
Molecular Mechanism of Blood Pressure Regulation through the Atrial Natriuretic Peptide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
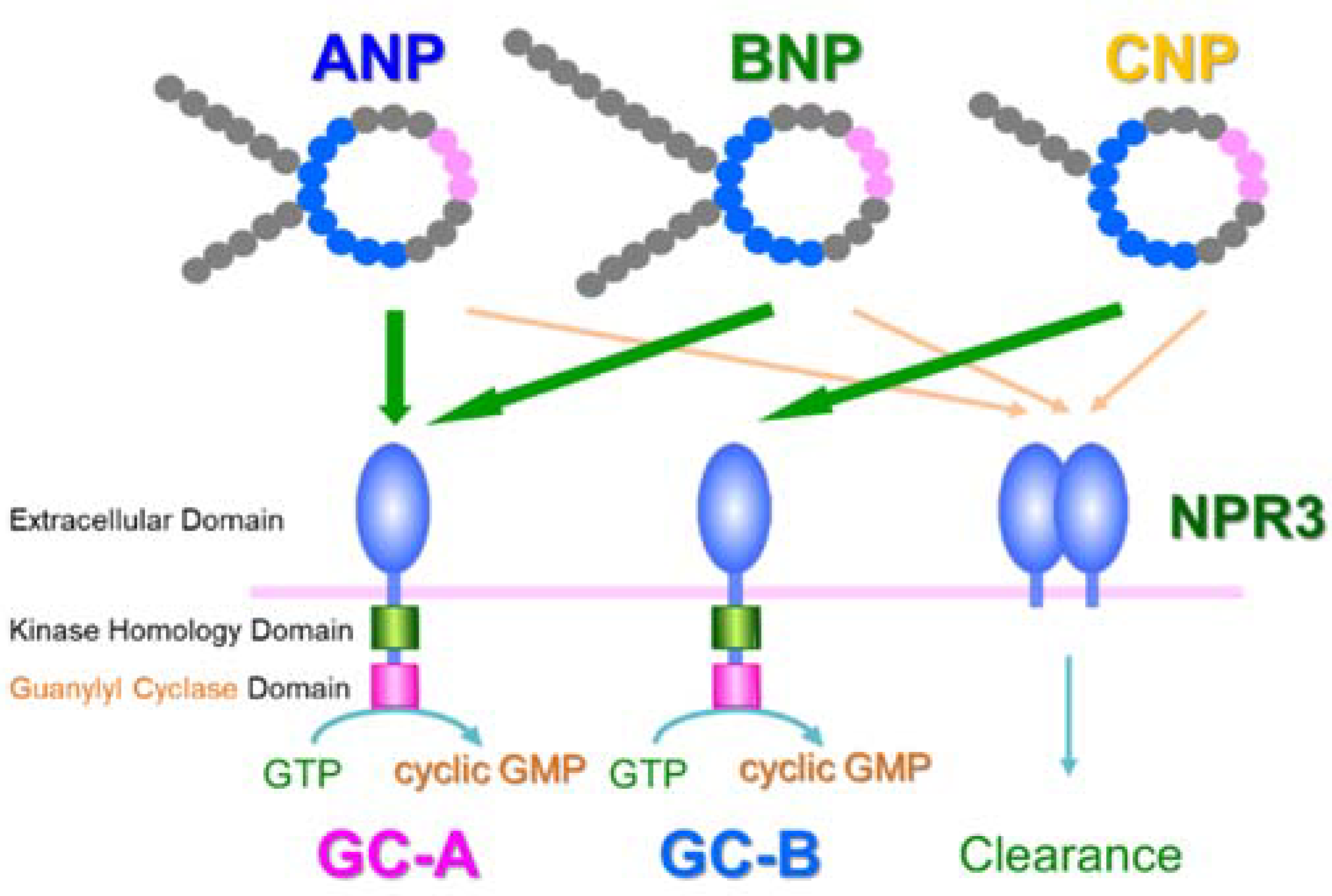
2. Discovery of ANP and Its Receptor
3. Translational Research of ANP
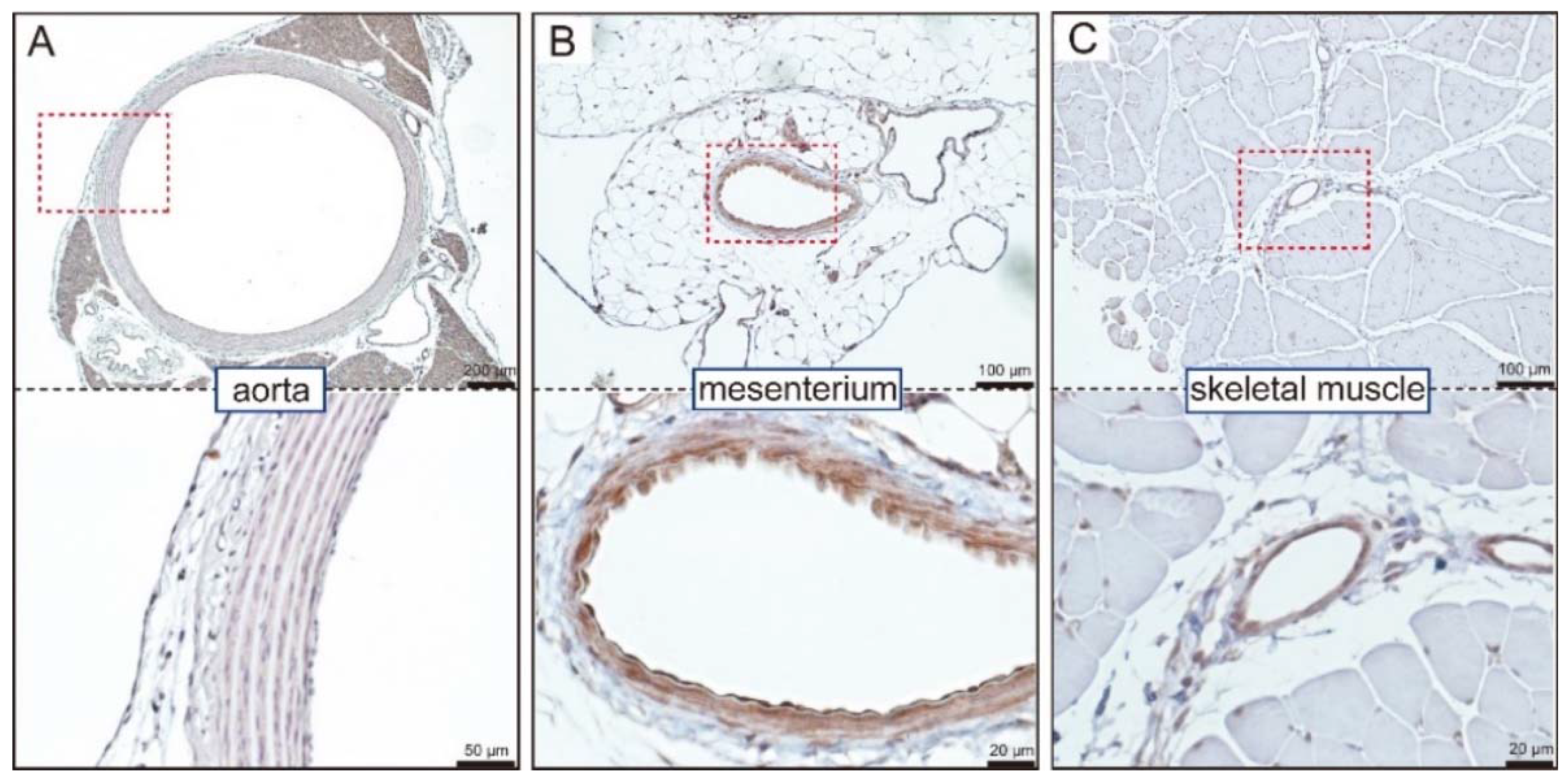
4. GC-A Expression in Blood Vessels
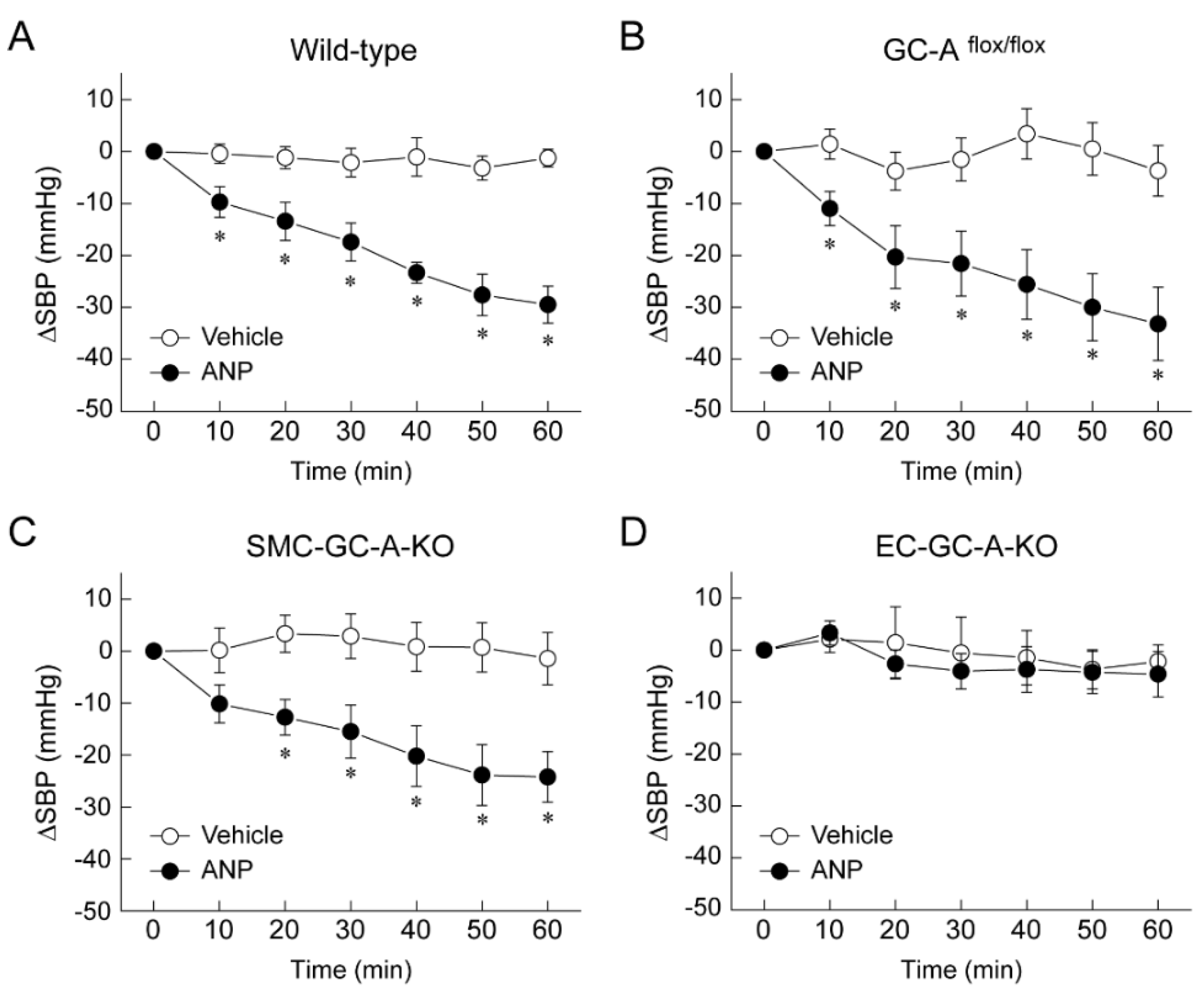
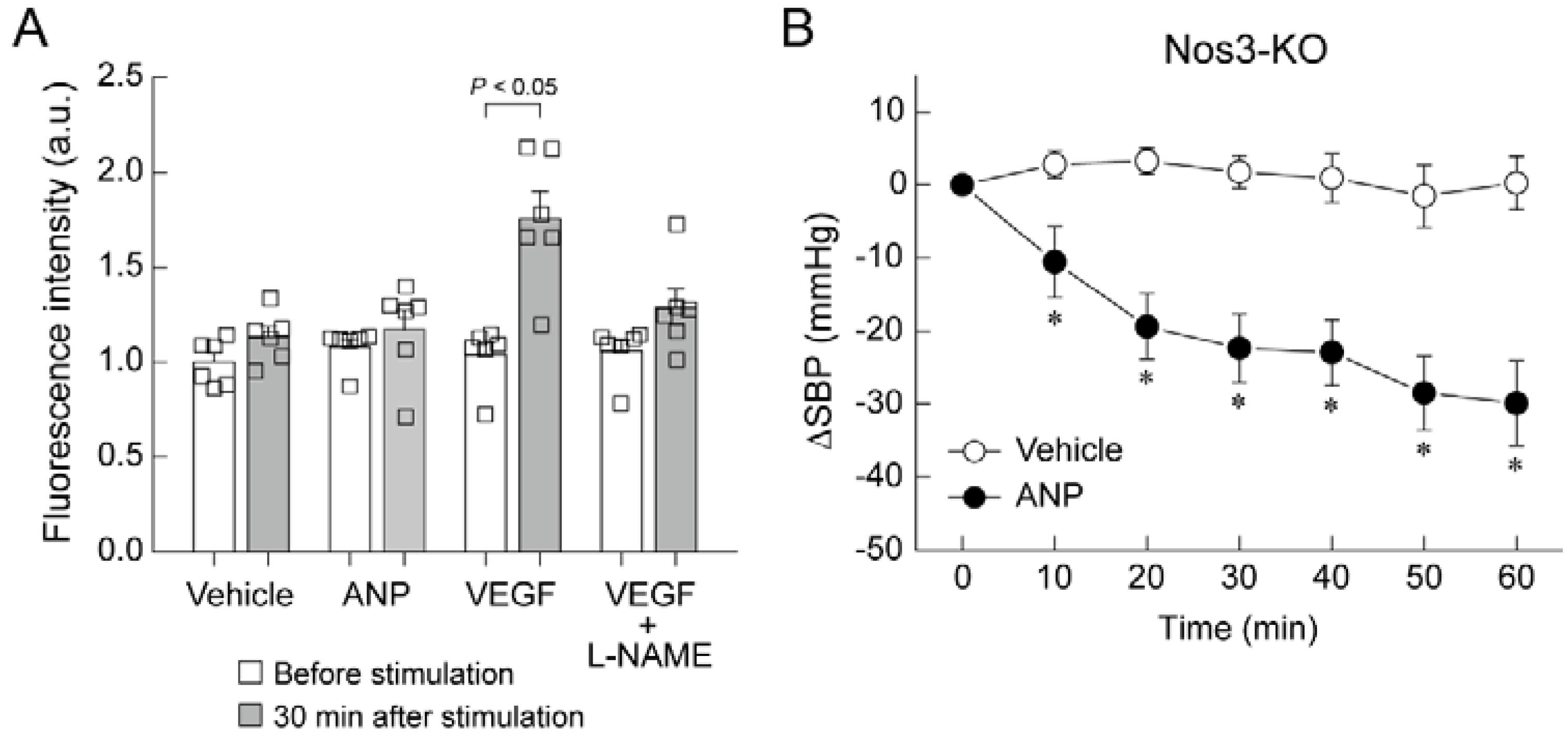
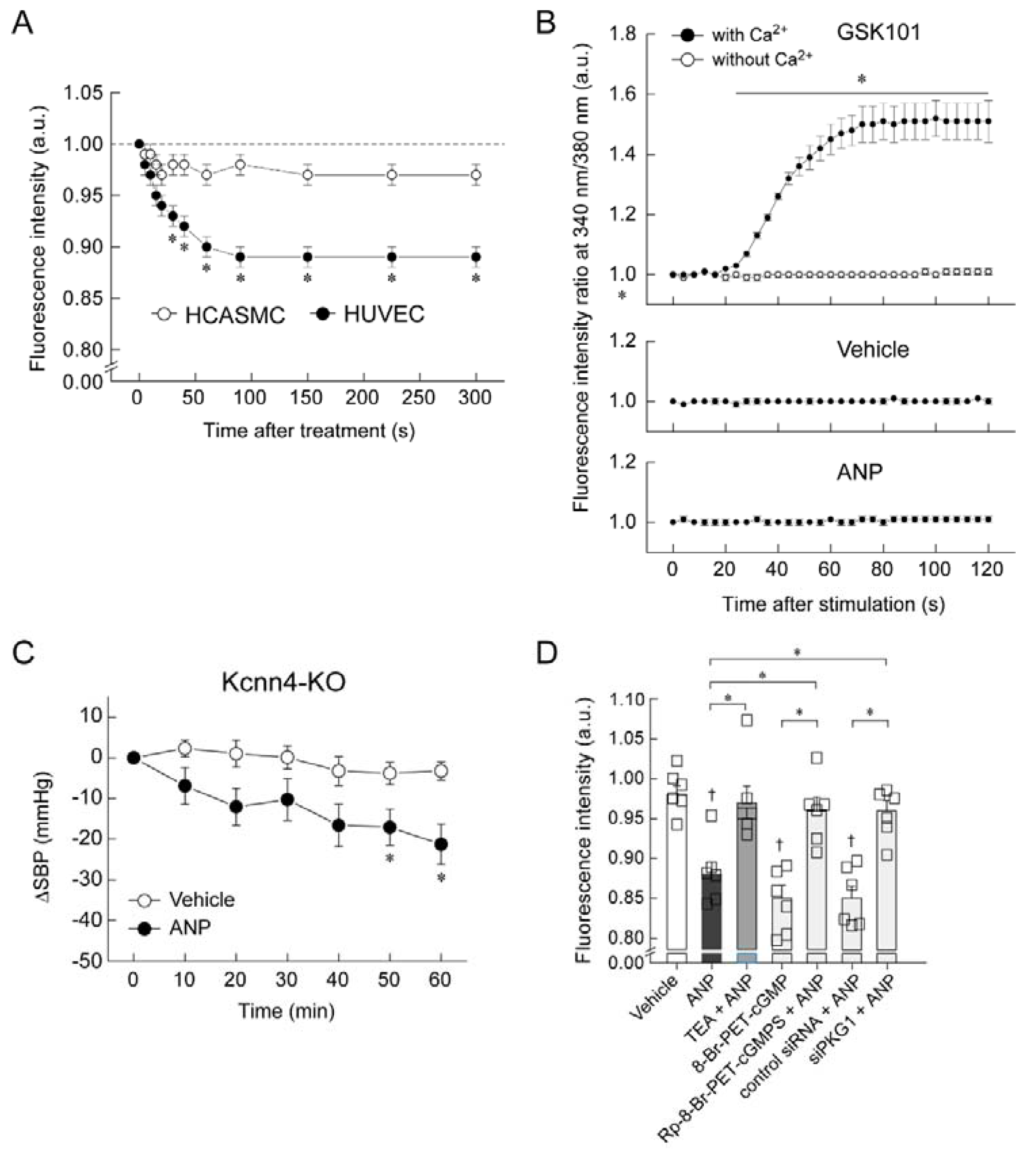
5. Mechanism of Blood Pressure Reduction by Intravenous ANP Infusion
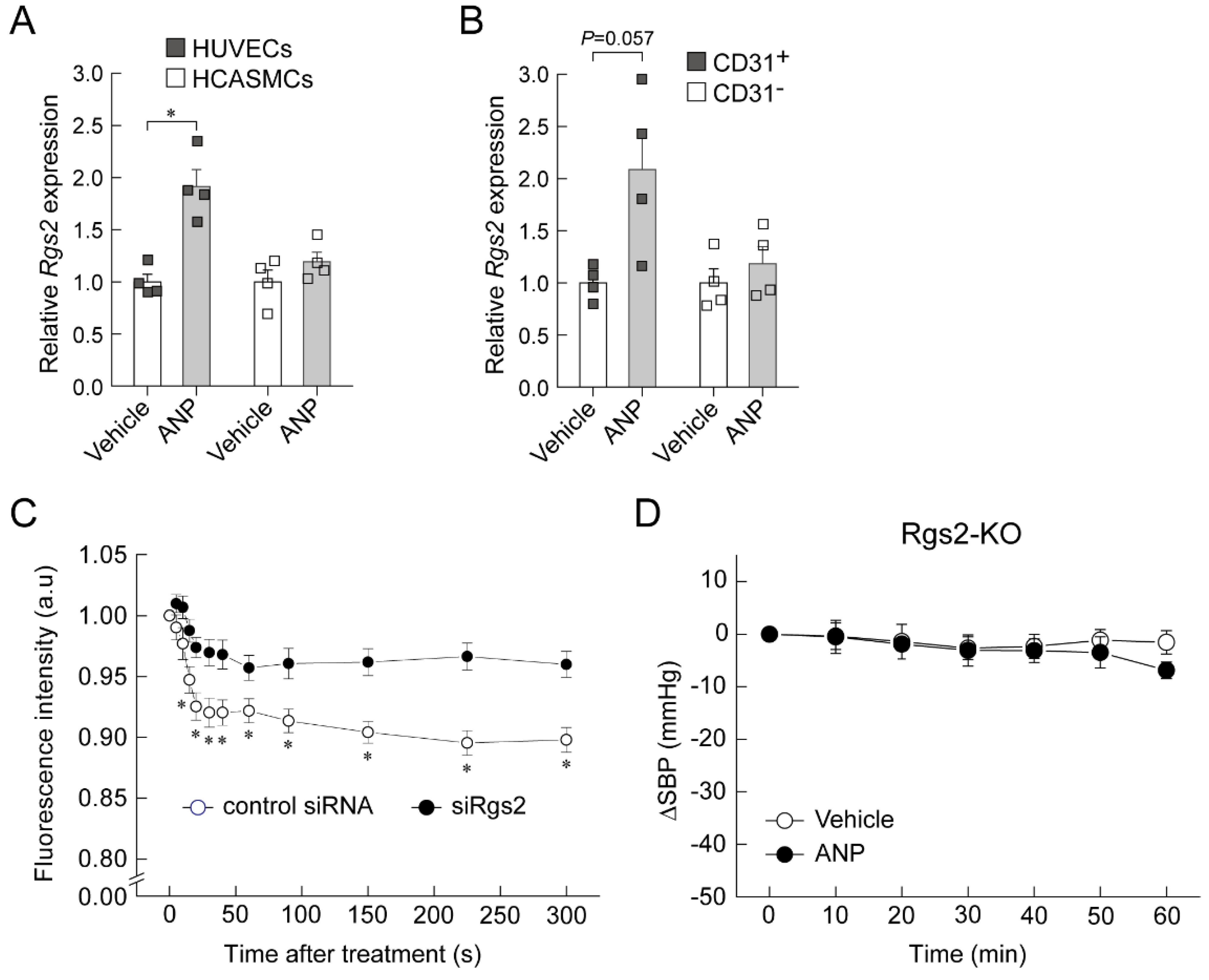
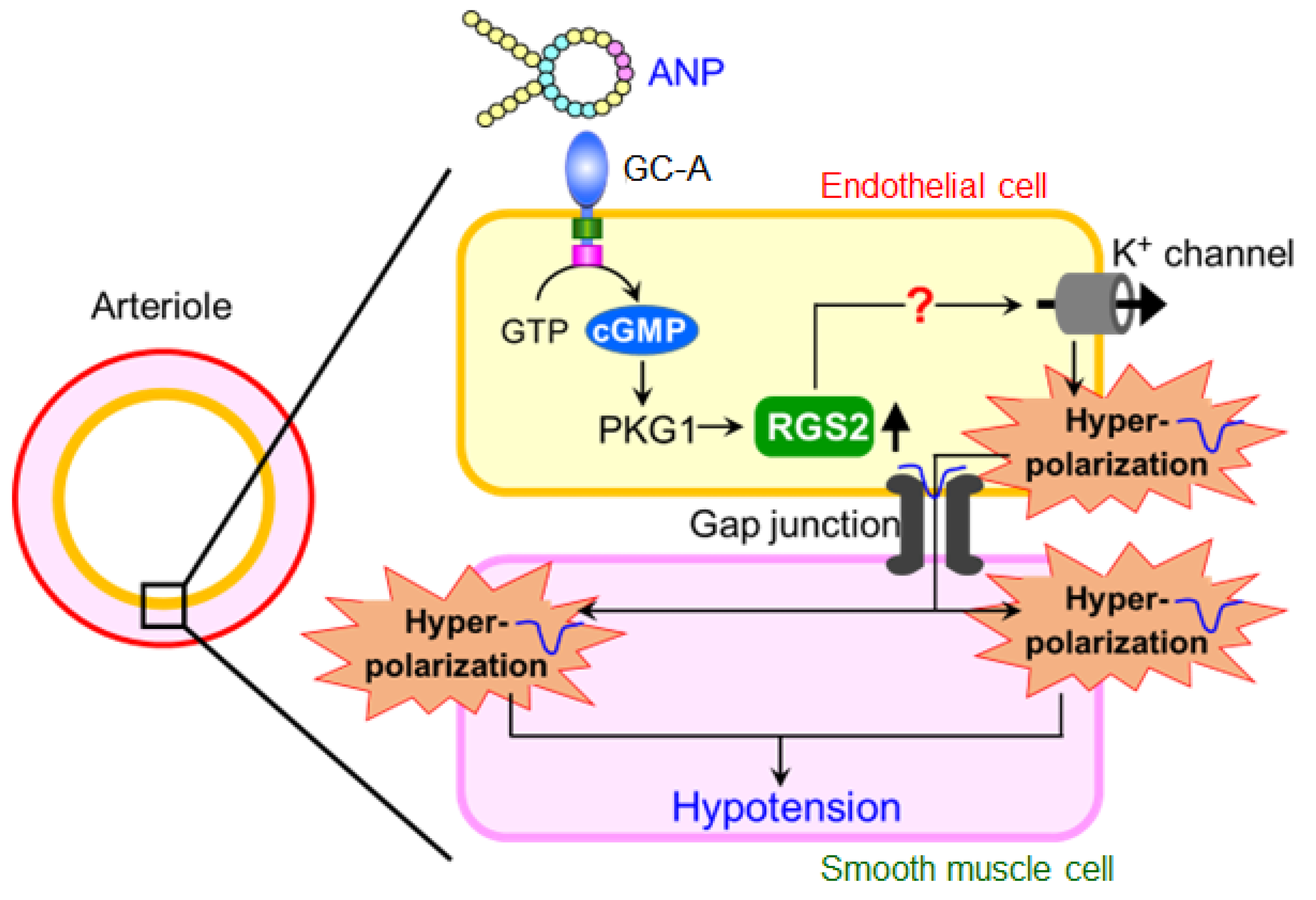
6. Regulator of G-Protein Signaling 2 (RGS2) Plays a Key Role in the Acute Hypotension Effect of ANP
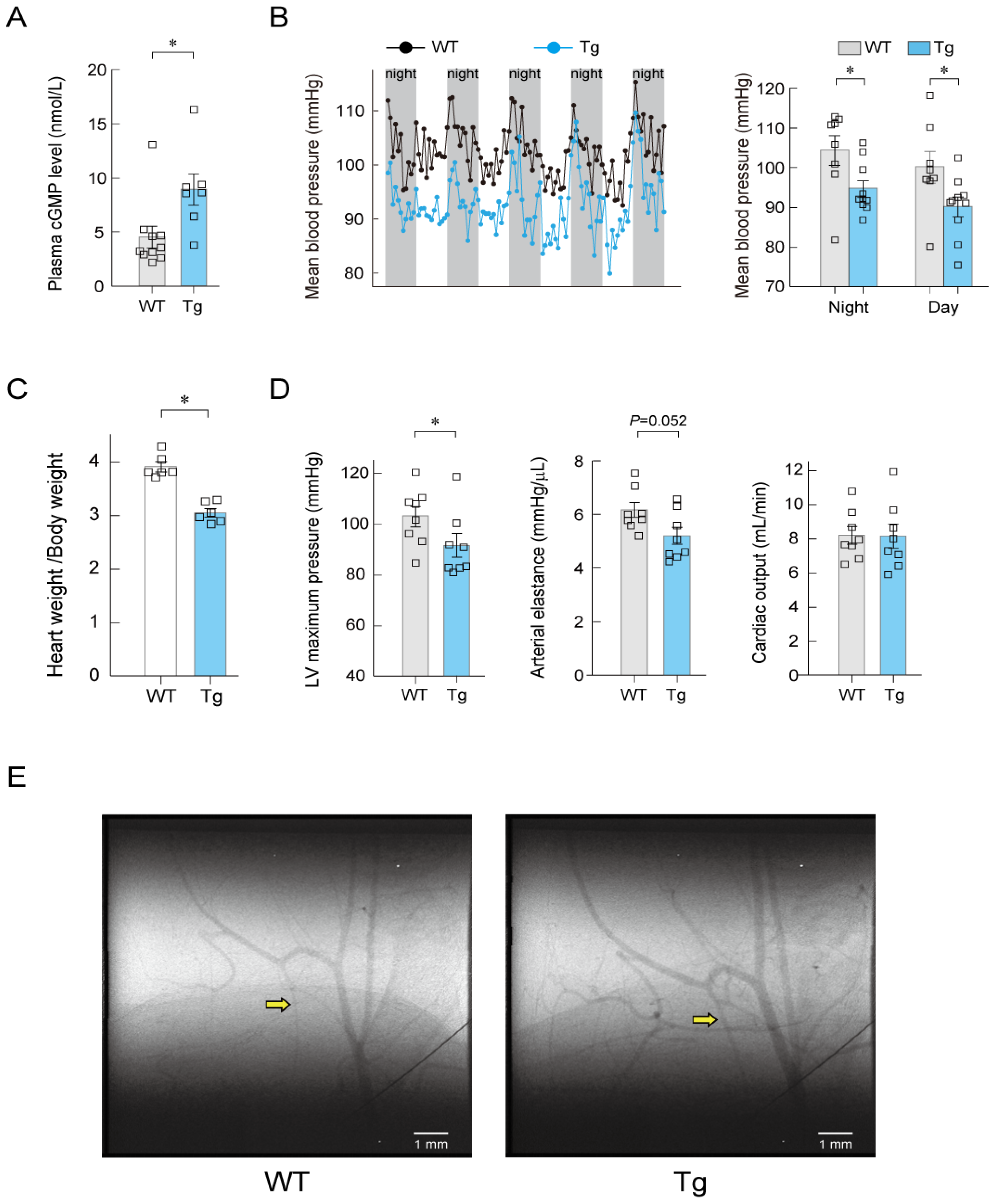
7. Long-Term Regulation of Blood Pressure by ANP
8. Summary and Conclusions
9. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev. 2006, 27, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, I.; Tokudome, T.; Nakao, K.; Kangawa, K. Natriuretic peptide system: An overview of studies using genetically engineered animal models. FEBS J. 2011, 278, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptor. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and brain natriuretic peptides: Hormones secreted from the heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kato, J. Natriuretic peptides and neprilysin inhibition in hypertension and hypertensive organ damage. Peptides 2020, 132, 170352. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Misono, K.S.; Philo, J.S.; Arakawa, T.; Ogata, C.M.; Qiu, Y.; Ogawa, H.; Young, H.S. Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J. 2011, 278, 1818–1829. [Google Scholar] [CrossRef]
- Pandey, K.N. Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genom. 2018, 50, 913–928. [Google Scholar] [CrossRef]
- Horio, T.; Tokudome, T.; Maki, T.; Yoshihara, F.; Suga, S.; Nishikimi, T.; Kojima, M.; Kawano, Y.; Kangawa, K. Gene expression, secretion, and autocrine action of C-type natriuretic peptide in cultured adult rat cardiac fibroblasts. Endocrinology 2003, 144, 2279–2284. [Google Scholar] [CrossRef]
- Soeki, T.; Kishimoto, I.; Okumura, H.; Tokudome, T.; Horio, T.; Mori, K.; Kangawa, K. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2005, 45, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Moyes, A.J.; Khambata, R.S.; Villar, I.; Bubb, K.J.; Baliga, R.S.; Lumsden, N.G.; Xiao, F.; Gane, P.J.; Rebstock, A.S.; Worthington, R.J.; et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J. Clin. Investig. 2014, 124, 4039–4051. [Google Scholar] [CrossRef] [PubMed]
- Moyes, A.J.; Hobbs, A.J. C-type natriuretic peptide: A multifaceted paracrine regulator in the heart and vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef]
- Saito, Y.; Nakao, K.; Nishimura, K.; Sugawara, A.; Okumura, K.; Obata, K.; Sonoda, R.; Ban, T.; Yasue, H.; Imura, H. Clinical application of atrial natriuretic polypeptide in patients with congestive heart failure: Beneficial effects on left ventricular function. Circulation 1987, 76, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Otani, K.; Chiba, A.; Nishimura, H.; Tokudome, T.; Takano-Watanabe, H.; Matsuo, A.; Ishikawa, H.; Shimamoto, K.; Fukui, H.; et al. A new secretory peptide of natriuretic peptide family, osteocrin, suppresses the progression of congestive heart failure after myocardial infarction. Circ. Res. 2018, 122, 742–751. [Google Scholar] [CrossRef]
- Winquist, R.J.; Faison, E.P.; Waldman, S.A.; Schwartz, K.; Murad, F.; Rapoport, R.M. Atrial natriuretic factor elicits an endothelium-independent relaxation and activates particulate guanylate cyclase in vascular smooth muscle. Proc. Natl. Acad. Sci. USA 1984, 81, 7661–7664. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.R. Drugs affecting renal excretory function. In Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th ed.; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill: New York, NY, USA, 2017; pp. 445–470. [Google Scholar]
- Holtwick, R.; Gotthardt, M.; Skryabin, B.; Steinmetz, M.; Potthast, R.; Zetsche, B.; Hammer, R.E.; Herz, J.; Kuhn, M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 2002, 99, 7142–7147. [Google Scholar] [CrossRef]
- Sabrane, K.; Kruse, M.N.; Fabritz, L.; Zetsche, B.; Mitko, D.; Skryabin, B.V.; Zwiener, M.; Baba, H.A.; Yanagisawa, M.; Kuhn, M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J. Clin. Investig. 2005, 115, 1666–1674. [Google Scholar] [CrossRef]
- Campbell, D.J. Long-term neprilysin inhibition-implications for ARNIs. Nat. Rev. Cardiol. 2017, 14, 171–186. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakajima, K.; Shimamori, Y.; Fujimoto, Y. Comparison of the hydrolysis of the three types of natriuretic peptides by human kidney neutral endopeptidase 24.11. Biochem. Mol. Med. 1997, 61, 47–51. [Google Scholar] [CrossRef]
- Jamieson, J.D.; Palade, G.E. Specific granules in atrial muscle cells. J. Cell Biol. 1964, 23, 151–172. [Google Scholar] [CrossRef] [Green Version]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Currie, M.G.; Geller, D.M.; Cole, B.R.; Boylan, J.G.; YuSheng, W.; Holmberg, S.W.; Needleman, P. Bioactive cardiac substances: Potent vasorelaxant activity in mammalian atria. Science 1983, 221, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Kangawa, K.; Matsuo, H. Purification and complete amino acid sequence of alpha-human atrial natriuretic polypeptide (alpha-hANP). Biochem. Biophys. Res. Commun. 1984, 118, 131–139. [Google Scholar] [CrossRef]
- Kangawa, K.; Fukuda, A.; Matsuo, H. Structural identification of β- and γ-human atrial natriuretic polypeptides. Nature 1985, 313, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Flynn, T.G.; de Bold, M.L.; de Bold, A.J. The amino acid sequence of an atrial peptide with potent diuretic and natriuretic properties. Biochem. Biophys. Res. Commun. 1983, 117, 859–865. [Google Scholar] [CrossRef]
- Yan, W.; Wu, F.; Morser, J.; Wu, Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc. Natl. Acad. Sci. USA 2000, 97, 8525–8529. [Google Scholar] [CrossRef]
- Wu, Q.; Xu-Cai, Y.O.; Chen, S.; Wang, W. Corin: New insights into the natriuretic peptide system. Kidney Int. 2009, 75, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Tomita, M.; Yoshimi, H.; Ikeda, M. Specific receptors for atrial natriuretic factor (ANF) in cultured vascular smooth muscle cells of rat aorta. Biochem. Biophys. Res. Commun. 1984, 125, 562–568. [Google Scholar] [CrossRef]
- Chinkers, M.; Garbers, D.L.; Chang, M.S.; Lowe, D.G.; Chin, H.M.; Goeddel, D.V.; Schulz, S. A membrane form of guanylate cyclase is an atrial natriuretic peptide receptor. Nature 1989, 338, 78–83. [Google Scholar] [CrossRef]
- Schulz, S.; Singh, S.; Bellet, R.A.; Singh, G.; Tubb, D.J.; Chin, H.; Garbers, D.L. The primary structure of a plasma membrane guanylate cyclase demonstrates diversity within this new receptor family. Cell 1989, 58, 1155–1162. [Google Scholar] [CrossRef]
- Sudoh, T.; Minamino, N.; Kangawa, K.; Matsuo, H. C-type natriuretic peptide (CNP): A new member of natriuretic peptide family identified in porcine brain. Biochem. Biophys. Res. Commun. 1990, 168, 863–870. [Google Scholar] [CrossRef]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef]
- Asakura, M.; Kitakaze, M. Cardioprotection in the clinical setting-lessons from J-WIND studies. Cardiovasc. Drugs Ther. 2010, 24, 289–295. [Google Scholar] [CrossRef]
- Kostis, J.B.; Packer, M.; Black, H.R.; Schmieder, R.; Henry, D.; Levy, E. Omapatrilat and enalapril in patients with hypertension: The Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am. J. Hypertens. 2004, 17, 103–111. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Tokudome, T.; Otani, K.; Mao, Y.; Jensen, L.J.; Arai, Y.; Miyazaki, T.; Sonobe, T.; Pearson, J.T.; Osaki, T.; Minamino, N.; et al. Endothelial natriuretic peptide receptor 1 play crucial role for acute and chronic blood pressure regulation by atrial natriuretic peptide. Hypertension 2022, 79, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Frees, A.; Assersen, K.B.; Jensen, M.; Hansen, P.B.L.; Vanhoutte, P.M.; Madsen, K.; Federlein, A.; Lund, L.; Toft, A.; Jensen, B.L. Natriuretic peptides relax human intrarenal arteries through natriuretic peptide receptor type-A recapitulated by soluble guanylyl cyclase agonists. Acta Physiol. 2021, 231, e13565. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor: Where are we now? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- de Wit, C.; Wölfle, S.E. EDHF and gap junctions: Important regulators of vascular tone within the microcirculation. Curr. Pharm. Biotechnol. 2007, 8, 11–25. [Google Scholar] [CrossRef]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Köhler, R. Endothelial Ca2+-activated K+ channels in normal and impaired EDHF-dilator responses—Relevance to cardiovascular pathologies and drug discovery. Br. J. Pharmacol. 2009, 157, 509–526. [Google Scholar] [CrossRef] [Green Version]
- Osei-Owusu, P.; Blumer, K.J. Regulator of G Protein Signaling 2: A Versatile Regulator of Vascular Function. Prog. Mol. Biol. Transl. Sci. 2015, 133, 77–92. [Google Scholar] [PubMed]
- Heximer, S.P.; Knutsen, R.H.; Sun, X.; Kaltenbronn, K.M.; Rhee, M.H.; Peng, N.; Oliveira-dos-Santos, A.; Penninger, J.M.; Muslin, A.J.; Steinberg, T.H.; et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Investig. 2003, 111, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Osei-Owusu, P.; Sabharwal, R.; Kaltenbronn, K.M.; Rhee, M.H.; Chapleau, M.W.; Dietrich, H.H.; Blumer, K.J. Regulator of G protein signaling 2 deficiency causes endothelial dysfunction and impaired endothelium-derived hyperpolarizing factor-mediated relaxation by dysregulating Gi/o signaling. J. Biol. Chem. 2012, 287, 12541–12549. [Google Scholar] [CrossRef]
- Lopez, M.J.; Wong, S.K.; Kishimoto, I.; Dubois, S.; Mach, V.; Friesen, J.; Garbers, D.L.; Beuve, A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 1995, 378, 65–68. [Google Scholar] [CrossRef]
- Oliver, P.M.; Fox, J.E.; Kim, R.; Rockman, H.A.; Kim, H.S.; Reddick, R.L.; Pandey, K.N.; Milgram, S.L.; Smithies, O.; Maeda, N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. USA 1997, 94, 14730–14735. [Google Scholar] [CrossRef]
- Ellmers, L.J.; Scott, N.J.; Piuhola, J.; Maeda, N.; Smithies, O.; Frampton, C.M.; Richards, A.M.; Cameron, V.A. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J. Mol. Endocrinol. 2007, 38, 245–257. [Google Scholar] [CrossRef]
- John, S.W.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 1995, 267, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Barbee, R.W.; Perry, B.D.; Ré, R.N.; Murgo, J.P.; Field, L.J. Hemodynamics in transgenic mice with overexpression of atrial natriuretic factor. Circ. Res. 1994, 74, 747–751. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokudome, T.; Otani, K. Molecular Mechanism of Blood Pressure Regulation through the Atrial Natriuretic Peptide. Biology 2022, 11, 1351. https://doi.org/10.3390/biology11091351
Tokudome T, Otani K. Molecular Mechanism of Blood Pressure Regulation through the Atrial Natriuretic Peptide. Biology. 2022; 11(9):1351. https://doi.org/10.3390/biology11091351
Chicago/Turabian StyleTokudome, Takeshi, and Kentaro Otani. 2022. "Molecular Mechanism of Blood Pressure Regulation through the Atrial Natriuretic Peptide" Biology 11, no. 9: 1351. https://doi.org/10.3390/biology11091351