
Intraspecific Comparative Analysis Reveals Genomic Variation of Didymella arachidicola and Pathogenicity Factors Potentially Related to Lesion Phenotype

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolates and Pathogenicity Assays
2.2. Genomic DNA, RNA Extraction, and Whole-Genome Sequencing
2.3. Genome Assembly and Hi-C Analysis
2.4. Genome Annotation
2.5. Genome Re-Sequencing and Variations Calling
2.6. Population Phylogeny Analysis
2.7. Identification of Potential Pathogenicity Factors
3. Results
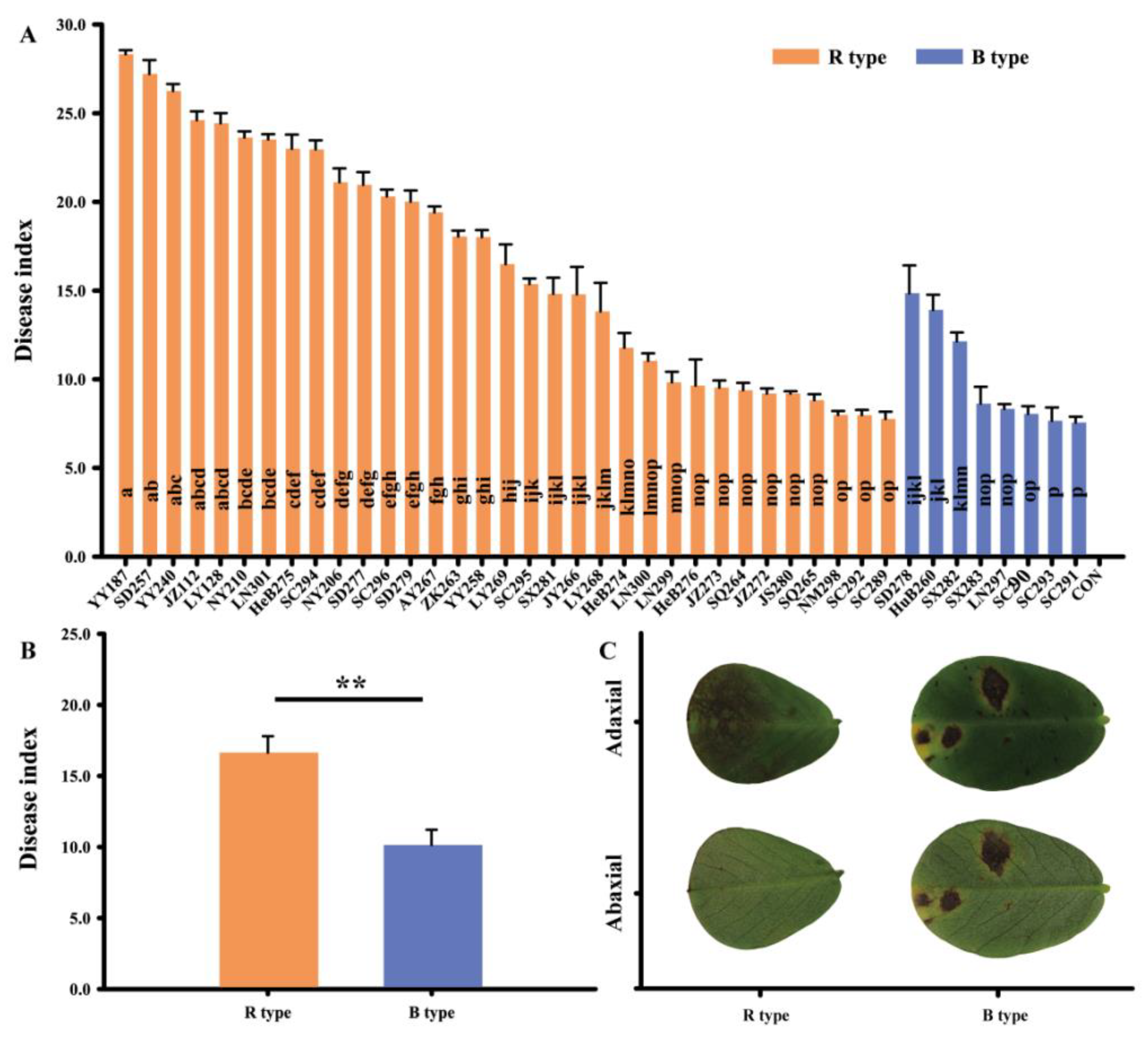
3.1. Pathogenicity Varied among D. arachidicola Isolates
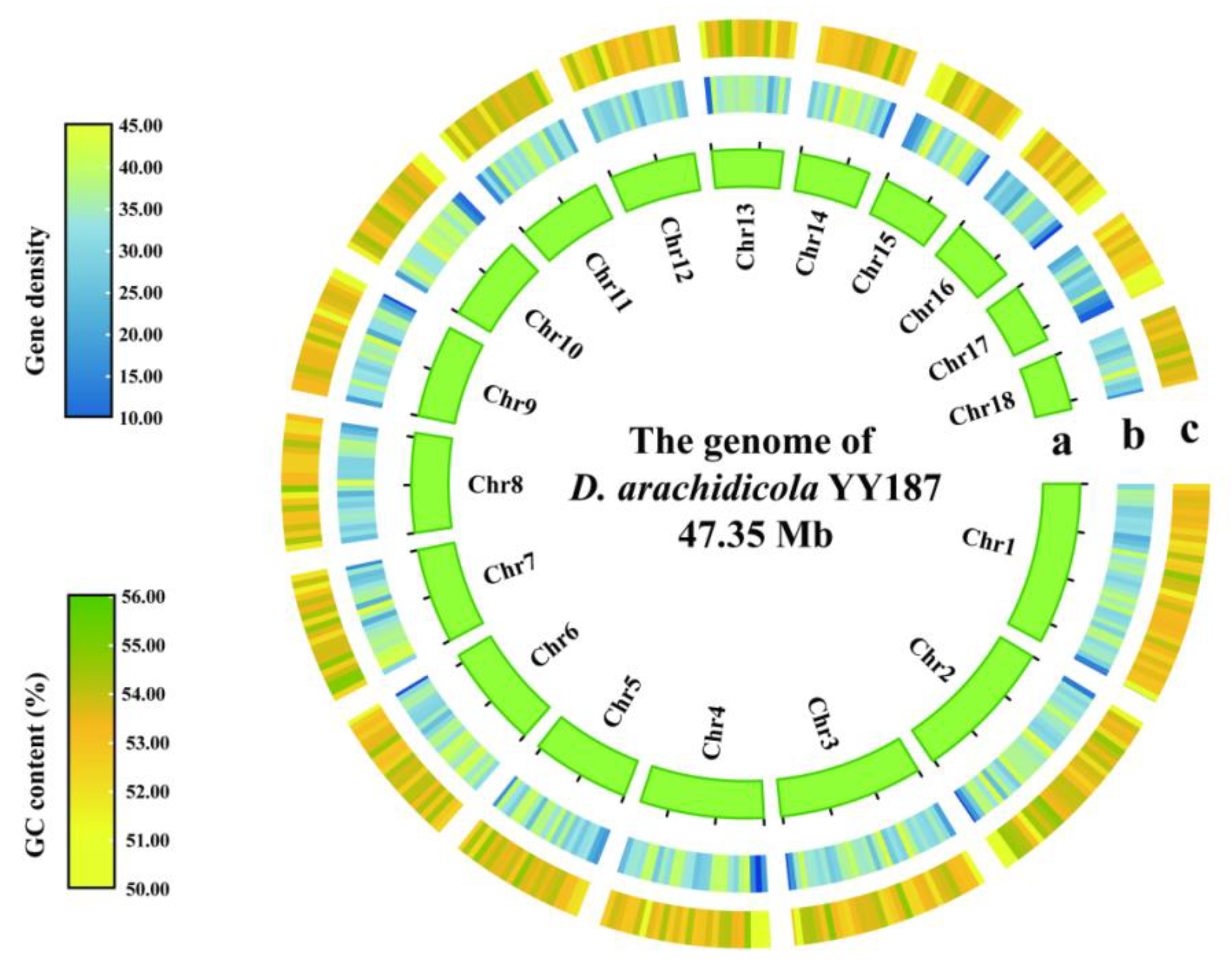
3.2. Genome Assembly and Annotation
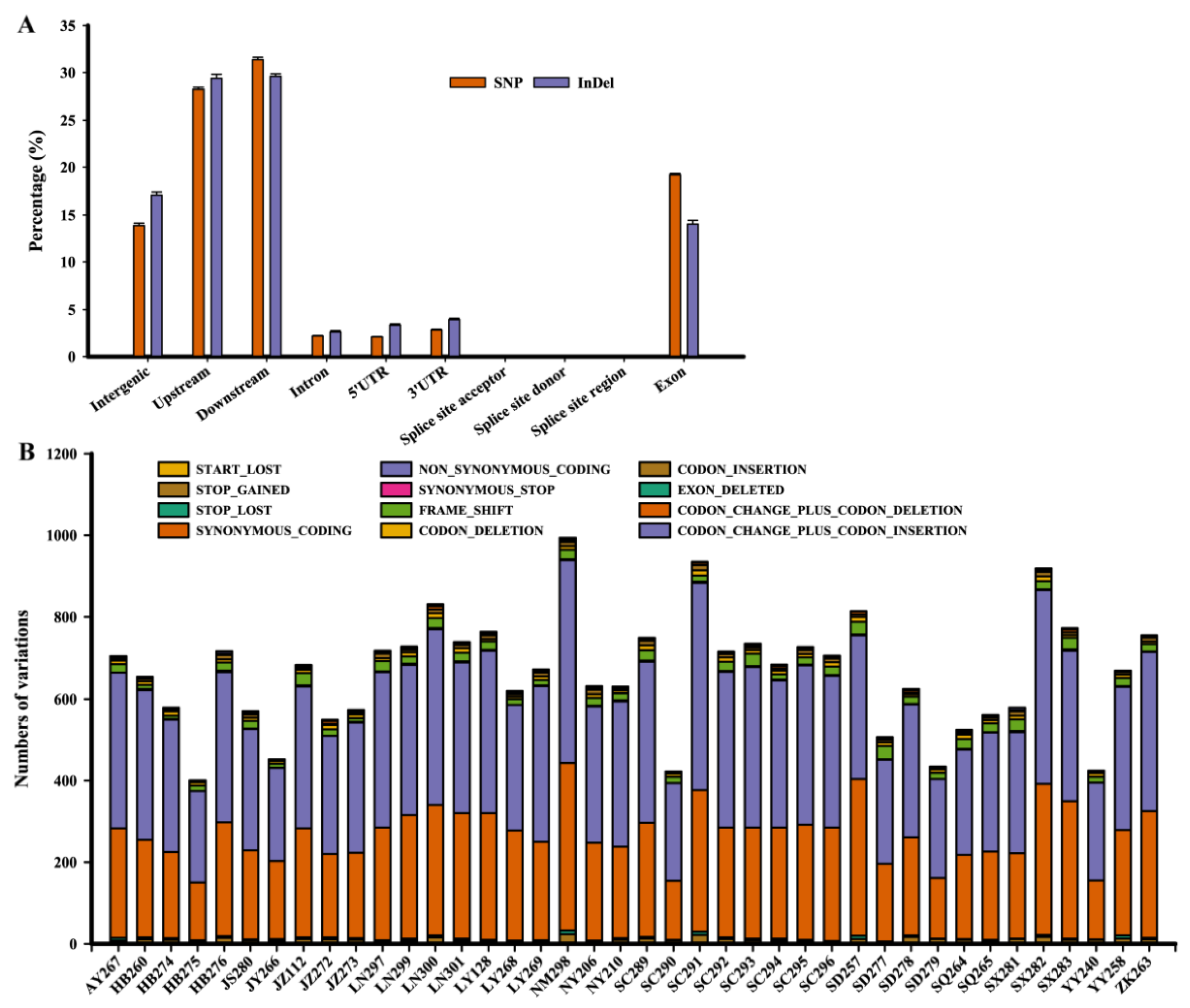
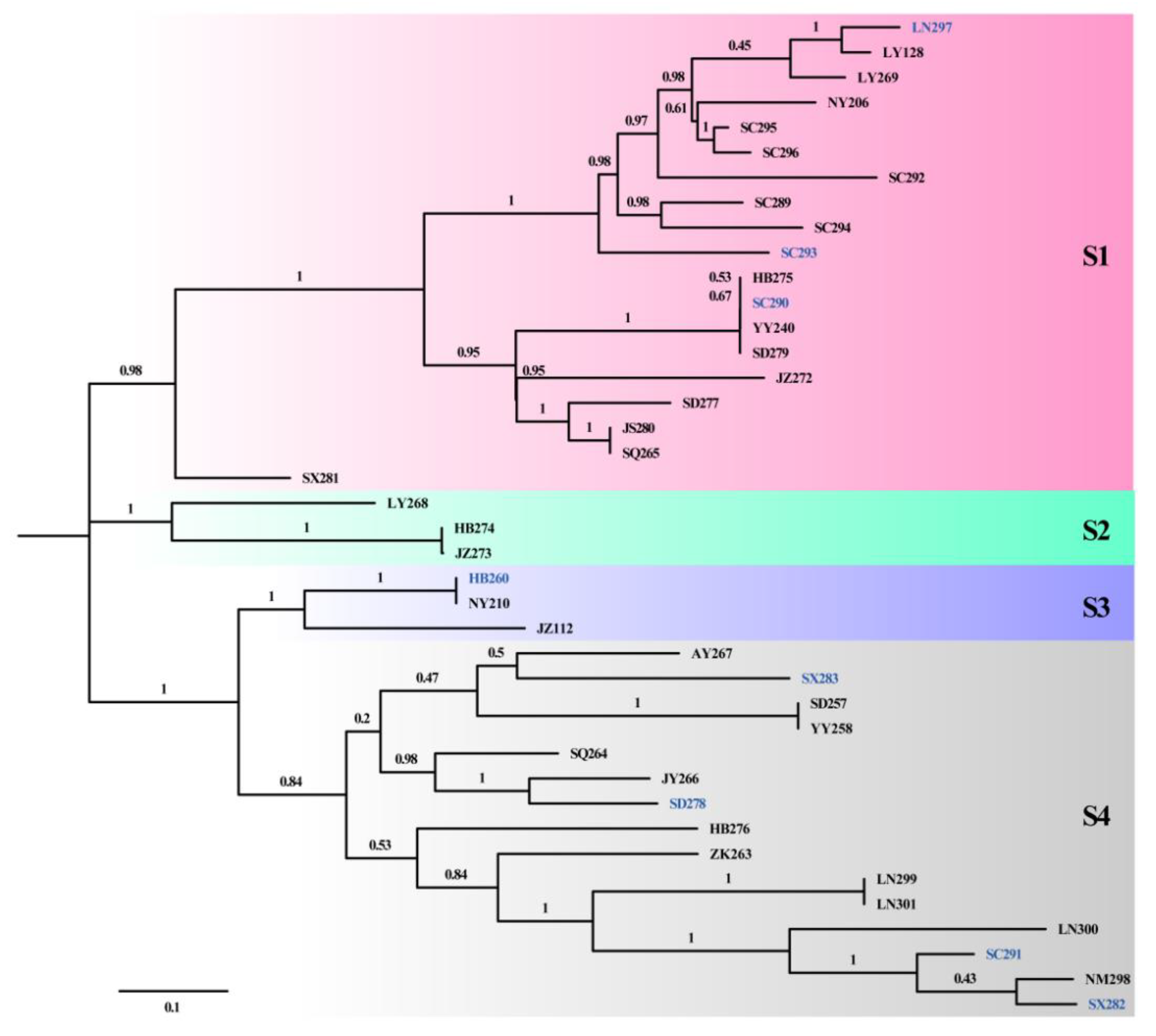
3.3. Genomic Variation and Phylogeny of D. arachidicola
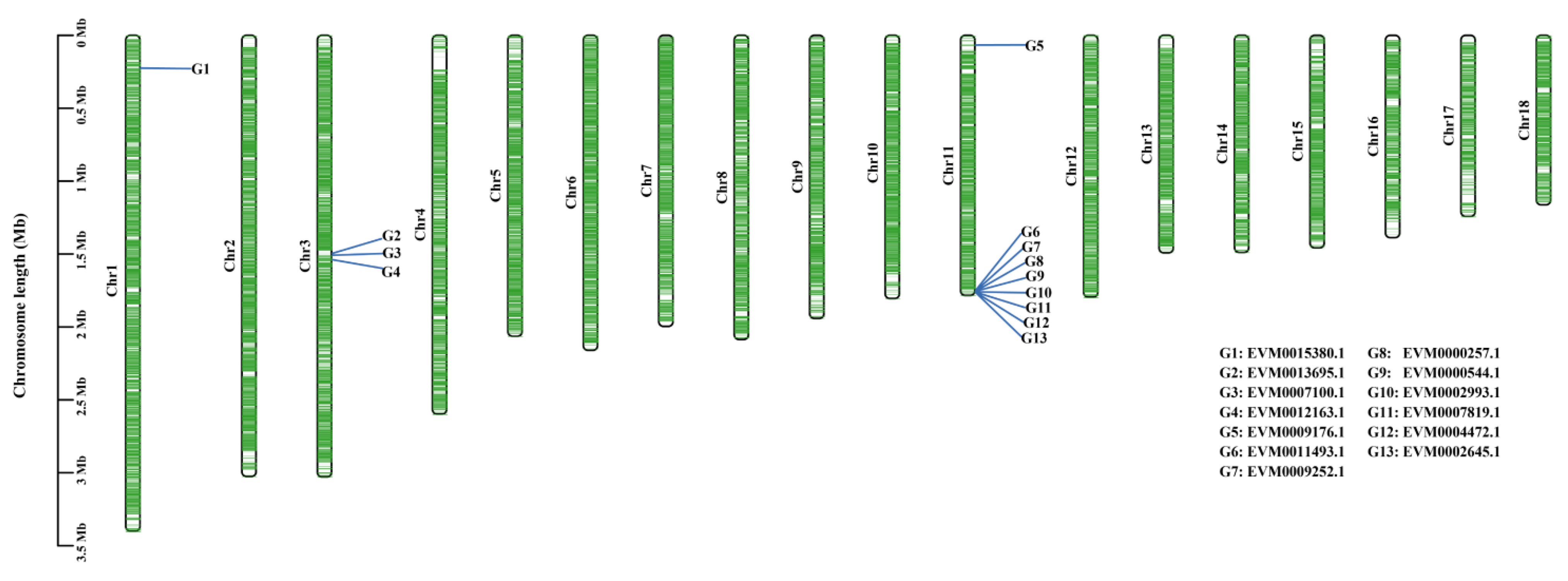
3.4. Identification of Pathogenicity Factors
3.5. Pathogenicity Factors Potentially Related to Different Lesion Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomilin, B.A. Opredelitel’ Gribov Roda Mycosphaerella Johansen; ‘Nauka’ Publishing House: Leningrad, Russia, 1979. [Google Scholar]
- Pettit, R.E.; Philley, G.L.; Smith, D.H.; Taber, R.A. Peanut Web Blotch: II Symptoms and Host Range of Pathogen1. Peanut Sci. 1986, 13, 27–30. [Google Scholar] [CrossRef]
- Fu, J.F.; Wang, D.Z.; Zhou, R.J.; Yang, F.Y.; Su, W.N. Occurrence and epidemic dynamics of peanut web blotch disease in Liaoning Province. Chin. J. Oil Crop Sci. 2013, 35, 80–83. [Google Scholar] [CrossRef]
- Xu, X.J.; Cui, F.G.; Shi, Y.M.; Xu, M.X.; Bi, G.J. Research on the peanut web blotch in China. Acta Phytopathol. Sin. 1995, 22, 70–74. [Google Scholar] [CrossRef]
- Lancaster, S.H.; Jordan, D.L.; York, A.C.; Wilcut, J.W.; Brandenburg, R.L.; Monks, D.W. Interactions of Late-Season Morningglory (Ipomoea spp.) Management Practices in Peanut (Arachis hypogaea). Weed Technol. 2005, 19, 803–808. [Google Scholar] [CrossRef]
- Aveskamp, M.M.; de Gruyter, J.; Woudenberg, J.H.C.; Verkley, G.J.M.; Crous, P.W. Highlights of the Didymellaceae: A polyphasic approach to characterise Phoma and related pleosporalean genera. Stud. Mycol. 2010, 65, 1–60. [Google Scholar] [CrossRef]
- Xie, J.H.; Lin, Y.; Zang, C.Q.; Pei, X.; Liang, C.H. Peanut net spot disease: Selection of chemical control agents and compound synergistic formulas. Chin. Agric. Sci. Bull. 2021, 23, 101–105. [Google Scholar] [CrossRef]
- Man-Lin, X.; Xia, Z.; Ju-Xiang, W.; Jing, Y.; Zhi-Qing, G.; Ying, L.; Hong-Feng, X.; De-Yun, T.; Ru-Jun, Z.; Yu-Cheng, C. Evaluation of peanut accessions resistance against Phoma arachidicola and relationship between disease-resistance and yield loss. Chin. J. Oil Crop Sci. 2021, 43, 731–735. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, J.R.; Zhang, G.Z.; Cai, L.; Crous, P.W. Resolving the Phoma enigma. Stud. Mycol. 2015, 82, 137–217. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Groenewald, J.; Pfenning, L.; Yarden, O.; Crous, P.; Cai, L. The phoma-like dilemma. Stud. Mycol. 2020, 96, 309–396. [Google Scholar] [CrossRef]
- Marasas, W.F.O.; Pauer, G.D.; Boerema, G.H. A serious leaf blotch disease of groundnuts (Arachis hypogaea L.) in southern Africa caused by Phoma arachidicola sp. nov. Phytophylactica 1974, 6, 195–202. [Google Scholar]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cox, M.P. Comparative genomics reveals a core gene toolbox for lifestyle transitions in Hypocreales fungi. Environ. Microbiol. 2021, 23, 3251–3264. [Google Scholar] [CrossRef] [PubMed]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- De Silva, N.I.; Lumyong, S.; Hyde, K.D.; Bulgakov, T.; Phillips, A.J.L.; Yan, J.Y. Mycosphere Essays 9: Defining biotrophs and hemibiotrophs. Mycosphere 2016, 7, 545–559. [Google Scholar] [CrossRef]
- Leuchtmann, A.; Schardl, C. The Epichloë Endophytes of Grasses and the Symbiotic Continuum. Mycology 2005, 23, 475–503. [Google Scholar] [CrossRef]
- Redman, R.S.; Freeman, S.; Clifton, D.R.; Morrel, J.; Brown, G.; Rodriguez, R.J. Biochemical Analysis of Plant Protection Afforded by a Nonpathogenic Endophytic Mutant of Colletotrichum magna. Plant Physiol. 1999, 119, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Redman, R.S.; Roossinck, M.J.; Maher, S.; Rodriguez, R.J. Field performance of cucurbit and tomato plants colonized with a nonpathogenic mutant of Colletotrichum magna (teleomorph: Glomerella magna; Jekins and Winstead). Symbiosis 2002, 32, 55–70. [Google Scholar]
- Hill, R.; Buggs, R.J.; Vu, D.T.; Gaya, E. Lifestyle Transitions in Fusarioid Fungi are Frequent and Lack Clear Genomic Signatures. Mol. Biol. Evol. 2022, 39, msac085. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Muszewska, A.; Stepniewska-Dziubinska, M.M.; Steczkiewicz, K.; Pawlowska, J.; Dziedzic, A.; Ginalski, K. Fungal lifestyle reflected in serine protease repertoire. Sci. Rep. 2017, 7, 9147. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Corwin, J.A.; Soltis, N.E.; Subedy, A.; Denby, K.J.; Kliebenstein, D.J. Whole genome resequencing of Botrytis cinerea isolates identifies high levels of standing diversity. Front. Microbiol. 2015, 6, 996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.; Urban, M.; Hammond-Kosack, M.C.U.; Hassani-Pak, K.; Hammond-Kosack, K.E. The completed genome sequence of the pathogenic ascomycete fungus Fusarium graminearum. BMC Genom. 2015, 16, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, L.; Pollastro, S.; Rotolo, C.; Romanazzi, G.; Faretra, F.; Angelini, R.M.D.M. Draft Genomic Resources for the Brown Rot Fungal Pathogen Monilinia laxa. Mol. Plant-Microbe Interact. 2019, 33, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.P.; Moon, S.; Yoo, S.-I.; Noh, J.-H.; Ko, H.-G.; Kim, H.A.; Ro, H.-S.; Cho, H.; Chung, J.-W.; Lee, H.-Y.; et al. Functional Analysis of a Novel ABL (Abnormal Browning Related to Light) Gene in Mycelial Brown Film Formation of Lentinula edodes. J. Fungi 2020, 6, 272. [Google Scholar] [CrossRef]
- Meile, L.; Peter, J.; Puccetti, G.; Alassimone, J.; McDonald, B.A.; Sánchez-Vallet, A. Chromatin Dynamics Contribute to the Spatiotemporal Expression Pattern of Virulence Genes in a Fungal Plant Pathogen. Mbio 2020, 11, e02343-20. [Google Scholar] [CrossRef] [PubMed]
- Hudec, C.; Biessy, A.; Novinscak, A.; St-Onge, R.; Lamarre, S.; Blom, J.; Filion, M. Comparative Genomics of Potato Common Scab-Causing Streptomyces spp. Displaying Varying Virulence. Front. Microbiol. 2021, 12, 716522. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Diao, Y.; Lu, Q.; Zhao, J.; Cui, S.; Xiong, X.; Lu, A.; Zhang, X.; Liu, H. Comparative Genomics Reveals Evolutionary Traits, Mating Strategies, and Pathogenicity-Related Genes Variation of Botryosphaeriaceae. Front. Microbiol. 2022, 13, 800981. [Google Scholar] [CrossRef]
- Marcet-Houben, M.; Villarino, M.; Vilanova, L.; De Cal, A.; van Kan, J.; Usall, J.; Gabaldón, T.; Torres, R. Comparative Genomics Used to Predict Virulence Factors and Metabolic Genes among Monilinia Species. J. Fungi 2021, 7, 464. [Google Scholar] [CrossRef]
- Gramaje, D.; Berlanas, C.; Martínez-Diz, M.; Diaz-Losada, E.; Antonielli, L.; Beier, S.; Gorfer, M.; Schmoll, M.; Compant, S. Comparative Genomic Analysis of Dactylonectria torresensis Strains from Grapevine, Soil and Weed Highlights Potential Mechanisms in Pathogenicity and Endophytic Lifestyle. J. Fungi 2020, 6, 255. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sánchez-García, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020, 11, 5125. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, B.; Zou, F.; Shen, D.; Yin, Z.; Wang, R.; He, F.; Wang, Y.; Tyler, B.M.; Fan, W.; et al. Whole Genome Re-sequencing Reveals Natural Variation and Adaptive Evolution of Phytophthora sojae. Front. Microbiol. 2019, 10, 2792. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Siddique, M.I.; Kwon, J.-K.; Kang, B.-C. Comparative Genomic Analysis Reveals Genetic Variation and Adaptive Evolution in the Pathogenicity-Related Genes of Phytophthora capsici. Front. Microbiol. 2021, 12, 694136. [Google Scholar] [CrossRef] [PubMed]
- Guttman, D.S.; McHardy, A.C.; Schulze-Lefert, P. Microbial genome-enabled insights into plant–microorganism interactions. Nat. Rev. Genet. 2014, 15, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Sun, H.; Vainio, E.J.; Raffaello, T.; Kovalchuk, A.; Morin, E.; Duplessis, S.; Asiegbu, F.O. Intraspecific comparative genomics of isolates of the Norway spruce pathogen (Heterobasidion parviporum) and identification of its potential virulence factors. BMC Genom. 2018, 19, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xu, M.; Wu, J.; Dong, W.; Chen, D.; Wang, L.; Chi, Y. Draft Genome Sequence of Phoma arachidicola Wb2 Causing Peanut Web Blotch in China. Curr. Microbiol. 2019, 76, 200–206. [Google Scholar] [CrossRef]
- Li, S.; Xue, X.; Gao, M.; Wang, N.; Cui, X.; Sang, S.; Fan, W.; Wang, Z. Genome Resource for Peanut Web Blotch Causal Agent Peyronellaea arachidicola Strain YY187. Plant Dis. 2021, 105, 1177–1178. [Google Scholar] [CrossRef]
- Guo, X.Q.; Li, X.; Zhao, Z.Q.; Li, X.; Ju, Q.; Jiang, X.J.; Qu, M.J. The research of different fungicides on control effects against peanut leaf spot and yield increase to peanut. J. Peanut Sci. 2014, 43, 56–60. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. Curr. Protoc. Bioinform. 2007, 18, e56. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.; Eddy, S.; Floden, E.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.; Esteicher, A.; Gasteiger, E. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2016, 32, 71–72. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Knoll, M.; Sirim, D.; Wagner, F.; Funke, S.; Pleiss, J. The Cytochrome P450 Engineering Database: A navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 2007, 23, 2015–2017. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas032. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. E ffector P: Predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2015, 210, 743–761. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Gawehns, F.; Ma, L.; Bruning, O.; Houterman, P.M.; Boeren, S.; Cornelissen, B.J.C.; Rep, M.; Takken, F.L.W. The effector repertoire of Fusarium oxysporum determines the tomato xylem proteome composition following infection. Front. Plant Sci. 2015, 6, 967. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Vallet, A.; Fouché, S.; Fudal, I.; Hartmann, F.E.; Soyer, J.L.; Tellier, A.; Croll, D. The Genome Biology of Effector Gene Evolution in Filamentous Plant Pathogens. Annu. Rev. Phytopathol. 2018, 56, 21–40. [Google Scholar] [CrossRef]
- Jones, D.A.B.; Rozano, L.; Debler, J.W.; Mancera, R.L.; Moolhuijzen, P.M.; Hane, J.K. An automated and combinative method for the predictive ranking of candidate effector proteins of fungal plant pathogens. Sci. Rep. 2021, 11, 19731. [Google Scholar] [CrossRef]
- Li, J.; Cornelissen, B.; Rep, M. Host-specificity factors in plant pathogenic fungi. Fungal Genet. Biol. 2020, 144, 103447. [Google Scholar] [CrossRef]
- Daverdin, G.; Rouxel, T.; Gout, L.; Aubertot, J.-N.; Fudal, I.; Meyer, M.; Parlange, F.; Carpezat, J.; Balesdent, M.-H. Genome Structure and Reproductive Behaviour Influence the Evolutionary Potential of a Fungal Phytopathogen. PLOS Pathog. 2012, 8, e1003020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkan, N.J.; Lydiate, D.J.; Parkin, I.A.P.; Nelson, M.N.; Epp, D.J.; Cowling, W.A.; Rimmer, S.R.; Borhan, M.H. The B rassica napus blackleg resistance gene LepR3 encodes a receptor-like protein triggered by the L eptosphaeria maculans effector AVRLM 1. New Phytol. 2013, 197, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.M.; Lukasiewicz, J.; Farrer, R.; Dam, P.; Bertoldo, C.; Rep, M. Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom-2 resistance gene in melon. New Phytol. 2016, 209, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouin, A.; Legeai, F.; Nouhaud, P.; Whibley, A.; Simon, J.-C.; Lemaitre, C. Whole-genome re-sequencing of non-model organisms: Lessons from unmapped reads. Heredity 2014, 114, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Laine, V.N.; Gossmann, T.I.; van Oers, K.; Visser, M.E.; Groenen, M.A.M. Exploring the unmapped DNA and RNA reads in a songbird genome. BMC Genom. 2019, 20, 19. [Google Scholar] [CrossRef] [Green Version]
- Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Bhardwaj, S.C.; Prasad, P.; Pal, D.; Devanna, B.N.; Sharma, T.R. Dissection of genomic features and variations of three pathotypes of Puccinia striiformis through whole genome sequencing. Sci. Rep. 2017, 7, srep42419. [Google Scholar] [CrossRef] [Green Version]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2012, 13, 59–69. [Google Scholar] [CrossRef]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef]
- Caracuel, Z.; Martínez-Rocha, A.L.; Di Pietro, A.; Madrid, M.P.; Roncero, M.I.G. Fusarium oxysporum gas1 Encodes a Putative β-1, 3-Glucanosyltransferase Required for Virulence on Tomato Plants. Mol. Plant-Microbe Interact. 2005, 18, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Schirawski, J.; Böhnert, H.U.; Steinberg, G.; Snetselaar, K.; Adamikowa, L.; Kahmann, R. Endoplasmic Reticulum Glucosidase II Is Required for Pathogenicity of Ustilago maydis [W]. Plant Cell 2005, 17, 3532–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, V.; Vélëz, H.; Tzelepis, G. The Role of Glycoside Hydrolases in Phytopathogenic Fungi and Oomycetes Virulence. Int. J. Mol. Sci. 2021, 22, 9359. [Google Scholar] [CrossRef]
- Pemberton, C.L.; Salmond, G.P.C. The Nep1-like proteins-a growing family of microbial elicitors of plant necrosis. Mol. Plant Pathol. 2004, 5, 353–359. [Google Scholar] [CrossRef]
- Qutob, D.; Kemmerling, B.; Brunner, F.; Küfner, I.; Engelhardt, S.; Gust, A.A.; Luberacki, B.; Seitz, H.U.; Stahl, D.; Rauhut, T.; et al. Phytotoxicity and Innate Immune Responses Induced by Nep1-Like Proteins. Plant Cell 2006, 18, 3721–3744. [Google Scholar] [CrossRef] [Green Version]
- Seidl, M.F.; Van Den Ackerveken, G. Activity and Phylogenetics of the Broadly Occurring Family of Microbial Nep1-like Proteins. Annu. Rev. Phytopathol. 2019, 57, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Pirc, K.; Hodnik, V.; Snoj, T.; Lenarčič, T.; Caserman, S.; Podobnik, M.; Böhm, H.; Albert, I.; Kotar, A.; Plavec, J.; et al. Nep1-like proteins as a target for plant pathogen control. PLOS Pathog. 2021, 17, e1009477. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.; Schirawski, J.; Müller, P.; Kahmann, R. An unusual MAP kinase is required for efficient penetration of the plant surface by Ustilago maydis. EMBO J. 2003, 22, 2199–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román, E.; Arana, D.M.; Nombela, C.; Monge, R.A.; Pla, J. MAP kinase pathways as regulators of fungal virulence. Trends Microbiol. 2007, 15, 181–190. [Google Scholar] [CrossRef]
- Son, H.; Seo, Y.-S.; Min, K.; Park, A.R.; Lee, J.; Jin, J.-M.; Lin, Y.; Cao, P.; Hong, S.-Y.; Kim, E.-K.; et al. A Phenome-Based Functional Analysis of Transcription Factors in the Cereal Head Blight Fungus, Fusarium graminearum. PLOS Pathog. 2011, 7, e1002310. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Lynch, V.J. The gene regulatory logic of transcription factor evolution. Trends Ecol. Evol. 2008, 23, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Shelest, E. Transcription Factors in Fungi: TFome Dynamics, Three Major Families, and Dual-Specificity TFs. Front. Genet. 2017, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Scaffold number | 26 |
| Total scaffold length (Mb) | 47.35 |
| Scaffold N50 (Mb) | 2.17 |
| Scaffold N90 (Mb) | 1.39 |
| Maximum scaffold length (Mb) | 6.56 |
| GC content (%) | 56.37 |
| Chromosomal located sequence length (Mb) | 36.03 |
| Number of chromosomes | 18 |
| Percentage of repeats (%) | 9.89 |
| Number of predicted genes | 16,629 |
| Average gene length (Kb) | 1.99 |
| Average CDS number | 2.47 |
| Number of non-coding RNAs | tRNA (286), rRNA (81), other ncRNA (95) |
| Gene ID | Brief Description | GO ID | Pfam Annotation |
|---|---|---|---|
| EVM0000257.1 a | -- | GO:0000981; GO:0003677; GO:0005634; GO:0006351; GO:0006355; GO:0008270 | Fungal Zn(2)-Cys(6) binuclear cluster domain |
| EVM0000544.1 a | Hypothetical protein | GO:0003700; GO:0008270 | AAA domain |
| EVM0002645.1 a, b | Carminomycin 4-O-methyltransferase | GO:0008171 | O-methyltransferase domain |
| EVM0002993.1 | -- | GO:0003676 | Zinc-finger double domain |
| EVM0004472.1 a, b | 3-oxoacyl-[acyl-carrier-protein] reductase | GO:0004316; GO:0006633; GO:0051287; GO:0102131; GO:0102132 | Enoyl-(Acyl carrier protein) reductase |
| EVM0007100.1 a, b, c | Alpha-glucosidase | GO:0004553; GO:0005975; GO:0030246 | Glycosyl hydrolases family 31 |
| EVM0007819.1 a, b, f | NPP1 family protein | - | Necrosis inducing protein (NPP1) |
| EVM0009176.1 | Isopenicillin N synthase family oxygenase | GO:0016491; GO:0046872; GO:0055114 | non-haem dioxygenase in morphine synthesis N-terminal |
| EVM0009252.1 e | Agmatinase | GO:0016813; GO:0046872 | Arginase family |
| EVM0011493.1 | Hypothetical protein | - | Heterokaryon incompatibility protein Het-C |
| EVM0012163.1 | Hypothetical protein | GO:0004843; GO:0006511; GO:0016579; GO:0036459 | Ubiquitin carboxyl-terminal hydrolase |
| EVM0013695.1 a | AAA family ATPase | GO:0005524 | ATPase family associated with various cellular activities (AAA) |
| EVM0015380.1 a, b, d | Hypothetical protein | GO:0004672; GO:0005524; GO:0006468 | Protein kinase domain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Wang, Z.; Gao, M.; Li, T.; Cui, X.; Zu, J.; Sang, S.; Fan, W.; Zhang, H. Intraspecific Comparative Analysis Reveals Genomic Variation of Didymella arachidicola and Pathogenicity Factors Potentially Related to Lesion Phenotype. Biology 2023, 12, 476. https://doi.org/10.3390/biology12030476
Li S, Wang Z, Gao M, Li T, Cui X, Zu J, Sang S, Fan W, Zhang H. Intraspecific Comparative Analysis Reveals Genomic Variation of Didymella arachidicola and Pathogenicity Factors Potentially Related to Lesion Phenotype. Biology. 2023; 12(3):476. https://doi.org/10.3390/biology12030476
Chicago/Turabian StyleLi, Shaojian, Zhenyu Wang, Meng Gao, Tong Li, Xiaowei Cui, Junhuai Zu, Suling Sang, Wanwan Fan, and Haiyan Zhang. 2023. "Intraspecific Comparative Analysis Reveals Genomic Variation of Didymella arachidicola and Pathogenicity Factors Potentially Related to Lesion Phenotype" Biology 12, no. 3: 476. https://doi.org/10.3390/biology12030476