamics and Model Validation
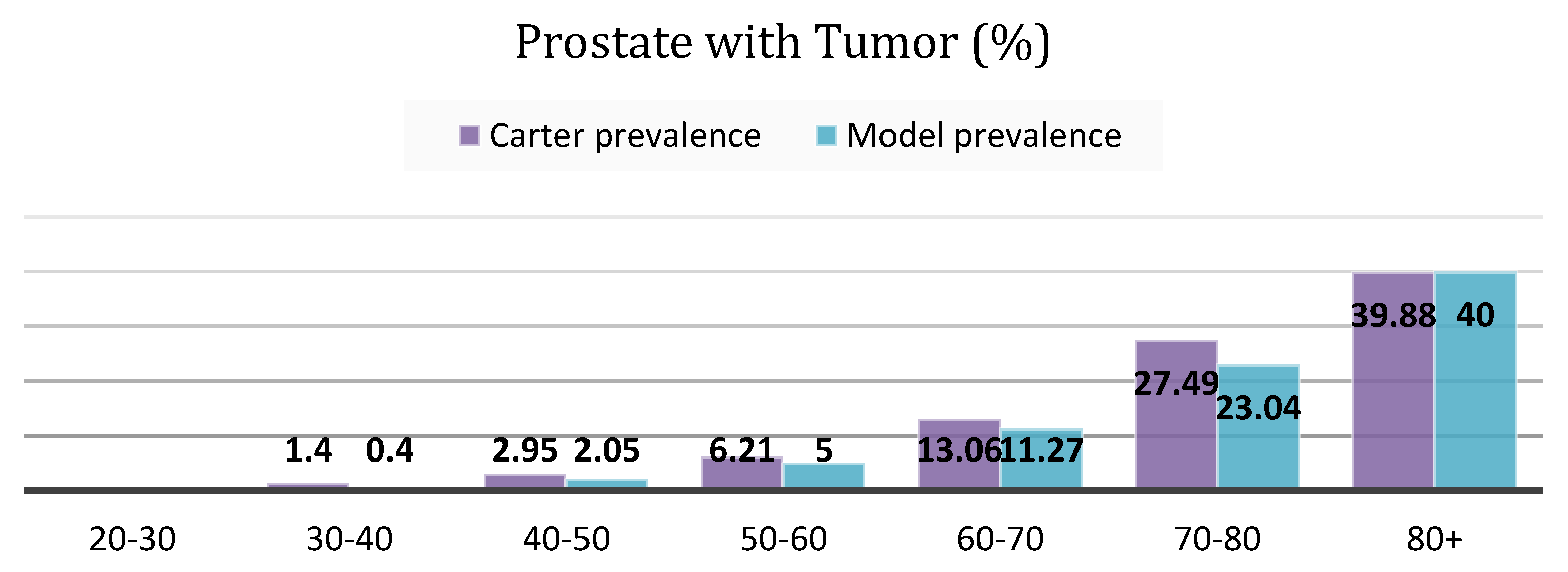
4.1.1. Cancer Prevalence
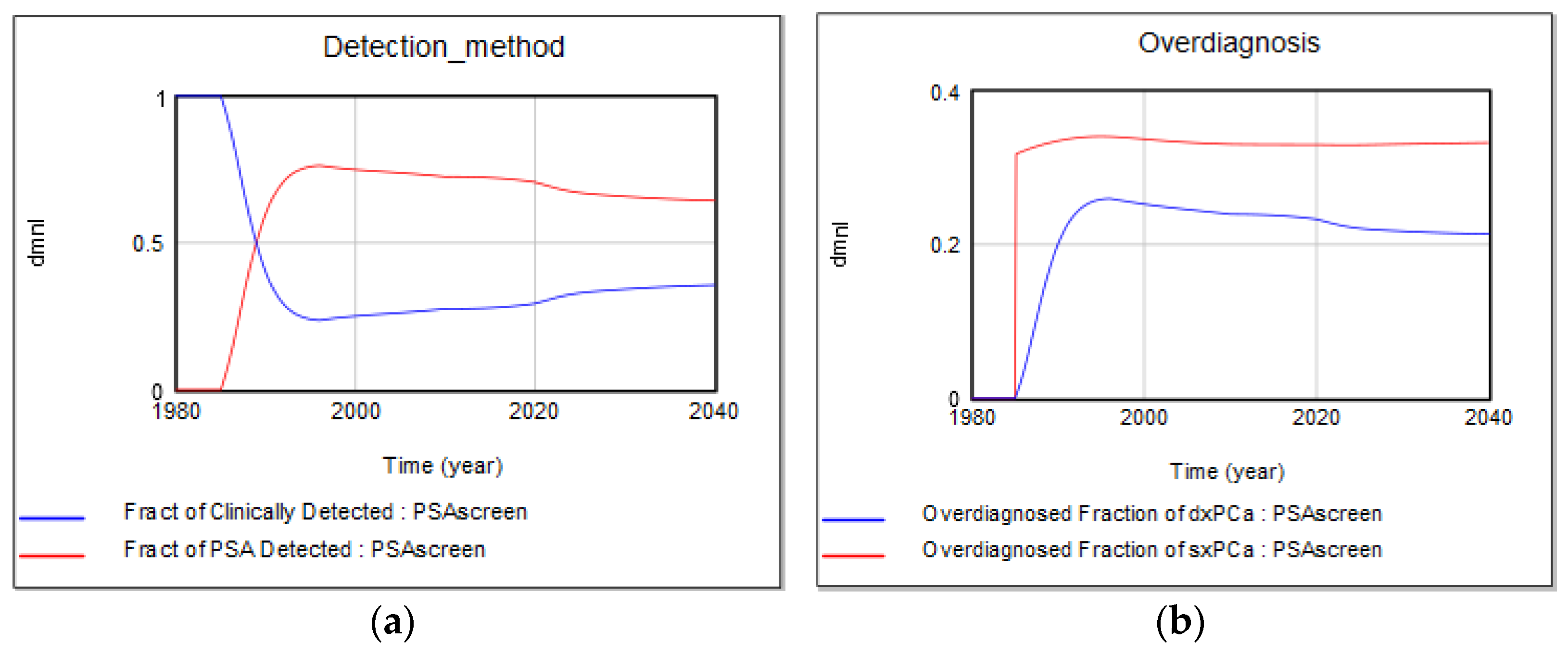
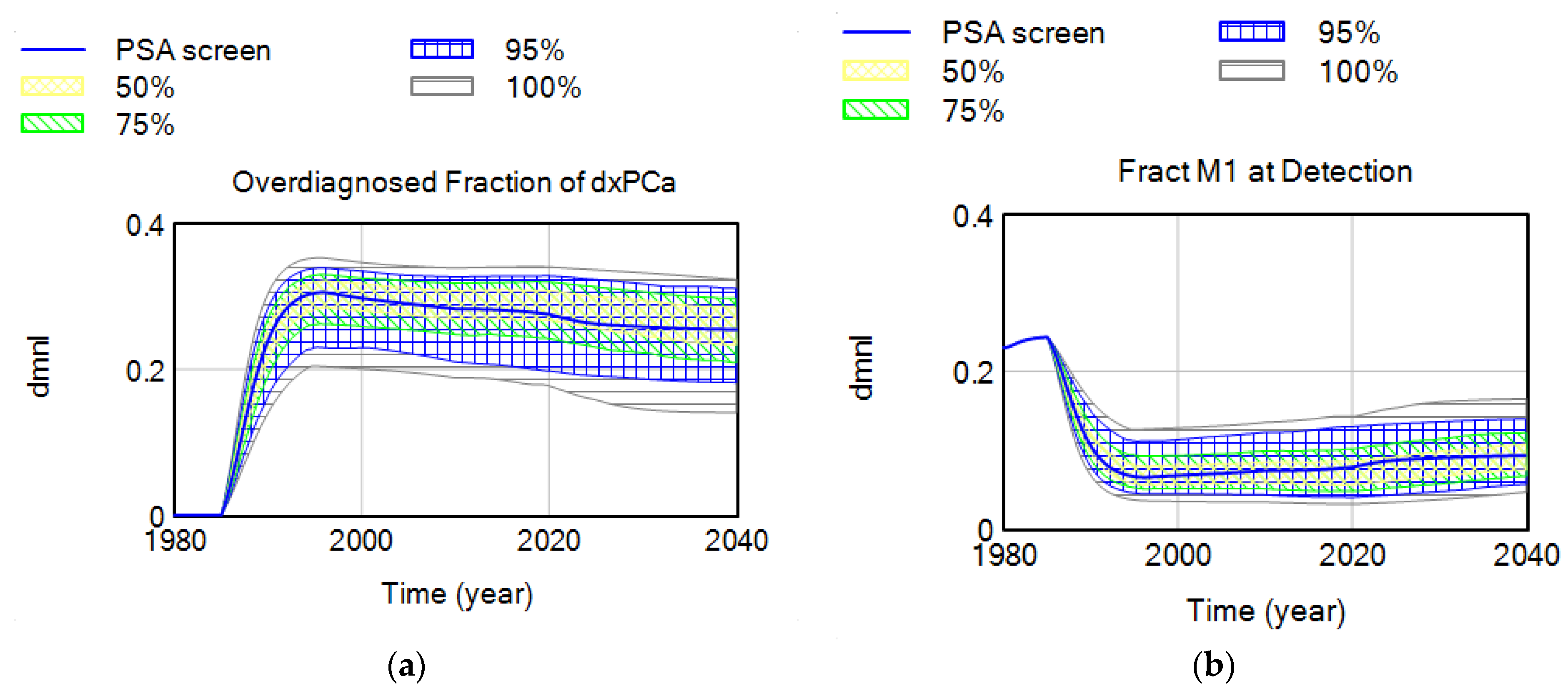
4.1.2. Screen vs. Clinical Detection and Overdiagnosis Rates
4.2. Sensitivity Testing
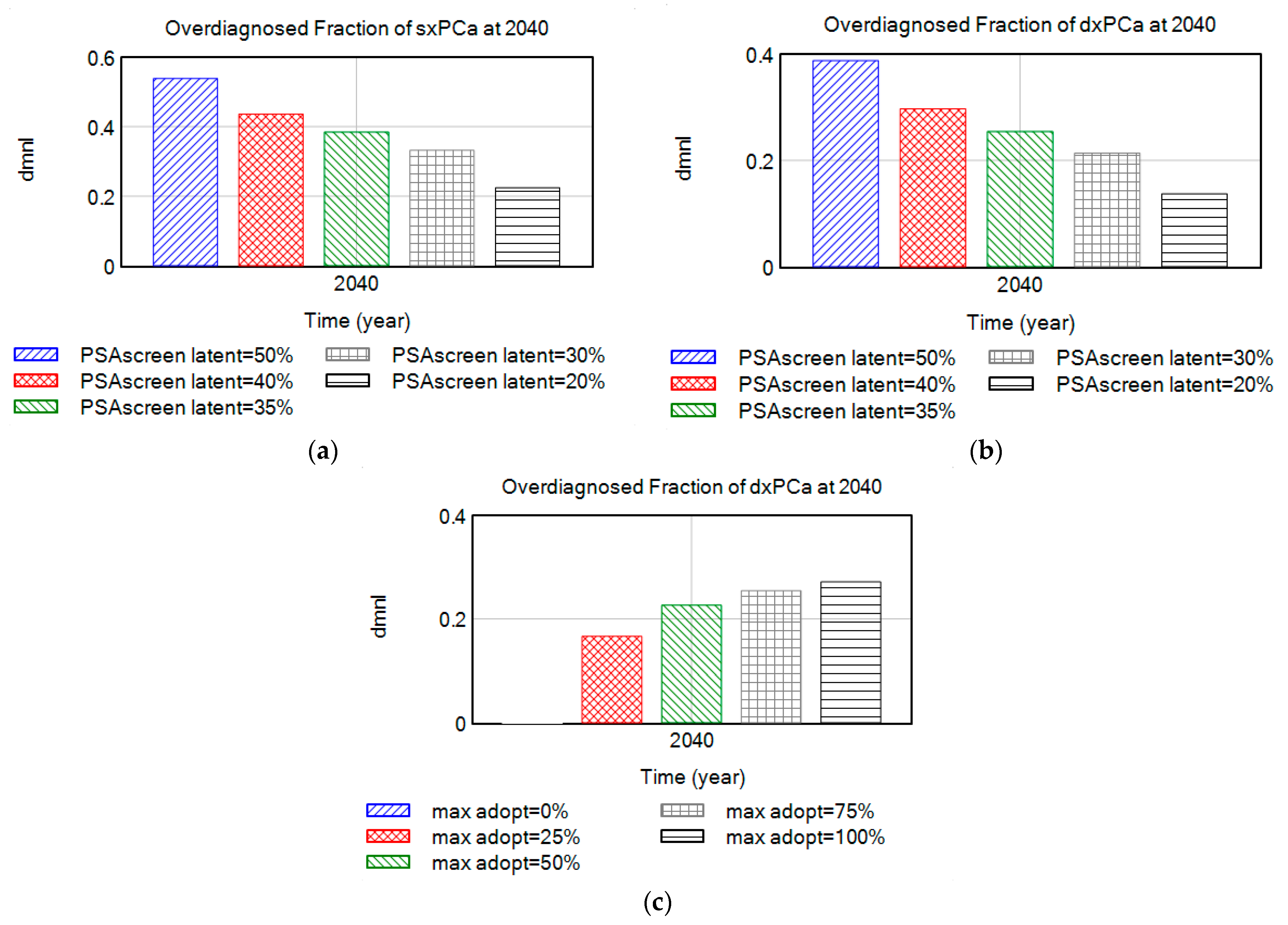
4.2.1. Over-Diagnosis Due to Large Pool of Indolent (Latent) Disease
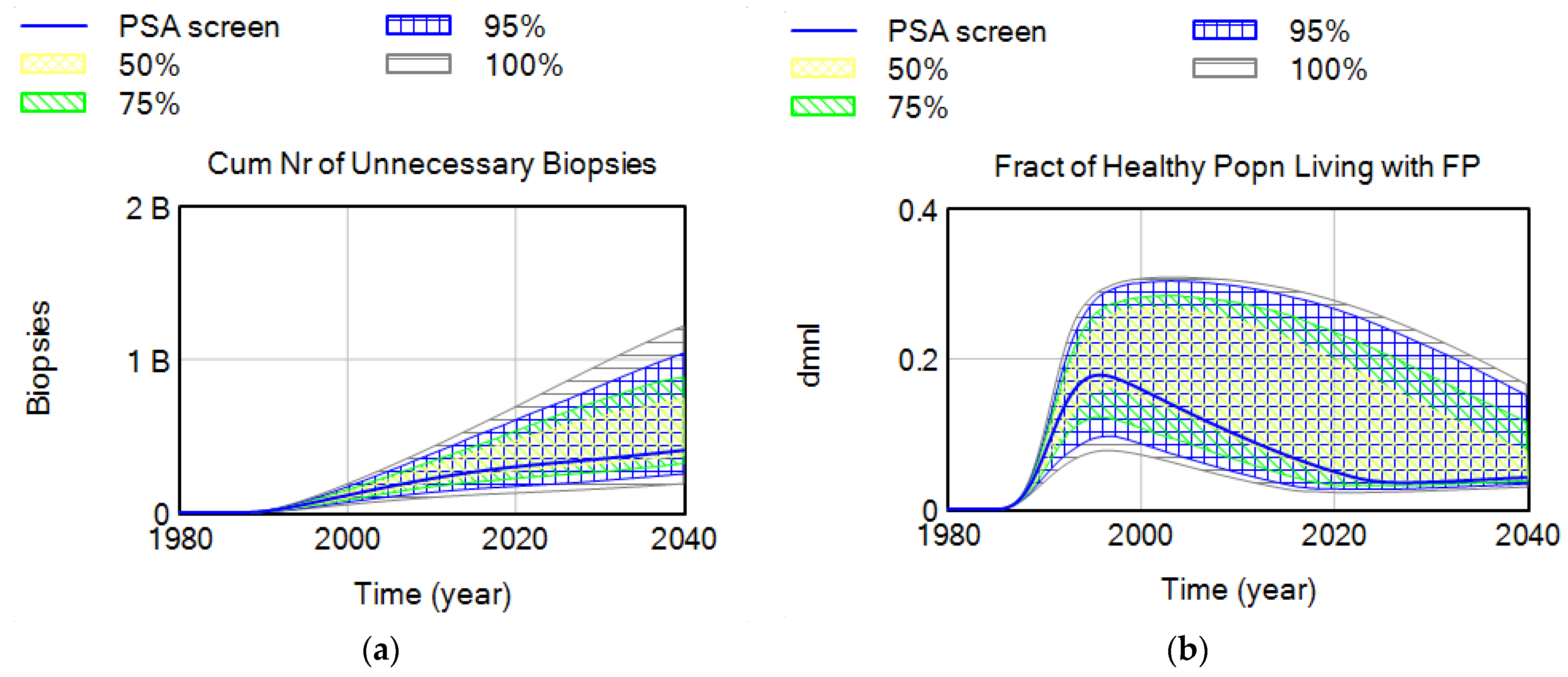
4.2.2. Parameter Set Exploration for Benefits and Harms of Screening
5. Discussion
5.1. Strategic Insights
- One of the important contributions of this study is the introduction of a more realistic yet flexible structure for routine medical screening that allows keeping track of critical stocks that have been generally overlooked in previous modeling studies. Existing modeling studies do not explicitly define some of these population stocks in the screening process, including men who currently live with an FP test result, which has implications for anxiety and depression. In this study, we use the flexibility of the SD modeling stock-flow structure to keep track and account for all critical stocks in the cancer screening problem, while their values are not readily measured in the literature. Simulations show that the fraction of healthy men who live with an FP may vary between 5 and 15% in most situations, depending on screening criteria, or breadth indications of disease, and may increase up to 30% with lower biopsy compliance rates. The value of the FP stock relative to the healthy population (i.e., “the fraction of healthy male population currently living with an FP”) may be an important indicator for policy making. Another variable for which we were not able to find historical data includes the the “fraction of disease in target screening population”. These metrics are potentially very important, yet not readily measured or considered in existing medical and modeling literature. Simulations show that the fraction of false positives in the healthy male population may have increased to as high as 18% in the 1990s when screening was overused. At the same time, the real diseased fraction of the target population must have dropped down to its historical minimum. We do not aim to suggest optimal estimates for these variables but would like to highlight the importance of having a better understanding of their dynamics by additional data collection, rather than excluding them from our “mental” models, or making the constancy assumption [52].
- The addition of an “indolent/latent” disease category is a novel addition in this modeling study, facilitating to make of inferences about the real (yet unknown) occult disease prevalence in the population. One aspect that increases the reported cancer prevalence is the existence of a silent pool of indolent diseases, which varies among different types of cancers. These are “TP” cases where the disease identified has uncertain significance, and where men would never become aware of their disease if they were not tested for it, as evidenced by the silent reservoirs of undetected thyroid, breast, and prostate [41] cancers. Our interview data for the PCa case study confirms the importance of the size of this latent pool of disease: “…If you take enough time to understand what this means, if I tell a patient, “Look I’m 47, my probability to have a prostate cancer histologically under the microscope right now as I sit here, is about 30%. Period.” That’s a start, so there’s a pool of prostate cancer that we all carry, most of them they’ll never become symptomatic, some of us have to have bad cards. Do we understand who have bad cards and who don’t? No, we don’t. There’s a residual risk that there’s something going on. “—Peter Juni. MD-PhD, Director, Applied Health Research Centre, St. Michael’s Hospital, and Professor of Medicine, University of Toronto, Previous: University of Bern, Director of the Institute of Primary Health Care, Professor and Chair of Primary Health Care and Clinical Epidemiology in the Faculty of Medicine, Switzerland.
- We endogenize variables that are mostly taken as constants in other studies. These include the breadth of indications of screening (including the biopsy threshold and the starting age to screen), the prevalence of disease in the screening population, the sensitivity and specificity of the test, and the harm reduction technology. It also separates the formal decision thresholds for screening from the decision thresholds that are implemented, showing their interdependency to each other as well as to the diagnostics of the test. For example, existing studies usually assume a constant PSA level as the trigger for biopsy, which stays constant over time, but this is not an accurate reflection of the clinical practice. Endogenizing such variables allows us to show how they are changing over time, affecting the target screening prevalence, and, hence, the screening diagnostics themselves which are also taken as constants in most studies. To be specific, the model endogenizes the adoption and diffusion of the screening process and defines the different components of screen detection explicitly. These include the fraction of the population that receives the screening test, the sensitivity of the test, biopsy compliance, and biopsy detection. The test sensitivity and currently screened fraction are endogenous to the model, while biopsy compliance and detection are exogenous. Subjects are eligible to receive regular screenings if their doctor adopted the PSA screening test at the time, and if they are around the age-eligible range for the test. Interview results confirm that one of the main determinants of screening is the doctor’s opinion: “Access to care, coverage, and I also think it is how the screening is presented by their doctor….a lot of medicine is sales, and if a doctor presents something as either optional or a bad idea like, “You don’t really want to do that, do you?” the patient’s going to say no. But if their doctor’s enthusiastic about it and believes in it, then they’re probably more likely to go ahead and get it done…”—MD, PhD Erin Hofstatter, Medical Oncologist, Yale School of Medicine.
- Karanfil and Sterman show that the “formal” recommended starting age to screen varies, both over time and between different guideline-issuing organizations [32]. The recommended “formal” biopsy threshold for PSA testing stayed constant at 4 ng/mL throughout the initial years of screening dissemination, after which it starts to vary in the 2000s. The informal, “practice” threshold, however, has reportedly been lower than the formal one, suggesting poor compliance with recommendations. The real pattern for the average biopsy threshold is unknown, but it is generally accepted to be 2.5 ng/mL between 1990 and 2000 [39]. In addition, Pinsky et al. [53] have shown that biopsy frequencies of men with PSAs between 2.5 and 4 ng/mL were of the same order of magnitude as for men with a PSA higher than 4 ng/mL. The actual starting age data are also not available, but they presumably follow the same pattern as the biopsy threshold, where formal indications first expand in the early years of screening and then start to narrow as harms and the evidence for harms accumulate over time.
- Since the test diagnostics are directly derived from the underlying probability distributions for diseased and healthy people, the model can as well be used to estimate the real prevalence of the disease.
5.2. Limitations and Further Research
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AACR | American Association for Cancer Research |
| CDC | Centers for Disease Control and Prevention |
| CISNET | Intervention and Surveillance Modeling Network |
| COUHES | Committee on the Use of Humans as Experimental Subjects |
| CPG | Clinical Practice Guidelines |
| DRE | Digital Rectal Exam |
| FN | False Negative |
| FP | False Positive |
| HBR | Harms and Benefits Ratio |
| HRT | Harm Reduction Technology |
| NCHS | National Center for Health Statistics |
| NCI | National Cancer Institute |
| NHANES | National Health and Nutrition Examination Survey. |
| NHIS | National Health Interview Survey |
| PCa | Prostate Cancer |
| PLCO | Prostate, Lung, Colorectal and Ovarian Trial |
| PSA | Prostate Specific Antigen |
| SEER | Surveillance, Epidemiology, and End Results |
| USPSTF | U.S. Preventive Services Task Force |
| TP | True Positive |
| TN | True Negative |
References
- Hoffman, J.R.; Cooper, R.J. Overdiagnosis of Disease. Arch. Intern. Med. 2012, 172, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Esserman, L.J.; Thompson, I.M.; Reid, B.; Nelson, P.; Ransohoff, D.F.; Welch, H.G.; Hwang, S.; Berry, D.A.; Kinzler, K.W.; Black, W.C.; et al. Addressing overdiagnosis and overtreatment in cancer: A prescription for change. Lancet Oncol. 2014, 15, e234–e242. [Google Scholar] [CrossRef] [PubMed]
- Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018; Available online: https://www.cancer.org/research/cancer-facts-statistics/global.html (accessed on 12 April 2023).
- Seidman, H.; Mushinski, M.H.; Gelb, S.K.; Silverberg, E. Probabilities of Eventually Developing or Dying of Cancer--United States, 1985. CA Cancer J. Clin. 1985, 35, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Kosary, C.L.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatalovich, Z.; Cho, H.; et al. SEER Cancer Statistics Review, 1975–2007. 2010. Available online: http://seer.cancer.gov/csr/1975_2007/ (accessed on 12 April 2023).
- Faiena, I.; Holden, S.; Cooperberg, M.R.; Soule, H.R.; Simons, J.; Morgan, T.M.; Penson, D.; Morgans, A.K.; Hussain, M. Prostate Cancer Screening and the Goldilocks Principle: How Much Is Just Right? J. Clin. Oncol. 2018, 36, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Ebell, M.H.; Thai, T.N.; Royalty, K.J. Cancer screening recommendations: An international comparison of high income countries. Public Health Rev. 2018, 39, 7. [Google Scholar] [CrossRef]
- US Preventive Services Task Force. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1901–1913. [Google Scholar] [CrossRef]
- SEER. Cancer of the Prostate—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 3 March 2023).
- Sterman, J.D. Business Dynamics: Systems Thinking and Modeling for a Complex World; Irwin/McGraw-Hill: Boston, MA, USA, 2000. [Google Scholar]
- Homer, J.B.; Hirsch, G.B. System Dynamics Modeling for Public Health: Background and Opportunities. Am. J. Public Health 2006, 96, 452–458. [Google Scholar] [CrossRef]
- Sterman, J.D. Learning from Evidence in a Complex World. Am. J. Public Health 2006, 96, 505–514. [Google Scholar] [CrossRef]
- Homer, J.B. A diffusion model with application to evolving medical technologies. Technol. Forecast. Soc. Change 1987, 31, 197–218. [Google Scholar] [CrossRef]
- Hirsch, G.; Trogdon, J.; Wile, K.; Orenstein, D. Using Simulation to Compare 4 Categories of Intervention for Reducing Cardiovascular Disease Risks. Am. J. Public Health 2014, 104, 1187–1195. [Google Scholar] [CrossRef]
- Milstein, B.; Homer, J.; Hirsch, G. Analyzing National Health Reform Strategies with a Dynamic Simulation Model. Am. J. Public Health 2010, 100, 811–819. [Google Scholar] [CrossRef]
- Yarnoff, B.; Honeycutt, A.; Bradley, C.; Khavjou, O.; Bates, L.; Bass, S.; Kaufmann, R.; Barker, L.; Briss, P. Validation of the Prevention Impacts Simulation Model (PRISM). In Prev. Chronic Dis.; 2021; 18, p. E09. Available online: www.cdc.gov/pcd/issues/2021/20_0225.htm (accessed on 12 April 2023). [CrossRef]
- Atun, R.A.; Lebcir, R.M.; McKee, M.; Habicht, J.; Coker, R.J. Impact of joined-up HIV harm reduction and multidrug resistant tuberculosis control programmes in Estonia: System dynamics simulation model. Health Policy 2006, 81, 207–217. [Google Scholar] [CrossRef]
- Burns, W.J.; Slovic, P. The Diffusion of Fear: Modeling Community Response to a Terrorist Strike (2006). Available online: https://ssrn.com/abstract=912736; http://dx.doi.org/10.2139/ssrn.912736 (accessed on 12 April 2023).
- Ghaffarzadegan, N.; Rahmandad, H. Simulation-based estimation of the early spread of COVID-19 in Iran: Actual versus confirmed cases. Syst. Dyn. Rev. 2020, 36, 101–129. [Google Scholar] [CrossRef]
- Rahmandad, H.; Sterman, J. Quantifying the COVID-19 endgame: Is a new normal within reach? Syst. Dyn. Rev. 2022, 38, 329–353. [Google Scholar] [CrossRef]
- Lim, T.Y.; Stringfellow, E.J.; Stafford, C.A.; DiGennaro, C.; Homer, J.B.; Wakeland, W.; Eggers, S.L.; Kazemi, R.; Glos, L.; Ewing, E.G.; et al. Modeling the Evolution of the US Opioid Crisis for National Policy Development. Proc. Natl. Acad. Sci. USA 2022, 119, e2115714119. [Google Scholar] [CrossRef]
- Darabi, N.; Hosseinichimeh, N. System dynamics modeling in health and medicine: A systematic literature review. Syst. Dyn. Rev. 2020, 36, 29–73. [Google Scholar] [CrossRef]
- Rahmandad, H.; Sterman, J.D. Reporting guidelines for simulation-based research in social sciences. Syst. Dyn. Rev. 2012, 28, 396–411. [Google Scholar] [CrossRef]
- Martinez-Moyano, I.J. Documentation for model transparency. Syst. Dyn. Rev. 2012, 28, 199–208. [Google Scholar] [CrossRef]
- Richmond, B. Towards a Structural Theory of Cancer. D-Memos. D-4151. MIT Sloan School of Management, Cambridge, MA, USA, 1990. Available online: https://systemdynamics.org/d-memos-4000-4499/ (accessed on 12 April 2023).
- Fett, M.J. Developing Simulation Dynamic Models of Breast Cancer Screening; Wellington, New Zealand, 1999. Available online: https://proceedings.systemdynamics.org/1999/PAPERS/PARA47.PDF (accessed on 12 April 2023).
- Fett, M. Computer modelling of the Swedish two county trial of mammographic screening and trade offs between participation and screening interval. J. Med. Screen. 2001, 8, 39–45. [Google Scholar] [CrossRef]
- Royston, G.; Dost, A.; Townshend, J.; Turner, H. Using System Dynamics to Help Develop and Implement Policies and Programmes in Health Care in England. Syst. Dyn. Rev. 1999, 15, 293–313. [Google Scholar] [CrossRef]
- Sheldrick, R.C.; Breuer, D.J.; Hassan, R.; Chan, K.; Polk, D.E.; Benneyan, J. A System Dynamics Model of Clinical Decision Thresholds for the Detection of Developmental-Behavioral Disorders. Implement. Sci. 2016, 11, 156. [Google Scholar] [CrossRef]
- Sheldrick, R.C.; Garfinkel, D. Is a Positive Developmental-Behavioral Screening Score Sufficient to Justify Referral? A Review of Evidence and Theory. Acad. Pediatr. 2017, 17, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.; Lounsbury, D.W.; Schlecht, N.F.; Agalliu, I. A System Dynamics Model of Serum Prostate-Specific Antigen Screening for Prostate Cancer. Am. J. Epidemiol. 2015, 183, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Karanfil, Ö.; Sterman, J. “Saving lives or harming the healthy?” Overuse and fluctuations in routine medical screening. Syst. Dyn. Rev. 2020, 36, 294–329. [Google Scholar] [CrossRef]
- Karanfil, Ö. Why Clinical Practice Guidelines Shift over Time: A Dynamic Model with Application to Prostate Cancer Screening. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2016. Available online: http://dspace.mit.edu/handle/1721.1/107531 (accessed on 12 April 2023).
- United States Department of Health and Human Services (US DHHS), Centers for Disease Control and Prevention (CDC), National Center for Health Statistics (NCHS), Compressed Mortality File (CMF) on CDC WONDER Online Database. Available online: https://wonder.cdc.gov/mortsql.html (accessed on 12 April 2023).
- Life Tables for the United States Social Security Area 1900–2100. August 2025. Available online: https://www.ssa.gov/oact/NOTES/as120/TOC.html (accessed on 12 April 2023).
- Cowen, M.E.; Chartrand, M.; Weitzel, W.F. A Markov model of the natural history of prostate cancer. J. Clin. Epidemiol. 1994, 47, 3–21. [Google Scholar] [CrossRef]
- Etzioni, R.; Cha, R.; Cowen, M.E. Serial prostate specific antigen screening for prostate cancer: A computer model evaluates competing strategies. J. Urol. 1999, 162, 741–748. [Google Scholar] [CrossRef]
- Tsodikov, A.; Szabo, A.; Wegelin, J. A population model of prostate cancer incidence. Stat. Med. 2006, 25, 2846–2866. [Google Scholar] [CrossRef]
- Gulati, R.; Inoue, L.; Katcher, J.; Hazelton, W.; Etzioni, R. Calibrating disease progression models using population data: A critical precursor to policy development in cancer control. Biostatistics 2010, 11, 707–719. [Google Scholar] [CrossRef]
- Ayer, T.; Alagoz, O.; Stout, N.K. OR Forum—A POMDP Approach to Personalize Mammography Screening Decisions. Oper. Res. 2012, 60, 1019–1034. [Google Scholar] [CrossRef]
- Jahn, J.L.; Giovannucci, E.L.; Stampfer, M.J. The high prevalence of undiagnosed prostate cancer at autopsy: Implications for epidemiology and treatment of prostate cancer in the Prostate-specific Antigen-era. Int. J. Cancer 2015, 137, 2795–2802. [Google Scholar] [CrossRef]
- Bell, K.J.; Del Mar, C.; Wright, G.; Dickinson, J.; Glasziou, P. Prevalence of incidental prostate cancer: A systematic review of autopsy studies. Int. J. Cancer 2015, 137, 1749–1757. [Google Scholar] [CrossRef]
- Haas, G.P.; Delongchamps, N.; Brawley, O.W.; Wang, C.Y.; de la Roza, G. The Worldwide Epidemiology of Prostate Cancer: Perspectives from Autopsy Studies. Can. J. Urol. 2008, 15, 3866–3871. [Google Scholar]
- Scardino, P.T.; Beck, J.R.; Miles, B.J. Conservative management of prostate cancer. N. Engl. J. Med. 1994, 330, 1831; author reply 1831–1832. [Google Scholar]
- Andriole, G.L.; Crawford, E.D.; Grubb, R.L.; Buys, S.S.; Chia, D.; Church, T.R.; Fouad, M.N.; Isaacs, C.; Kvale, P.A.; Reding, D.J.; et al. Prostate Cancer Screening in the Randomized Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial: Mortality Results after 13 Years of Follow-up. Gynecol. Oncol. 2012, 104, 125–132. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Etzioni, R.; Krapcho, M.; Feuer, E.J. Reconstructing PSA testing patterns between black and white men in the US from Medicare claims and the National Health Interview Survey. Cancer 2007, 109, 1877–1886. [Google Scholar] [CrossRef]
- Messing, E.M.; Manola, J.; Yao, J.; Kiernan, M.; Crawford, D.; Wilding, G.; di’SantAgnese, P.A.; Trump, D. Immediate versus deferred androgen deprivation treatment in patients with node-positive prostate cancer after radical prostatectomy and pelvic lymphadenectomy. Lancet Oncol. 2006, 7, 472–479. [Google Scholar] [CrossRef]
- Aus, G.; Robinson, D.; Rosell, J.; Sandblom, G.; Varenhorst, E. Survival in prostate carcinoma—Outcomes from a prospective, population-based cohort of 8887 men with up to 15 years of follow-up. Cancer 2005, 103, 943–951. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Harlan, L.C.; Klabunde, C.N.; Gilliland, F.D.; Stephenson, R.A.; Hunt, W.C.; Potosky, A.L. Racial differences in initial treatment for clinically localized prostate cancer. J. Gen. Intern. Med. 2003, 18, 845–853. [Google Scholar] [CrossRef]
- Carter, H.B.; Piantadosi, S.; Isaacs, J.T. Clinical Evidence for and Implications of the Multistep Development of Prostate Cancer. J. Urol. 1990, 143, 742–746. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Zeliadt, S.B. The Cautionary Tale of Psa Testing: Comment on ‘risk Profiles and Treatment Patterns among Men Diagnosed as Having Prostate Cancer and a Prostate-Specific Antigen Level below 4.0 Ng/mL. Arch. Intern. Med. 2010, 170, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Forrester, J.W. 14 ‘Obvious Truths’. Syst. Dyn. Rev. 1987, 3, 156–159. [Google Scholar] [CrossRef]
- Pinsky, P.F.; Andriole, G.L.; Kramer, B.S.; Hayes, R.; Prorok, P.C.; Gohagan, J.K. Prostate biopsy following a positive screen in the prostate, lung, colorectal and ovarian cancer screening trial. J. Urol. 2005, 173, 746–751; discussion 750–751. [Google Scholar] [CrossRef] [PubMed]
- Castellano, T.; Moore, K.; Ting, J.; Washington, C.; Yildiz, Y.; Surinach, A.; Sonawane, K.; Chhatwal, J.; Ayer, T. Cervical cancer geographical burden analyzer: An interactive, open-access tool for understanding geographical disease burden in patients with recurrent or metastatic cervical cancer. Gynecol. Oncol. 2022, 169, 113–117. [Google Scholar] [CrossRef]
- Cancer Progress Report. AACR Cancer Progress Report. Available online: https://cancerprogressreport.aacr.org/progress/ (accessed on 9 April 2023).










| Extreme Condition Test | Qualitative Behavior |
|---|---|
| Screening switch turned off | PSA screening tests go to zero, % Ever had PSA goes to zero, % of Screen detected cancer goes to zero, % of Clinically detected cancer goes to 100%, Reported PCa prevalence goes down, % of men healthy with a FP goes to zero, no detection, and treatment of latent (indolent) disease |
| Clinical detection switch turned off | % of Cancer clinically detected goes to zero, All cancer detection is through PSA screening |
| Both screen and clinical detection switches turned off | Reported PCa incidence goes to zero, Reported PCa prevalence goes to zero, no new PCa cancer survivors |
| Treatment switch turned off | % Ever treated goes to zero, There are no survivors with primary treatment |
| Treatment is 100% effective | No one dies of prostate cancer, M0 and M1 PCa deaths go to zero |
| Metastasis switch turned off | M0 loco-regional disease doesn’t get metastasized, no distant M1 cases, no M1 prostate cancer deaths |
| All-cause mortality turned off | Mean population age increases, only deaths are PCa deaths |
| Decrease in mortality trend is removed | Overall deaths increase, population’s mean age goes down |
| All disease is indolent | No prostate cancer deaths, 100% overdiagnosis |
| Other logic tests | |
| % PSA detected | % of disease detected by screening is 100% for indolent disease (Latent cancer cannot get detected clinically) |
| PCa incidence/prevalence | Reported PCa incidence is higher for older age groups |
| % Loco-regional at detection | 100% for latent disease, as latent disease cannot get metastasized to M1 disease |
| % Distant at detection | 0% for latent disease, higher for higher grade cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karanfil, Ö. Dynamics of Medical Screening: A Simulation Model of PSA Screening for Early Detection of Prostate Cancer. Systems 2023, 11, 252. https://doi.org/10.3390/systems11050252
Karanfil Ö. Dynamics of Medical Screening: A Simulation Model of PSA Screening for Early Detection of Prostate Cancer. Systems. 2023; 11(5):252. https://doi.org/10.3390/systems11050252
Chicago/Turabian StyleKaranfil, Özge. 2023. "Dynamics of Medical Screening: A Simulation Model of PSA Screening for Early Detection of Prostate Cancer" Systems 11, no. 5: 252. https://doi.org/10.3390/systems11050252