
Genetic Markers for Metabarcoding of Freshwater Microalgae: Review

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
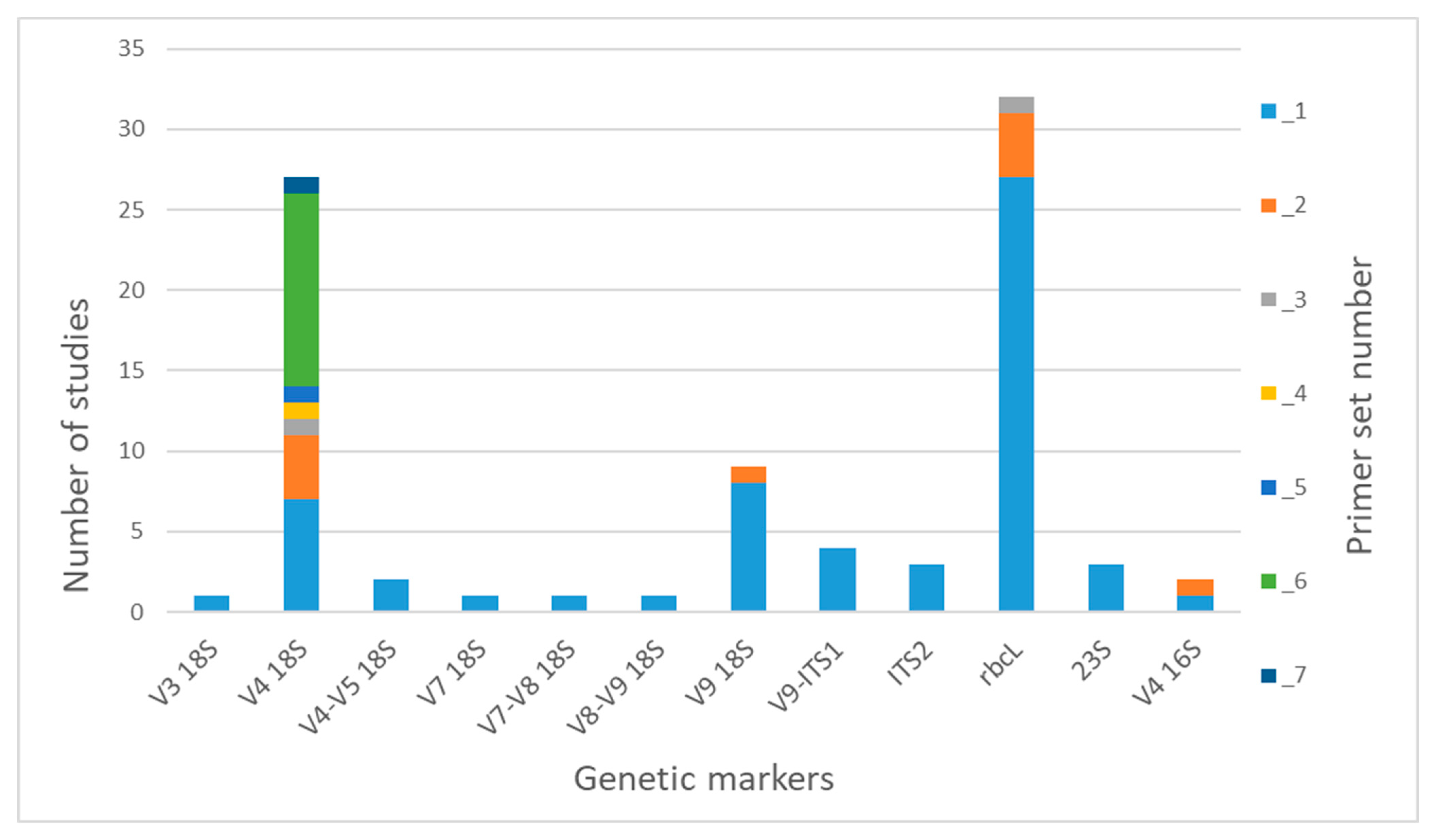
3.1. Gene Markers and Primer Sets for Freshwater Microalgae Metabarcoding
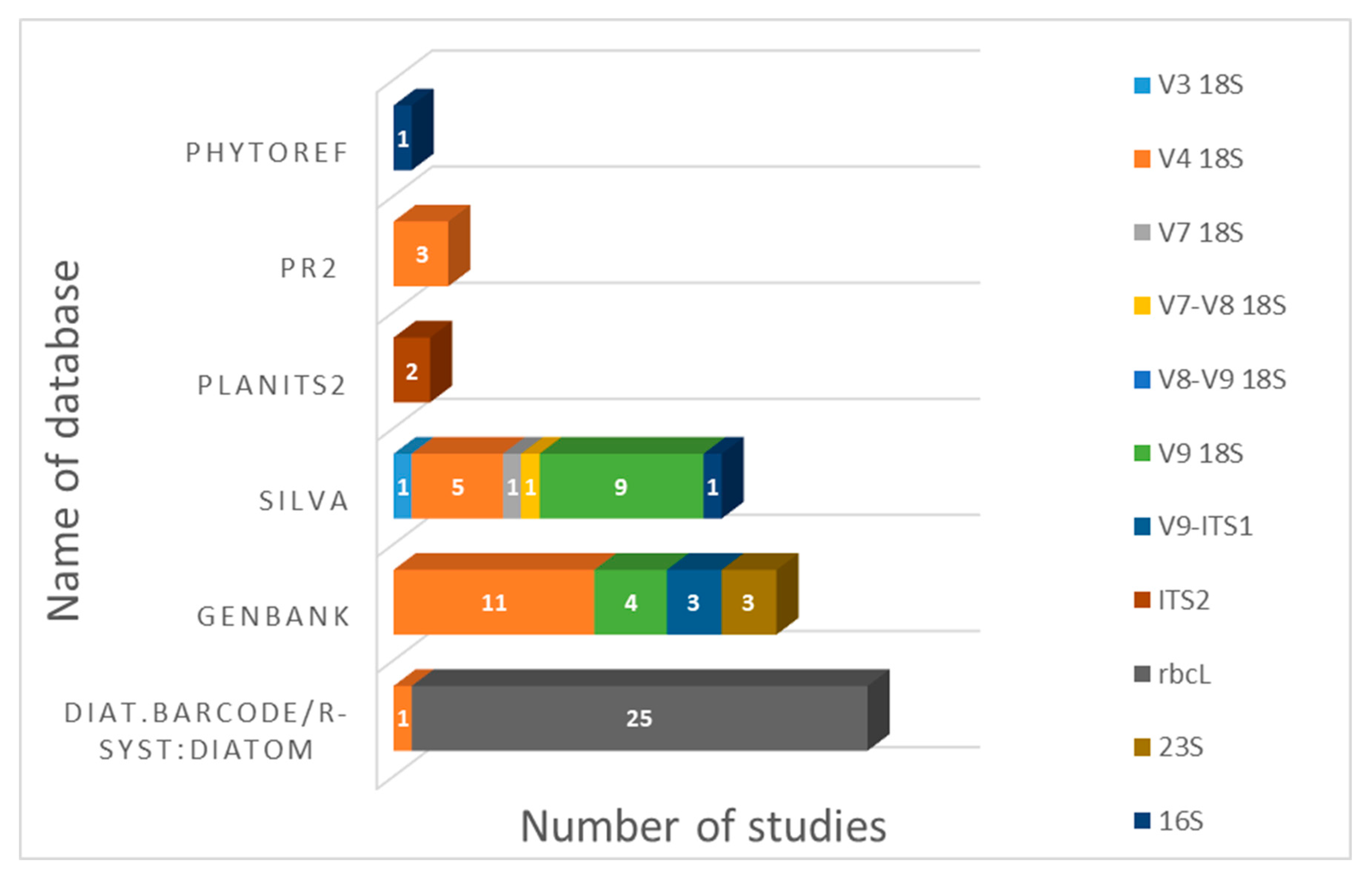
3.2. Reference Databases for Sequence Interpretation
3.3. First Works on Testing Genetic Markers on Monoclonal Microalgal Cultures Provide Insight on the Effectiveness of Amplification and the Resolution of Species Differentiation
3.4. 18S—Choosing a Variable Barcode Region for Eukaryotes In Silico
3.5. 18S rRNA Gene Metabarcoding: V4 vs. V9
3.6. Internal Transcribed Spacer Ribosomal DNA (ITS) in Metabarcoding Researches
3.7. Gene Markers for Diatoms
3.8. Specific Primers Targeted to rbcL Region Detected a High Diversity of Eustigmatophyceae
3.9. Comparison of rbcL and 18S Markers for Freshwater Diatoms Biomonitoring
3.10. A 23S rDNA Plastid Marker for Simultaneous Detection of Eukaryotic Algae and Cyanobacteria
3.11. The 16S rRNA Gene as a Marker for Simultaneous Detection of Prokaryotes and Eukaryotes
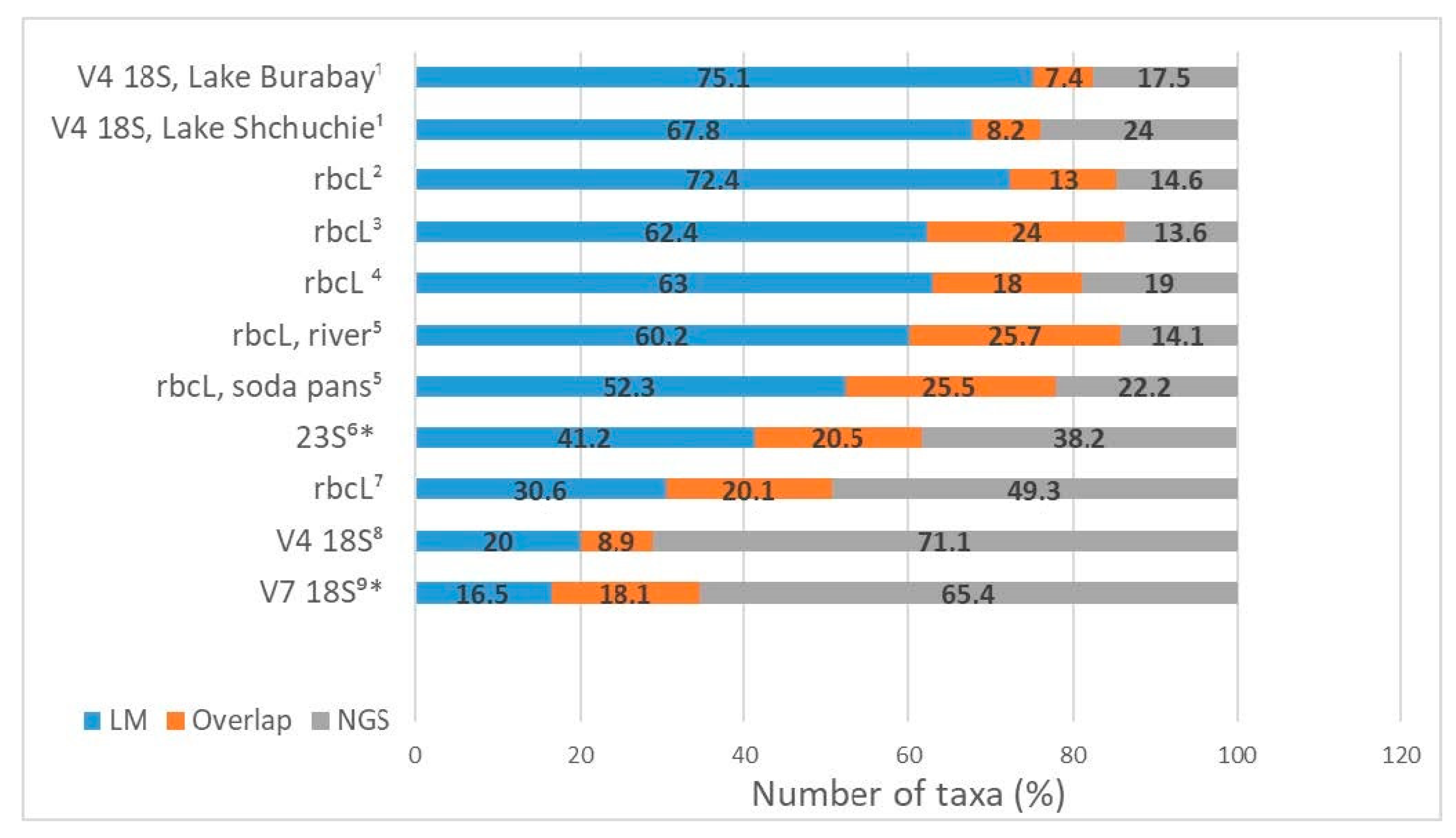
3.12. Comparing Approaches: Metabarcoding vs. Morphological Identification (Congruency between Methods)
- The natural intraspecific and intragenomic variabilities of the barcoding marker (single taxon has multiple genotypes at the barcoding region, and members of that taxon might cluster into different Molecular Operational Taxonomic Units (MOTUs)) [35].
- MOTU richness can be artificially inflated through technical errors at different steps of sample processing during amplification and sequencing [35].
- Complete absence of amplification on the whole due to a mismatch of the primer set used. For example, Salmaso et al. [27] did not find any species belonging to the Euglenales in the HTS results (with universal eukaryotic primers (TAReuk454FWD1 and TAReukREV3) for V4 18S), although they were present in LM. Hanžek et al. [66] reported that the taxa that contributed most to the biomass (Actinotaenium/Mesotaenium sp. and the species Cosmarium tenue, Pantocsekiella comensis, Sphaerocystis schroeteri and Synedropsis roundii) were not identified by eDNA metabarcoding (V9 18S region was amplified using the universal primer pair 1391F and EukB). Proeschöld and Darienko [140] noted that, although Stichococcus-like organisms are widely distributed in almost all habitats, they are not recorded in environmental studies based on HTS approaches, because the V4 or V9 regions of the SSU contain introns that obstruct amplification. Groendahl et al. [42] reported that Monorhaphidium sp., Selenastrum sp. and Trachelomonas sp. detected using the morphology-based approach were not identified by the metabarcoding approach, despite the fact that all three genera are included in the reference database.
- Uncertainties and lack of sensitivity of reference databases for the selected DNA markers [27].
- The proportion of live diatoms found in environmental samples varies greatly, ranging from 2 to 98% [35].
- Small-celled species and pico-sized cells are often overlooked or underestimated by the morphological approach. For example, the valves of Fistulifera saprophila tend to dissolve during sample processing, which can explain why this species is often missed during morphological identification [78,80,82,99].
- The different sample volumes settled for microscopy and metabarcoding [143].
- A short barcode gene fragment may have limited the taxonomic resolution [143]. For example, the resolution of the V4 18S region does not allow to unambiguously identify some species of Navicula [32]. For the V7–9 18S marker, a lack of intergenus taxonomic resolution was found (the MOTUs matched multiple genera, e.g., Alexandrium pseudogonyaulax and A. hiranoi, Chaetoceros neogracile and C. curvisetus and Thalassiosira eccentrica and T. antarctica) [144]. In some Chlamydomonas, the V9 region is very similar to that of prasinophytes clade VII A5 [122].
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adl, S.M.; Simpson, A.G.B.; Farmer, M.A.; Andersen, R.A.; Anderson, O.R.; Barta, J.R.; Bowser, S.S.; Brugerolle, G.; Fensome, R.A.; Fredericq, S.; et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot. Microbiol. 2005, 52, 399–451. [Google Scholar] [CrossRef]
- Adl, S.M.; Simpson, A.G.B.; Lane, C.E.; Lukeš, J.; Bass, D.; Bowser, S.S.; Brown, M.W.; Burki, F.; Dunthorn, M.; Hampl, V.; et al. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 2012, 59, 429–514. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Park, J.H.; Bhattacharya, D.; Yoon, H.S. Applications of next-generation sequencing to unravelling the evolutionary history of algae. Int. J. Syst. Evol. Microbiol. 2014, 64, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Burki, F. The convoluted evolution of eukaryotes with complex plastids. In Secondary Endosymbioses; Hirakawa, Y., Ed.; Elsevier: Tsukuba, Japan, 2017; Volume 84, pp. 1–30. [Google Scholar] [CrossRef]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G. The new tree of eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gololobova, M.A.; Belyakova, G.A. Position of Algae on the Tree of Life. Dokl. Biol. Sci. 2022, 507, 312–326. [Google Scholar] [CrossRef]
- Fawley, M.W.; Fawley, K.P. Identification of Eukaryotic Microalgal Strains. J. Appl. Phycol. 2020, 32, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Leliaert, F.; Verbruggen, H.; Vanormelingen, P.; Steen, F.; López-Bautista, J.M.; Zuccarello, G.C.; De Clerck, O. DNA-based species delimitation in algae. Eur. J. Phycol. 2014, 49, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Leray, M.; Knowlton, N. Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 2017, 3, e3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahoon, A.B.; Huffman, A.G.; Krager, M.M.; Crowell, R.M. A meta-barcoding census of freshwater planktonic protists in Appalachia—Natural Tunnel State Park, Virginia, USA. Metabarcoding Metagenom. 2018, 2, e26939. [Google Scholar] [CrossRef] [Green Version]
- Marcelino, V.; Verbruggen, H. Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 2016, 6, 31508. [Google Scholar] [CrossRef] [Green Version]
- van der Loos, L.M.; Nijland, R. Biases in bulk: DNA metabarcoding of marine communities and the methodology involved. Mol. Ecol. 2021, 30, 3270–3288. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA Gene for Designing Universal Eukaryote Specific Primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [Green Version]
- Bailet, B.; Apothéloz-Perret-Gentil, L.; Baričević, A.; Chonova, T.; Franc, A.; Frigerio, J.; Kelly, M.; Mora, D.; Pfannkuchen, M.; Proft, S.; et al. Diatom DNA metabarcoding for ecological assessment: Comparison among bioinformatics pipelines used in six European countries reveals the need for standardization. Sci. Total Environ. 2020, 745, 140948. [Google Scholar] [CrossRef] [PubMed]
- Czech, L.; Stamatakis, A.; Dunthorn, M.; Barbera, P. Metagenomic Analysis Using Phylogenetic Placement-A Review of the First Decade. Front. Bioinform. 2022, 26, 871393. [Google Scholar] [CrossRef] [PubMed]
- Vasselon, V.; Domaizon, I.; Rimet, F.; Kahlert, M.; Bouchez, A. Application of high-throughput sequencing (HTS) metabarcoding to diatom biomonitoring: Do DNA extraction methods matter? Freshw. Sci. 2017, 36, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.; Boonham, N.; Juggins, S.; Kille, P.; Mann, D.; Pass, D.; Sapp, M.; Sato, S.; Glover, R. A DNA based diatom metabarcoding approach for classification of rivers. In Science Report SC140024/R; Environment Agency: Bristol, UK, 2018; p. 157. [Google Scholar]
- Kelly, M.; Boonham, N.; Juggins, S.; Mann, D.; Glover, R. Further development of a DNA based metabarcoding approach to assess diatom communities in rivers. Chief Scientist’s Group report. In Version: SC160014/R; Environment Agency: Bristol, UK, 2020; p. 133. [Google Scholar]
- Ruppert, K.M.; Kline, R.J.; Rahman, M.S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 2019, 17, e00547. [Google Scholar] [CrossRef]
- Bruce, K.; Blackman, R.; Bourlat, S.J.; Hellström, A.M.; Bakker, J.; Bista, I.; Bohmann, K.; Bouchez, A.; Brys, R.; Clark, K.; et al. A Practical Guide to DNA-Based Methods for Biodiversity Assessment; Pensoft Advanced Books: Sofia, Bulgaria, 2021. [Google Scholar] [CrossRef]
- Pawlowski, J.; Bruce, K.; Panksep, K.; Aguirre, F.; Amalfitano, S.; Apothéloz-Perret-Gentil, L.; Baussant, T.; Bouchez, A.; Carugati, L.; Cermakova, K.; et al. Environmental DNA metabarcoding for benthic monitoring: A review of sediment sampling and DNA extraction methods. Sci. Total Environ. 2022, 818, 151783. [Google Scholar] [CrossRef]
- Baricevic, A.; Chardon, C.; Kahlert, M.; Karjalainen, S.M.; Pfannkuchen, D.M.; Pfannkuchen, M.; Rimet, F.; Tankovic, M.S.; Trobajo, R.; Vasselon, V.; et al. Recommendations for the preservation of environmental samples in diatom metabarcoding studies. Metabarcoding Metagenom. 2022, 6, e85844. [Google Scholar] [CrossRef]
- Kelly, M.G.; Juggins, S.; Mann, D.G.; Sato, S.; Glover, R.; Boonham, N.; Sapp, M.; Lewis, E.; Hany, U.; Kille, P.; et al. Development of a novel metric for evaluating diatom assemblages in rivers using DNA metabarcoding. Ecol. Indic. 2020, 118, 106725. [Google Scholar] [CrossRef]
- Pawlowski, J.; Kelly-Quinn, M.; Altermatt, F.; Apothéloz-Perret-Gentil, L.; Beja, P.; Boggero, A.; Borja, A.; Bouchez, A.; Cordier, T.; Domaizon, I.; et al. The future of biotic indices in the ecogenomic era: Integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. Sci. Total. Environ. 2018, 637–638, 1295–1310. [Google Scholar] [CrossRef]
- Sagova-Mareckova, M.; Boenigk, J.; Bouchez, A.; Cermakova, K.; Chonova, T.; Cordier, T.; Eisendle, U.; Elersek, T.; Fazi, S.; Fleituch, T.; et al. Expanding ecological assessment by integrating microorganisms into routine freshwater biomonitoring. Water Res. 2021, 191, 116767. [Google Scholar] [CrossRef]
- Pawlowski, J.; Apothéloz-Perret-Gentil, L.; Altermatt, F. Environmental DNA: What’s behind the term? Clarifying the terminology and recommendations for its future use in biomonitoring. Mol. Ecol. 2020, 29, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, N.; Vasselon, V.; Rimet, F.; Vautier, M.; Elersek, T.; Boscaini, A.; Donati, C.; Moretto, M.; Pindo, M.; Riccioni, G.; et al. DNA sequence and taxonomic gap analyses to quantify the coverage of aquatic cyanobacteria and eukaryotic microalgae in reference databases: Results of a survey in the Alpine region. Sci. Total. Environ. 2022, 834, 155175. [Google Scholar] [CrossRef] [PubMed]
- Feist, S.M.; Lance, R.F. Genetic detection of freshwater harmful algal blooms: A review focused on the use of environmental DNA (eDNA) in Microcystis aeruginosa and Prymnesium parvum. Harmful Algae 2021, 110, 102124. [Google Scholar] [CrossRef]
- Bartolo, A.G.; Zammit, G.P.; Akira, F.; Küpper, F.C. The current state of DNA barcoding of macroalgae in the Mediterranean Sea: Presently lacking but urgently required. Bot. Mar. 2020, 63, 253–272. [Google Scholar] [CrossRef]
- Mikhailov, I.S.; Zakharova, Y.R.; Bukin, Y.S.; Galachyants, Y.; Petrova, D.; Sakirko, M.; Likhoshway, Y. Co-occurrence Networks Among Bacteria and Microbial Eukaryotes of Lake Baikal During a Spring Phytoplankton Bloom. Microb. Ecol. 2019, 77, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Nolte, V.; Pandey, R.V.; Jost, S.; Medinger, R.; Ottenwälder, B.; Boenigk, J.; Schlötterer, C. Contrasting seasonal niche separation between rare and abundant taxa conceals the extent of protist diversity. Mol. Ecol. 2010, 19, 2908–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visco, J.A.; Apothéloz-Perret-Gentil, L.; Cordonier, A.; Esling, P.; Pillet, L.; Pawlowski, J. Environmental Monitoring: Inferring the Diatom Index from Next-Generation Sequencing Data. Environ. Sci. Technol. 2015, 49, 7597–7605. [Google Scholar] [CrossRef]
- Apothéloz-Perret-Gentil, L.; Bouchez, A.; Cordier, T.; Cordonier, A.; Guéguen, J.; Rimet, F.; Vasselon, V.; Pawlowski, J. Monitoring the ecological status of rivers with diatom eDNA metabarcoding: A comparison of taxonomic markers and analytical approaches for the inference of a molecular diatom index. Mol. Ecol. 2021, 30, 2959–2968. [Google Scholar] [CrossRef]
- Bailet, B.; Bouchez, A.; Franc, A.; Frigerio, J.-M.; Keck, F.; Karjalainen, S.-M.; Rimet, F.; Schneider, S.; Kahlert, M. Molecular versus morphological data for benthic diatoms biomonitoring in Northern Europe freshwater and consequences for ecological status. Metabarcoding Metagenom. 2019, 3, e34002. [Google Scholar] [CrossRef] [Green Version]
- Mora, D.; Abarca, N.; Proft, S.; Grau, J.H.; Enke, N.; Carmona, J.; Skibbe, O.; Jahn, R.; Zimmermann, J. Morphology and metabarcoding: A test with stream diatoms from Mexico highlights the complementarity of identification methods. Freshw. Sci. 2019, 38, 448–464. [Google Scholar] [CrossRef]
- Kutty, S.N.; Loh, R.K.; Bannister, W.; Taylor, D. Evaluation of a diatom eDNA-based technique for assessing water quality variations in tropical lakes and reservoirs. Ecol. Indic. 2022, 141, 109108. [Google Scholar] [CrossRef]
- Zimmermann, J.; Jahn, R.; Gemeinholzer, B. Barcoding diatoms: Evaluation of the V4 subregion on the 18S rRNA gene, including new primers and protocols. Org. Divers. Evol. 2011, 11, 173–192. [Google Scholar] [CrossRef]
- Zimmermann, J.; Abarca, N.; Enke, N.; Skibbe, O.; Kusber, W.-H.; Jahn, R. Taxonomic reference libraries for environmental barcoding: A best practice example from diatom research. PLoS ONE 2014, 9, e108793. [Google Scholar] [CrossRef] [PubMed]
- Apothéloz-Perret-Gentil, L.; Cordonier, A.; Straub, F.; Iseli, J.; Esling, P.; Pawlowski, J. Taxonomy-free molecular diatom index for high-throughput eDNA biomonitoring. Mol. Ecol. Resour. 2017, 17, 1231–1242. [Google Scholar] [CrossRef]
- Malashenkov, D.; Dashkova, V.; Zhakupova, K.; Vorobjev, I.; Barteneva, N. Comparative analysis of freshwater phytoplankton communities in two lakes of Burabay National Park using morphological and molecular approaches. Sci. Rep. 2021, 11, 16130. [Google Scholar] [CrossRef] [PubMed]
- Hugerth, L.; Muller, E.; Hu, Y.; Lebrun, L.; Roume, H.; Wilmes, L.; Andersson, A. Systematic Design of 18S rRNA Gene Primers for Determining Eukaryotic Diversity in Microbial Consortia. PLoS ONE 2014, 9, e95567. [Google Scholar] [CrossRef] [PubMed]
- Groendahl, S.; Kahlert, M.; Fink, P. The best of both worlds: A combined approach for analyzing microalgal diversity via metabarcoding and morphology-based methods. PLoS ONE 2017, 12, e0172808. [Google Scholar] [CrossRef] [Green Version]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.; Breiner, H.W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 1, 21–31. [Google Scholar] [CrossRef]
- Filker, S.; Sommaruga, R.; Vila, I.; Stoeck, T. Microbial eukaryote plankton communities of high-mountain lakes from three continents exhibit strong biogeographic patterns. Mol. Ecol. 2016, 25, 2286–2301. [Google Scholar] [CrossRef] [Green Version]
- Kammerlander, B.; Breiner, H.W.; Filker, S.; Sommaruga, R.; Sonntag, B.; Stoeck, T. High diversity of protistan plankton communities in remote high mountain lakes in the European Alps and the Himalayan mountains. FEMS Microbiol. Ecol. 2015, 91, fiv010. [Google Scholar] [CrossRef] [Green Version]
- Seymour, M.; Edwards, F.; Cosby, B.; Kelly, M.; Bruyn, M.; Carvalho, G.; Creer, S. Executing multi-taxa eDNA ecological assessment via traditional metrics and interactive networks. Sci. Total. Environ. 2020, 729, 138801. [Google Scholar] [CrossRef] [PubMed]
- Annenkova, N.V.; Giner, C.R.; Logares, R. Tracing the Origin of Planktonic Protists in an Ancient Lake. Microorganisms 2020, 1, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakata, Y.; Hatamoto, M.; Oshiki, M.; Watari, T.; Kuroda, K.; Araki, N.; Yamaguchi, T. Temporal variation of eukaryotic community structures in UASB reactor treating domestic sewage as revealed by 18S rRNA gene sequencing. Sci. Rep. 2019, 9, 12783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elersek, T. (Ed.) Technical Guidelines for eDNA Monitoring in Alpine Waters for Stakeholders and End-Users. 2021. Available online: https://www.alpine-space.eu/project/eco-alpswater/ (accessed on 22 May 2023).
- Brandani, J.; Peter, H.; Busi, S.B.; Kohler, T.J.; Fodelianakis, S.; Ezzat, L.; Michoud, G.; Bourquin, M.; Pramateftaki, P.; Roncoroni, M.; et al. Spatial patterns of benthic biofilm diversity among streams draining proglacial floodplains. Front. Microbiol. 2022, 13, 948165. [Google Scholar] [CrossRef]
- Yang, N.; Wang, L.; Lin, L.; Li, Y.; Zhang, W.; Niu, L.; Zhang, H.; Wang, L. Pelagic-benthic coupling of the microbial food web modifies nutrient cycles along a cascade-dammed river. Front. Environ. Sci. Eng. 2022, 16, 50. [Google Scholar] [CrossRef]
- Liang, D.; Xia, J.; Song, J.; Sun, H.; Xu, W. Using eDNA to Identify the Dynamic Evolution of Multi-Trophic Communities under the Eco-Hydrological Changes in River. Front. Environ. Sci. 2022, 853, 929541. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.S. Comparative analyses of the V4 and V9 regions of 18S rDNA for the extant eukaryotic community using the Illumina platform. Sci. Rep. 2020, 10, 6519. [Google Scholar] [CrossRef] [Green Version]
- Bradley, I.M.; Pinto, A.J.; Guest, J.S. Design and evaluation of Illumina MiSeq-compatible, 18S rRNA gene-specific primers for improved characterization of mixed phototrophic communities. Appl. Environ. Microbiol. 2016, 82, 5878–5891. [Google Scholar] [CrossRef] [Green Version]
- Carles, L.; Wullschleger, S.; Joss, A.; Eggen, R.I.; Schirmer, K.; Schuwirth, N.; Stamm, C.; Tlili, A. Impact of wastewater on the microbial diversity of periphyton and its tolerance to micropollutants in an engineered flow-through channel system. Water Res. 2021, 203, 117486. [Google Scholar] [CrossRef]
- Carles, L.; Wullschleger, S.; Joss, A.; Eggen, R.; Schirmer, K.; Schuwirth, N.; Stamm, C.; Tlili, A. Wastewater microorganisms impact microbial diversity and important ecological functions of stream periphyton. Water Res. 2022, 225, 119119. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Li, X.; Xi, B.; Zhang, H.; Ma, C.; He, Z. Combining morphological and metabarcoding approaches reveals the freshwater eukaryotic phytoplankton community. Environ. Sci. Eur. 2020, 32, 37. [Google Scholar] [CrossRef] [Green Version]
- Gast, R.J.; Dennett, M.R.; Caron, D.A. Characterization of Protistan assemblages in the Ross Sea, Antarctica, by denaturing gradient gel electrophoresis. Appl. Environ. Microb. 2004, 70, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Amaral-Zettler, L.A.; McCliment, E.A.; Ducklow, H.W.; Huse, S.M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE 2009, 4, e6372. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–147. [Google Scholar]
- The Earth Microbiome Project. Available online: http://www.earthmicrobiome.org (accessed on 22 May 2023).
- Abad, D.; Albaina, A.; Aguirre, M.; Laza-Martínez, A.; Uriarte, I.; Iriarte, A.; Villate, F.; Estonba, A. Is metabarcoding suitable for estuarine plankton monitoring? A comparative study with microscopy. Mar. Biol. 2016, 163, 149. [Google Scholar] [CrossRef]
- Yi, Z.; Berney, C.; Hartikainen, H.; Mahamdallie, S.; Gardner, M.; Boenigk, J.; Cavalier-Smith, T.; Bass, D. High-throughput sequencing of microbial eukaryotes in Lake Baikal reveals ecologically differentiated communities and novel evolutionary radiations. FEMS Microbiol. Ecol. 2017, 93, fix073. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Álvarez, R.; Triadó-Margarit, X.; Camarero, L.; Casamayor, E.; Catalan, J. High planktonic diversity in mountain lakes contains similar contributions of autotrophic, heterotrophic and parasitic eukaryotic life forms. Sci. Rep. 2018, 8, 4457. [Google Scholar] [CrossRef] [Green Version]
- Minerovic, A.D.; Potapova, M.G.; Sales, C.M.; Price, J.R.; Enache, M.D. 18S-V9 DNA metabarcoding detects the effect of water-quality impairment on stream biofilm eukaryotic assemblages. Ecol. Indic. 2020, 113, 106225. [Google Scholar] [CrossRef]
- Hanžek, N.; Udovič, M.G.; Kajan, K.; Borics, G.; Várbíró, G.; Stoeck, T.; Žutinić, P.; Orlić, S.; Stanković, I. Assessing ecological status in karstic lakes through the integration of phytoplankton functional groups, morphological approach and environmental DNA metabarcoding. Ecol. Indic. 2021, 131, 108166. [Google Scholar] [CrossRef]
- Medlin, L.; Elwood, H.J.; Stickel, S.; Sogin, M.L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 1988, 71, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Boenigk, J.; Wodniok, S.; Bock, C.; Beisser, D.; Hempel, C.; Grossmann, L.; Lange, A.; Jensen, M. Geographic distance and mountain ranges structure freshwater protist communities on a European scale. Metabarcoding Metagenom. 2018, 2, e21519. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.; Jensen, M.; Forster, D.; Marks, S.; Nuy, J.; Psenner, R.; Beisser, D.; Boenigk, J. Factors shaping community patterns of protists and bacteria on a European scale. Environ. Microbiol. 2020, 22, 2243–2260. [Google Scholar] [CrossRef] [PubMed]
- Olefeld, J.L.; Bock, C.; Jensen, M.; Vogt, J.; Sieber, G.; Albach, D.; Boenigk, J. Centers of endemism of freshwater protists deviate from pattern of taxon richness on a continental scale. Sci. Rep. 2020, 10, 14431. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Olefeld, J.L.; Vogt, J.C.; Albach, D.; Boenigk, J. Phylogenetic and functional diversity of Chrysophyceae in inland waters. Org. Divers. Evol. 2022, 2022, 327–341. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Câmara, P.E.; Menezes, G.C.; Pinto, O.H.; Silva, M.C.; Convey, P.; Rosa, L.H. Using metabarcoding to assess Viridiplantae sequence diversity present in Antarctic glacial ice. Annu. Acad. Bras. Cienc. 2022, 94, e20201736. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Câmara, P.E.A.S.; Ogaki, M.B.; Pinto, O.; Lirio, J.; Coria, S.; Vieira, R.; Carvalho-Silva, M.; Amorim, E.; Convey, P.; et al. Green algae (Viridiplantae) in sediments from three lakes on Vega Island, Antarctica, assessed using DNA metabarcoding. Mol. Biol. Rep. 2022, 49, 179–188. [Google Scholar] [CrossRef]
- Rimet, F.; Vasselon, V.; A.-Keszte, B.; Bouchez, A. Do we similarly assess diversity with microscopy and high-throughput sequencing? Case of microalgae in lakes. Org. Divers. Evol. 2018, 18, 51–62. [Google Scholar] [CrossRef]
- Câmara, P.E.A.S.; Carvalho-Silva, M.; Pinto, O.H.B.; Amorim, E.; Henriques, D.; Holanda da Silva, T.; Pellizzari, F.; Convey, P.; Rosa, L. Diversity and Ecology of Chlorophyta (Viridiplantae) Assemblages in Protected and Non-protected Sites in Deception Island (Antarctica, South Shetland Islands) Assessed Using an NGS Approach. Microb. Ecol. 2021, 81, 323–334. [Google Scholar] [CrossRef]
- Rimet, F.; Abarca, N.; Bouchez, A.; Kusber, W.-H.; Jahn, R.; Kahlert, M.; Keck, F.; Kelly, M.; Mann, D.; Piuz, A.; et al. The potential of High-Throughput Sequencing (HTS) of natural samples as a source of primary taxonomic information for reference libraries of diatom barcodes. Fottea 2018, 18, 37–54. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.F.; Vasselon, V.; Ballorain, K.; Carpentier, A.; Wetzel, C.; Ector, L.; Bouchez, A.; Rimet, F. DNA metabarcoding and microscopic analyses of sea turtles biofilms: Complementary to understand turtle behavior. PLoS ONE 2018, 13, e0195770. [Google Scholar] [CrossRef]
- Maitland, V.C.; Robinson, C.V.; Porter, T.M.; Hajibabaei, M. Freshwater diatom biomonitoring through benthic kick-net metabarcoding. PLoS ONE 2020, 15, e0242143. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Burillo, J.; Trobajo, R.; Leira, M.; Keck, F.; Rimet, F.; Sigró, J.; Mann, D.G. DNA metabarcoding reveals differences in distribution patterns and ecological preferences among genetic variants within some key freshwater diatom species. Sci. Total Environ. 2021, 798, 149029. [Google Scholar] [CrossRef]
- Smucker, N.J.; Pilgrim, E.M.; Nietch, C.T.; Darling, J.A.; Johnson, B.R. DNA metabarcoding effectively quantifies diatom responses to nutrients in streams. Ecol. Appl. 2020, 30, e02205. [Google Scholar] [CrossRef]
- Pissaridou, P.; Cantonati, M.; Bouchez, A.; Tziortzis, I.; Dörflinger, G.; Vasquez, M.I. How can integrated morphotaxonomy- and metabarcoding-based diatom assemblage analyses best contribute to the ecological assessment of streams? Metabarcoding Metagenom. 2021, 5, e68438. [Google Scholar] [CrossRef]
- Borrego-Ramos, M.; Bécares, E.; García, P.; Nistal, A.; Blanco, S. Epiphytic Diatom-Based Biomonitoring in Mediterranean Ponds: Traditional Microscopy versus Metabarcoding Approaches. Water 2021, 13, 1351. [Google Scholar] [CrossRef]
- Rimet, F.; Pinseel, E.; Bouchez, A.; Japoshvili, B.; Mumladze, L. Diatom endemism and taxonomic turnover: Assessment in high-altitude alpine lakes covering a large geographical range. Sci. Total. Environ. 2023, 871, 161970. [Google Scholar] [CrossRef]
- Kahlert, M.; Karjalainen, S.M.; Keck, F.; Kelly, M.; Ramon, M.; Rimet, F.; Schneider, S.; Tapolczai, K.; Zimmermann, J. Co-occurrence, ecological profiles and geographical distribution based on unique molecular identifiers of the common freshwater diatoms Fragilaria and Ulnaria. Ecol. Indic. 2022, 141, 109114. [Google Scholar] [CrossRef]
- Pérez-Burillo, J.; Mann, D.G.; Trobajo, R. Evaluation of two short overlapping rbcL markers for diatom metabarcoding of environmental samples: Effects on biomonitoring assessment and species resolution. Chemosphere 2022, 307, 135933. [Google Scholar] [CrossRef]
- Yuan, L.L.; Mitchell, R.M.; Pollard, A.I.; Nietch, C.T.; Pilgrim, E.M.; Smucker, N.J. Understanding the effects of phosphorus on diatom richness in rivers and streams using taxon–environment relationships. Freshw. Biol. 2023, 68, 473–486. [Google Scholar] [CrossRef]
- Bíró, T.; Duleba, M.; Földi, A.; Kiss, K.T.; Orgoványi, P.; Trábert, Z.; Vadkerti, E.; Wetzel, C.E.; Ács, É. Metabarcoding as an effective complement of microscopic studies in revealing the composition of the diatom community—A case study of an oxbow lake of Tisza River (Hungary) with the description of a new Mayamaea species. Metabarcoding Metagenom. 2022, 6, e87497. [Google Scholar] [CrossRef]
- Robinson, C.; Porter, T.; Maitland, V.; Wright, M.; Hajibabaei, M. Multi-marker metabarcoding resolves subtle variations in freshwater condition: Bioindicators, ecological traits, and trophic interactions. Ecol. Indic. 2022, 145, 109603. [Google Scholar] [CrossRef]
- Vasselon, V.; Rimet, F.; Tapolczai, K.; Bouchez, A. Assessing ecological status with diatoms DNA metabarcoding: Scaling-up on a WFD monitoring network (Mayotte island, France). Ecol. Indic. 2017, 82, 1–12. [Google Scholar] [CrossRef]
- Duleba, M.; Földi, A.; Micsinai, A.; Várbíró, G.; Mohr, A.; Sipos, R.; Szabó, G.; Buczkó, K.; Trábert, Z.; Kiss, K.; et al. Applicability of diatom metabarcoding in the ecological status assessment of Hungarian lotic and soda pan habitats. Ecol. Indic. 2021, 130, 108105. [Google Scholar] [CrossRef]
- Vasselon, V.; Rimet, F.; Domaizon, I.; Monnier, O.; Reyjol, Y.; Bouchez, A. Assessing pollution of aquatic environments with diatoms’ DNA metabarcoding: Experience and developments from France Water Framework Directive networks. Metabarcoding Metagenom. 2019, 3, e39646. [Google Scholar] [CrossRef]
- Mortágua, A.; Vasselon, V.; Oliveira, R.; Elias, C.; Chardon, C.; Bouchez, A.; Rimet, F.; Feio, M.; Almeida, S. Applicability of DNA metabarcoding approach in the bioassessment of Portuguese rivers using diatoms. Ecol. Indic. 2019, 106, 105470. [Google Scholar] [CrossRef]
- Tapolczai, K.; Selmeczy, G.B.; Szabó, B.; B.-Béres, V.; Keck, F.; Bouchez, A.; Rimet, F.; Padisák, J. The potential of exact sequence variants (ESVs) to interpret and assess the impact of agricultural pressure on stream diatom assemblages revealed by DNA metabarcoding. Ecol. Ind. 2021, 122, 107322. [Google Scholar] [CrossRef]
- Baker, L.A.; Beauger, A.; Kolovi, S.; Voldoire, O.; Allain, E.; Breton, V.; Chardon, P.; Miallier, D.; Bailly, C.; Montavon, G.; et al. Diatom DNA metabarcoding to assess the effect of natural radioactivity in mineral springs on ASV of benthic diatom communities. Sci. Total Environ. 2023, 873, 162270. [Google Scholar] [CrossRef]
- Kang, W.; Anslan, S.; Börner, N.; Schwarz, A.; Schmidt, R.; Künzel, S.; Rioual, P.; Echeverría-Galindo, P.; Vences, M.; Wang, J.; et al. Diatom metabarcoding and microscopic analyses from sediment samples at Lake Nam Co, Tibet: The effect of sample-size and bioinformatics on the identified communities. Ecol. Indic. 2021, 121, 107070. [Google Scholar] [CrossRef]
- Fawley, M.W.; Fawley, K.P.; Cahoon, A.B. Finding needles in a haystack—Extensive diversity in the eustigmatophyceae revealed by community metabarcode analysis targeting the rbcL gene using lineage-directed primers. J. Phycol. 2021, 57, 1636–1647. [Google Scholar] [CrossRef]
- Qiao, L.; Chang, Z.; Li, J.; Chen, Z.; Yang, L.; Luo, Q. Phytoplankton community structure and diversity in the indoor industrial aquaculture system for Litopenaeus vannamei revealed by high-throughput sequencing and morphological identification. Aquac. Res. 2019, 50, 2563–2576. [Google Scholar] [CrossRef]
- Brown, P.D.; Craine, J.M.; Richards, D.; Chapman, A.; Marden, B. DNA metabarcoding of the phytoplankton of Great Salt Lake’s Gilbert Bay: Spatiotemporal assemblage changes and comparisons to microscopy. J. Great Lakes Res. 2022, 48, 110–124. [Google Scholar] [CrossRef]
- Sherwood, A.R.; Presting, G.G. Universal primers amplify a 23S rDNA plastid marker in eukaryotic algae and cyanobacteria. J. Phycol. 2007, 43, 605–608. [Google Scholar] [CrossRef]
- Eiler, A.; Drakare, S.; Bertilsson, S.; Pernthaler, J.; Peura, S.; Rofner, C.; Simek, K.; Yang, Y.; Znachor, P.; Lindström, E. Unveiling Distribution Patterns of Freshwater Phytoplankton by a Next Generation Sequencing Based Approach. PLoS ONE 2013, 8, e53516. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Primers for marine microbiome studies. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Bonfantine, K.L.; Trevathan-Tackett, S.M.; Matthews, T.G.; Neckovic, A.; Gan, H.M. Dumpster diving for diatom plastid 16S rRNA genes. PeerJ. 2021, 9, e11576. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.; Turnbaugh, P.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Rimet, F.; Gusev, E.; Kahlert, M.; Kelly, M.; Kulikovskiy, M.; Maltsev, Y.; Mann, D.; Pfannkuchen, M.; Trobajo, R.; Vasselon, V.; et al. Diat. barcode, an open-access curated barcode library for diatoms. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Banchi, E.; Ametrano, C.G.; Greco, S.; Stanković, D.; Muggia, L.; Pallavicini, A. PLANiTS: A curated sequence reference dataset for plant ITS DNA metabarcoding. Database 2020, 2020, baz155. [Google Scholar] [CrossRef] [Green Version]
- Decelle, J.; Romac, S.; Stern, R.F.; Bendif, E.M.; Zingone, A.; Audic, S.; Guiry, M.; Guillou, L.; Tessier, D.; Le Gall, F.; et al. PhytoREF: A reference database of the plastidial 16S rRNA gene of photosynthetic eukaryotes with curated taxonomy. Mol. Ecol. Resour. 2015, 15, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Evans, K.M.; Wortley, A.H.; Mann, D.G. An assessment of potential diatom “barcode” genes (cox1, rbcL, 18S and ITS rDNA) and their effectiveness in determining relationships in Sellaphora (Bacillariophyta). Protist 2007, 158, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Behnke, A.; Friedl, T.; Chepurnov, V.A.; Mann, D.G. Reproductive Compatibility and rDNA Sequence Analyses in the Sellaphora Pupula Species Complex (Bacillariophyta). J. Phycol. 2004, 40, 193–208. [Google Scholar] [CrossRef]
- Moniz, M.B.J.; Kaczmarska, I. Barcoding diatoms: Is there a good marker? Mol. Ecol. Resour. 2009, 9, 65–74. [Google Scholar] [CrossRef]
- Moniz, M.B.J.; Kaczmarska, I. Barcoding of diatoms: Nuclear encoded ITS revisited. Protist 2010, 161, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Hamsher, S.E.; Evans, K.M.; Mann, D.G.; Poulickova, A.; Saunders, G.W. Barcoding diatoms: Exploring alternatives to COI-5P. Protist 2011, 162, 405–422. [Google Scholar] [CrossRef] [PubMed]
- MacGillivary, M.L.; Kaczmarska, I. Survey of the Efficacy of a Short Fragment of the rbcL Gene as a Supplemental DNA Barcode for Diatoms. J. Eukaryot. Microbiol. 2011, 58, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Kermarrec, L.; Franc, A.; Rimet, F.; Chaumeil, P.; Humbert, J.F.; Bouchez, A. Next-generation sequencing to inventory taxonomic diversity in eukaryotic communities: A test for freshwater diatoms. Mol. Ecol. Resour. 2013, 13, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.D.; Fučíková, K.; Lo, C.; Lewis, L.A.; Karol, K.G. An assessment of proposed DNA barcodes in freshwater green algae. Cryptogam. Algol. 2010, 31, 529–555. [Google Scholar]
- Litaker, R.W.; Vandersea, M.W.; Kibler, S.R.; Reece, K.S.; Stokes, N.A.; Lutzoni, F.M.; Yonish, F.M.; West, M.A.; Black, M.N.D. Recognizing dinoflagellate species using ITS rDNA sequences. J. Phycol. 2007, 43, 344–355. [Google Scholar] [CrossRef]
- Stern, R.; Horak, A.; Andrew, R.; Coffroth, M.; Andersen, R.; Andersen, R.; Küpper, F.; Jameson, I.; Hoppenrath, M.; Véron, B.; et al. Environmental Barcoding Reveals Massive Dinoflagellate Diversity in Marine Environments. PLoS ONE 2010, 5, e13991. [Google Scholar] [CrossRef]
- Stern, R.F.; Andersen, R.A.; Jameson, I.; Küpper, F.C.; Coffroth, M.A.; Vaulot, D.; Le Gall, F.; Véron, B.; Brand, J.J.; Skelton, H.; et al. Evaluating the ribosomal internal transcribed spacer (ITS) as a candidate dinoflagellate barcode marker. PLoS ONE 2012, 7, e42780. [Google Scholar] [CrossRef] [PubMed]
- La Jeunesse, T.C.; Thornhill, D.J. Improved Resolution of Reef-Coral Endosymbiont (Symbiodinium) Species Diversity, Ecology, and Evolution through psbA Non-Coding Region Genotyping. PLoS ONE 2011, 6, e29013. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, A.S.; Nagai, S.; Hida, K.; Yasuike, M.; Fujiwara, A.; Nakamura, Y.; Takano, Y.; Katakura, S. Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Mol. Ecol. Resour. 2016, 16, 402–414. [Google Scholar] [CrossRef]
- Tragin, M.; Zingone, A.; Vaulot, D. Comparison of coastal phytoplankton composition estimated from the V4 and V9 regions of the 18S rRNA gene with a focus on photosynthetic groups and especially Chlorophyta. Environ. Microbiol. 2018, 20, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Giner, C.R.; Forn, I.; Romac, S.; Logares, R.; de Vargas, C.; Massana, R. Environmental Sequencing Provides Reasonable Estimates of the Relative Abundance of Specific Picoeukaryotes. Appl. Environ. Microbiol. 2016, 82, 4757–4766. [Google Scholar] [CrossRef] [Green Version]
- Maritz, J.M.; Rogers, K.H.; Rock, T.M.; Liu, N.; Joseph, S.; Land, K.; Carlton, J. An 18S rRNA Workflow for Characterizing Protists in Sewage, with a Focus on Zoonotic Trichomonads. Microb. Ecol. 2017, 74, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; He, Z.; Wang, C.; Yan, Q.; Shu, L. Evaluation of different primers of the 18S rRNA gene to profile amoeba communities in environmental samples. Water Biol. Secur. 2022, 1, 100057. [Google Scholar] [CrossRef]
- Piredda, R.; Tomasino, M.P.; D’Erchia, A.M.; Manzari, C.; Pesole, G.; Montresor, M.; Kooistra, W.; Sarno, D.; Zingone, A. Diversity and temporal patterns of planktonic protist assemblages at a Mediterranean Long Term Ecological Research site. FEMS Microbiol. Ecol. 2017, 93, fiw200. [Google Scholar] [CrossRef] [Green Version]
- Buchheim, M.A.; Keller, A.; Koetschan, C.; Förster, F.; Merget, B.; Wolf, M. Internal Transcribed Spacer 2 (nu ITS2 rRNA) Sequence-Structure Phylogenetics: Towards an Automated Reconstruction of the Green Algal Tree of Life. PLoS ONE 2011, 6, e16931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Câmara, P.E.A.S.; de Menezes, G.C.A.; Oliveira, F.S.; Souza, C.D.; Amorim, E.T.; Schaefer, C.E.; Convey, P.; Pinto, O.H.; Carvalho-Silva, M.; Rosa, L.H. Diversity of Viridiplanta e DNA present on rock surfaces in the Ellsworth Mountains, continental Antarctica. Polar. Biol. 2022, 45, 637–646. [Google Scholar] [CrossRef]
- Wang, Y.-K.; Stevenson, R.J.; Metzmeier, L. Development and evaluation of a diatom-based Index of Biotic Integrity for the Interior Plateau Ecoregion, USA. J. North Am. Benthol. Soc. 2005, 24, 990–1008. [Google Scholar] [CrossRef]
- Chessman, B.C.; Bate, N.; Gell, P.A.; Newall, P. A diatom species index for bioassessment of Australian rivers. Mar. Freshw. Res. 2007, 58, 542–557. [Google Scholar] [CrossRef]
- Lavoie, I.; Hamilton, P.B.; Wang, Y.-K.; Dillon, P.J.; Campeau, S. A comparison of stream bioassessment in Québec (Canada) using six European and North American diatom-based indices. Nova Hedwig. 2009, 135, 37–56. [Google Scholar]
- Zimmermann, J.; Glöckner, G.; Jahn, R.; Enke, N.; Gemeinholzer, B. Metabarcoding vs. morphological identification to assess diatom diversity in environmental studies. Mol. Ecol. Resour. 2015, 15, 526–542. [Google Scholar] [CrossRef]
- Kermarrec, L.; Franc, A.; Rimet, F.; Chaumeil, P.; Frigerio, J.M.; Humbert, J.F.; Bouchez, A. A next-generation sequencing approach to river biomonitoring using benthic diatoms. Freshw. Sci. 2014, 33, 349–363. [Google Scholar] [CrossRef]
- Wolf, D.I.; Vis, M.L. Stream Algal Biofilm Community Diversity Along an Acid Mine Drainage Recovery Gradient Using Multimarker Metabarcoding. J Phycol. 2020, 11–22, 12935. [Google Scholar] [CrossRef]
- Jackson, E.E.; Hawes, I.; Jungblut, A.D. 16S rRNA gene and 18S rRNA gene diversity in microbial mat communities in meltwater ponds on the McMurdo Ice Shelf, Antarctica. Polar Biol. 2021, 44, 823–836. [Google Scholar] [CrossRef]
- Keck, F.; Couton, M.; Altermatt, F. Navigating the seven challenges of taxonomic reference databases in metabarcoding analyses. Mol. Ecol. Resour. 2022, 23, 742–755. [Google Scholar] [CrossRef]
- Mann, D.G.; Evans, K.M. The species concept and cryptic diversity, Moestrup, Ø, Eds. In Proceedings of the 12th International Conference on Harmful Algae, International Society for the Study of Harmful Algae and Intergovernmental Oceanographic Commission of UNESCO, Copenhagen, Sweden, 4–8 August 2008; pp. 262–268. [Google Scholar]
- Souffreau, C.; Vanormelingen, P.; Van de Vijver, B.; Isheva, T.; Verleyen, E.; Sabbe, K.; Vyverman, W. Molecular evidence for distinct Antarctic lineages in the cosmopolitan terrestrial diatoms Pinnularia borealis and Hantzschia amphioxys. Protist 2013, 164, 101–115. [Google Scholar] [CrossRef]
- Darienko, T.; Gustavs, L.; Eggert, A.; Wolf, W.; Pröschold, T. Evaluating the Species Boundaries of Green Microalgae (Coccomyxa, Trebouxiophyceae, Chlorophyta) Using Integrative Taxonomy and DNA Barcoding with Further Implications for the Species Identification in Environmental Samples. PLoS ONE 2015, 10, e0127838. [Google Scholar] [CrossRef]
- Proeschöld, T.; Darienko, T. The green puzzle Stichococcus (Trebouxiophyceae, Chlorophyta): New generic and species concept among this widely distributed genus. Phytotaxa 2020, 441, 113–142. [Google Scholar] [CrossRef]
- Irisarri, I.; Darienko, T.; Pröschold, T.; Fürst-Jansen, J.M.R.; Jamy, M.; de Vries, J. Unexpected cryptic species among streptophyte algae most distant to land plants. Proc. Biol. Sci. 2021, 288, 1963. [Google Scholar] [CrossRef] [PubMed]
- Hoef-Emden, K. Revision of the genus Chroomonas Hansgirg: The benefits of DNA-containing specimens. Protist 2018, 169, 662–681. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, P.W.; Garner, R.E.; Monchamp, M.È.; Walsh, D.; E Onana, V.; Kraemer, S.; Pick, F.; Beisner, B.; Agbeti, M.; Barbosa da Costa, N.; et al. Comparing microscopy and DNA metabarcoding techniques for identifying cyanobacteria assemblages across hundreds of lakes. Harmful Algae 2022, 113, 102187. [Google Scholar] [CrossRef] [PubMed]
- Dzhembekova, N.; Moncheva, S.; Ivanova, P.; Slabakova, N.; Nagai, S. Biodiversity of phytoplankton cyst assemblages in surface sediments of the Black Sea based on metabarcoding. Biotechnol. Biotechnol. Equip. 2018, 32, 1507–1513. [Google Scholar] [CrossRef]
- CEN/TR 17245:2018; Water Quality—Technical Report for the Routine Sampling of Benthic Diatoms from Rivers and Lakes Adapted for Metabarcoding Analyses. iTeh Standards: Newark, DE, USA, 2018.
- CEN/TR 17244:2018; Water Quality—Technical Report for the Management of Diatom Barcodes. iTeh Standards: Newark, DE, USA, 2018; CEN/TC 230/WG 23—Aquatic Macrophytes and Algae: 2018.
- Xie, Y.; Giesy, J.P. UofS-ETL-EDNA-30 Metabarcoding of Cyanobacteria Assembly; University of Saskatchewan: Saskatoon, SK, Canada, 2018; Version 1. [Google Scholar]
- Yarimizu, K.; Fujiyoshi, S.; Kawai, M.; Norambuena-Subiabre, L.; Cascales, E.-K.; Rilling, J.-I.; Vilugrón, J.; Cameron, H.; Vergara, K.; Morón-López, J.; et al. Protocols for monitoring harmful algal blooms for sustainable aquaculture and coastal fisheries in Chile. Int. J. Env. Res. Pub. Health 2020, 17, 7642. [Google Scholar] [CrossRef]
- Jerney, J.; Hällfors, H.; Oja, J.; Reunamo, A.; Suikkanen, S.; Lehtinen, S. Guidelines for using environmental DNA in Finnish marine phytoplankton monitoring–Improved biodiversity assessment through method complementation. Rep. Finn. Environ. Inst. 2022, 40, 69. [Google Scholar]
- Pawlowski, J.; Audic, S.; Adl, S.; Bass, D.; Belbahri, L.; Berney, C.; Bowser, S.; Cepicka, I.; Decelle, J.; Dunthorn, M.; et al. CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 2012, 10, e1001419. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Number of Primer Set | Gene Region | Target Group | Primer Name | Primer Sequence 5′ to 3′ (Primer Author Reference) | Forward/Reverse | References | PCR Cycling | |
|---|---|---|---|---|---|---|---|---|
| V3 18S | Eukaryotes | ATTAGGGTTCGATTCCGGAGAGG | forward | [30] | n.d. | |||
| CTGGAATTACCGCGGSTGCTG | reverse | |||||||
| [31] | ||||||||
| 1 | V4 18S | Diatoms | DIV4for: | GCGGTAATTCCAGCTCCAATAG | forward | [14,32,33,34,35,36] | 94 °C—2 min (35 cycles: 94 °C—45 s, 50 °C—45 s, 72 °C—1 min), 72 °C—10 min | |
| DIV4rev3 | CTCTGACAATGGAATACGAATA | reverse | ||||||
| [32] | ||||||||
| 2 | V4 18S | Protist, Diatoms | M13F–D512 | TGT AAA ACG ACG GCC AGT ATT CCA GCT CCA ATA GCG | forward | [10,37,38,39] | 94 °C—2 min, (5 cycles: 94 °C—45 s, 52/54 °C—45 s, 72 °C—1 min), (35 cycles: 94 °C—45 s, 50/52 °C—45 s, 72 °C—1 min), 72 °C—10 min. | |
| M13R–D978rev | CAG GAA ACA GCT ATG AC GAC TAC GAT GGT ATC TAATC | reverse | ||||||
| [37] | ||||||||
| 3 | V4 18S | Eukaryotes | F574 | GCGGTAATTCCAGCTCCAA [13] | forward | [40] | 95 °C—5 min, (25 cycles: 98 °C—1 min, 98 °C—20 s, 51 °C—20 s, 72 °C—12 s), 72 °C—1 min. | |
| 1132r | CCGTCAATTHCTTYAART [41] | reverse | ||||||
| 4 | V4 18S | Eukaryotes | AATTCCAGCTCCAATAGCGTATAT | forward | [42] | 98 °C—30 s, (30 cycles:98 °C—10 s, 59 °C—30 s, 72 °C—30 s), 72 °C—10 min. | ||
| TTTCAGCCTTGCGACCATAC | reverse | |||||||
| [42] | ||||||||
| 5 | V4 18S | Eukaryotes | F574 | GCGGTAATTCCAGCTCCAA | forward | [13] | PCR in silico, Tm 55.3 | |
| R952 | AAG ACG ATC AGA TAC C | reverse | ||||||
| [13] | ||||||||
| 6 | V4 18S | Eukaryotes | TAReuk454FWD1 | CCAGCA (G/C)C(C/T)GCGGTAATTCC [43] | forward | [10,27,44,45,46,47,48,49,50,51,52] * | 94 °C—5 min, (15 cycles: 94 °C—30 s, 53 °C—45 s, 72 °C—1 min), (20 cycles: 94 °C—of 30 s, 48 °C—45 s, 72 °C—1 min), 72 °C—10 min. | |
| TAReukREV3 | ACTTTCGTTCTTGAT(C/T)(A/G)A [43] | reverse | ||||||
| V4 forward | CCAGCAGCCGCGGTAATTCC [43] modfied primers from [43] | forward | [53] | |||||
| V4 reverse | ACTTTCGTTCTTGATTAA [43] modfied primers from [43] | reverse | [53] | |||||
| 7 | V4 18S | Eukaryotes | TAReuk454FWD1 | CCAGCA (G/C)C(C/T)GCGGTAATTCC [43] | forward | [54] | 95 °C—5 min, (10 cycles: 94 °C—30 s, 57 °C—45 s, 72 °C—1 min), (15 cycles: 94 °C—30 s, 47 °C—45 s, 72 °C—1 min), 72 °C—10 min. | |
| V4r | ACTTTCGTTCTTGAT [54] modfied primers from [43] | reverse | ||||||
| V4–V5 18S | Eukaryotes | 563f | GCCAGCAVCYGCGGTAAY | forward | [41,55,56] | |||
| 1132r | CCGTCAATTHCTTYAART | reverse | ||||||
| [41] | ||||||||
| V7 18S | Eukaryotic phytoplankton community | 960F | GGCTTAATTTGACTCAACRCG | forward | [57] | Two-step tailed PCR. Round 1: 95 °C for 3 min, (15 cycles: 95 °C—1 min, 55 °C—1 min, 72 °C—1 min), 72 °C—10 min (260 bp). Round 2: 98 °C—30 s, (10 cycles—98 °C—10 s, 55 °C—30 s, 72 °C—30 s), 72 °C—5 min. | ||
| NSR1438 | GGGCATCACAGACCTGTTAT | reverse | ||||||
| [58] | ||||||||
| V7–V8 18S | Eukaryotes | F-1183 | AAT TTG ACT CAA CAC GGG | forward | [13] | The annealing temperature of 52 °C | ||
| R-1631 | TAC AAA GGG CAG GGA CGT AAT | reverse | The annealing temperature of 59.1 °C | |||||
| [13] | ||||||||
| V8–V9 18S | Eukaryotes | V8f 1422 | ATAACAGGTCTGTGATGCCCT [54] | forward | [30,54] | 95 °C—3 min (25 cycles: 98 °C—20 s, 65 °C—15s и 72 °C—15 s), 72 °C—10 min. | ||
| 1510R | GCCTTGCCAGCCCGCTCAG (eukaryotic) [59] | reverse | ||||||
| 1 | V9 18S | Eukaryotes | 1391F | GTACACACCGCCCGTC [60] | forward | [10,48,61,62,63,64,65,66] | 92 °C—3 min, (30 cycles: 45-s—92 °C, 1-min—57 °C, 1.5-min—72 °C.) 10 min—72 °C. | |
| EukBr | TGATCCTTCTGCAGGTTCACCTAC [67] | reverse | ||||||
| 2 | V9 18S | Eukaryotes | 1380F | CCCTGCCHTTTGTACACAC (eukaryotic) | forward | [53] | 94 °C—3 min, 30 cycles: 94 °C—30 s, 57 °C—60 s, 72 °C—90 s), 72 °C—10 min 94 °C 10 min, (35 cycles: 94 °C—40 s, 58 °C—25 s, 72 °C—30 s), 72 °C—10 min. | |
| 1389F | TTGTACACACCGCCC (universal) | forward | ||||||
| 1510R | CCTTCYGCAGGTTCACCTAC (eukaryotic) | reverse | ||||||
| [59] | ||||||||
| V9-ITS1 | Protist | GTACACACCGCCCGTC | forward | [68,69,70,71] | 98 °C—3 min, (35 cycles: 98 °C—30 s, 52 °C—75 s, 72 °C—60 s), 72 °C—10 min. | |||
| ITS2_Dino; 10% | GCTGCGCCCTTCATCGKTG | reverse | ||||||
| ITS2_broad; 90% | GCTGCGTTCTTCATCGWTR | reverse | ||||||
| ITS2 | Chlorophyceae | ITS3 | GCATCGATGAAGAACGCAGC | forward | [72,73,74] | n.d. | ||
| ITS4 | TCCTCCGCTTATTGATATGC | reverse | ||||||
| [75] | ||||||||
| 1 | rbcL | Diatoms | Diat_ rbcL _708F_1 | AGGTGAAGTAAAAGGTTCWTACTTAAA | forward | [14,16,22,33,34,36] ** [76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95] | 95 °C—15 min, (30–40 cycles: 95 °C—45 s, 55 °C—45, 72 °C—45 s) (final extension). | |
| Diat_ rbcL _708F_2 | AGGTGAAGTTAAAGGTTCWTAYTTAAA | forward | ||||||
| Diat_ rbcL _708F_3 | AGGTGAAACTAAAGGTTCWTACTTAAA | forward | ||||||
| R3_1 | CCTTCTAATTTACCWACWACTG | reverse | ||||||
| R3_2 | CCTTCTAATTTACCWACAACAG | reverse | ||||||
| [16] | ||||||||
| 2 | rbcL | Diatoms | rbcL 646F | ATGCGTTGGAGAGARGTTTC | [17,46,86,96] | 95 °C—15 min, (32–35 cycles: 95 °C—20 s, 55 °C—45 s, 72 °C—60 s),72 °C—5 min. | ||
| rbcL 998R | GATCACCTTCTAATTTACCWACAACTG | |||||||
| [17] | ||||||||
| 3 | rbcL | Eustigmatophyceae | EU rbcL 500FA | GGNCGYGTWGTDTWYGAAGGT | forward | [97] | The annealing temperature of 53.5 °C | |
| Eustig rbcL-R900 | CACCWGCCATACGCATCC | reverse | ||||||
| [97] | ||||||||
| 23S | Protist | p23SrV_f1 | GGA CAG AAA GAC CCT ATG AA | forward | [10,98,99] | 94 °C—2 min, (35 cycles: 94 °C—20 s, 55 °C—30 s, and 72 °C—30 s) 72 °C—10 min. | ||
| p23SrV_r1 | TCA GCC TGT TAT CCC TAG AG | reverse | ||||||
| [100] | ||||||||
| 1 | V3–V4 16S | Freshwater phytoplankton | 341F | CCTACGGGNGGCWGCAG | forward | [101] | 95 °C—5 min, (25 cycles: 95 °C—40 s, 53 °C—40 s and 72 °C—1 min) 72 °C—7 min. | |
| 805R | GACTACHVGGGTATCTAATCC | reverse | ||||||
| 2 | V4 16S | Diatom plastid | 515F | GTGYCAGCMGCCGCGGTAA [102] | forward | [103] | 94 °C for 3 min, (30–35 cycles: 94 °C—30 s, 53 °C—40 s, 72 °C—1 min), 72 °C—5 min. | |
| 806R | GGA CTA CHV GGG TWTCTA AT [104] | reverse | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kezlya, E.; Tseplik, N.; Kulikovskiy, M. Genetic Markers for Metabarcoding of Freshwater Microalgae: Review. Biology 2023, 12, 1038. https://doi.org/10.3390/biology12071038
Kezlya E, Tseplik N, Kulikovskiy M. Genetic Markers for Metabarcoding of Freshwater Microalgae: Review. Biology. 2023; 12(7):1038. https://doi.org/10.3390/biology12071038
Chicago/Turabian StyleKezlya, Elena, Natalia Tseplik, and Maxim Kulikovskiy. 2023. "Genetic Markers for Metabarcoding of Freshwater Microalgae: Review" Biology 12, no. 7: 1038. https://doi.org/10.3390/biology12071038