
Putative Protein Discovery from Microalgal Genomes as a Synthetic Biology Protein Library for Heavy Metal Bio-Removal

 , and
, and
Abstract
:Simple Summary
Abstract
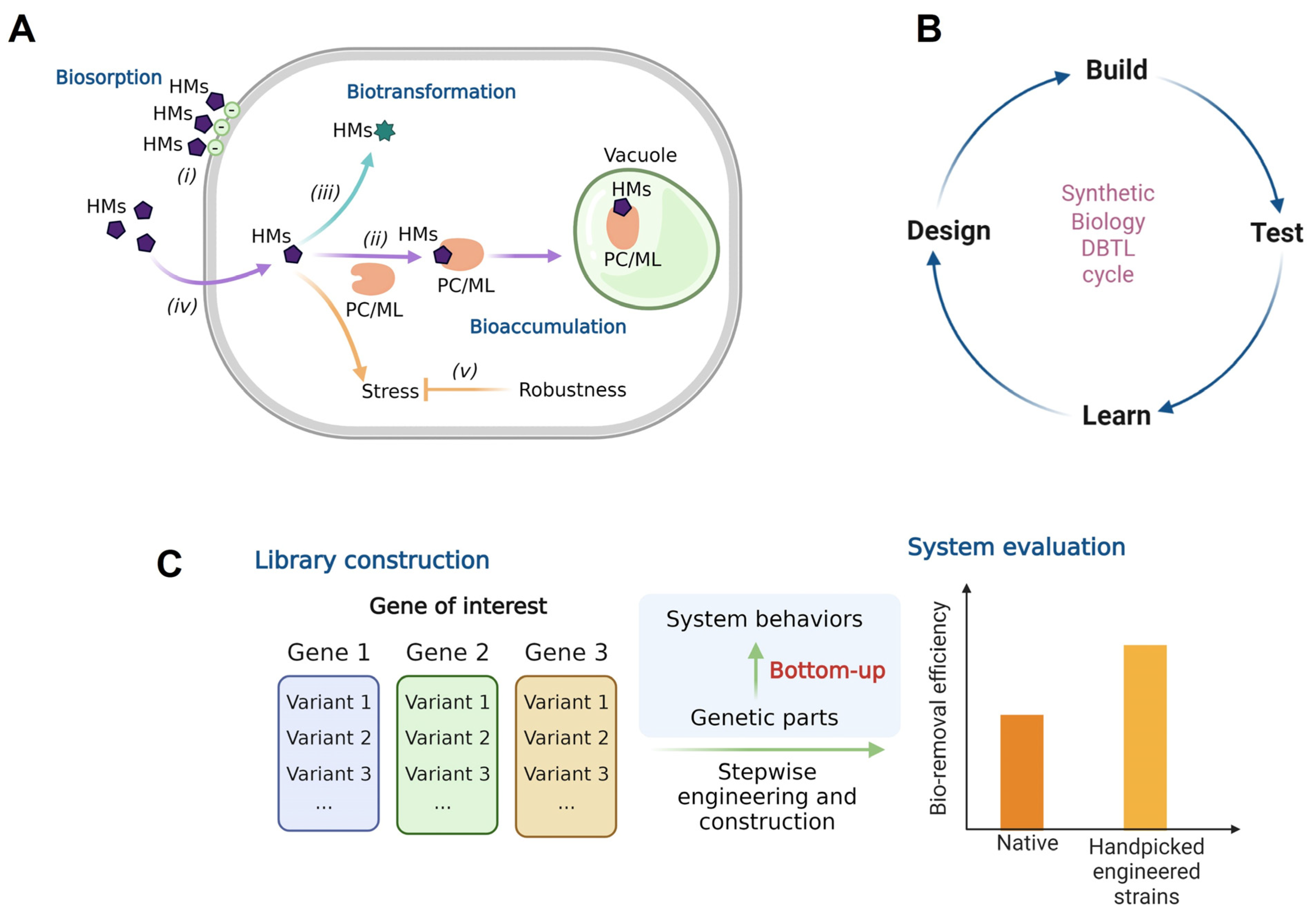
1. Introduction
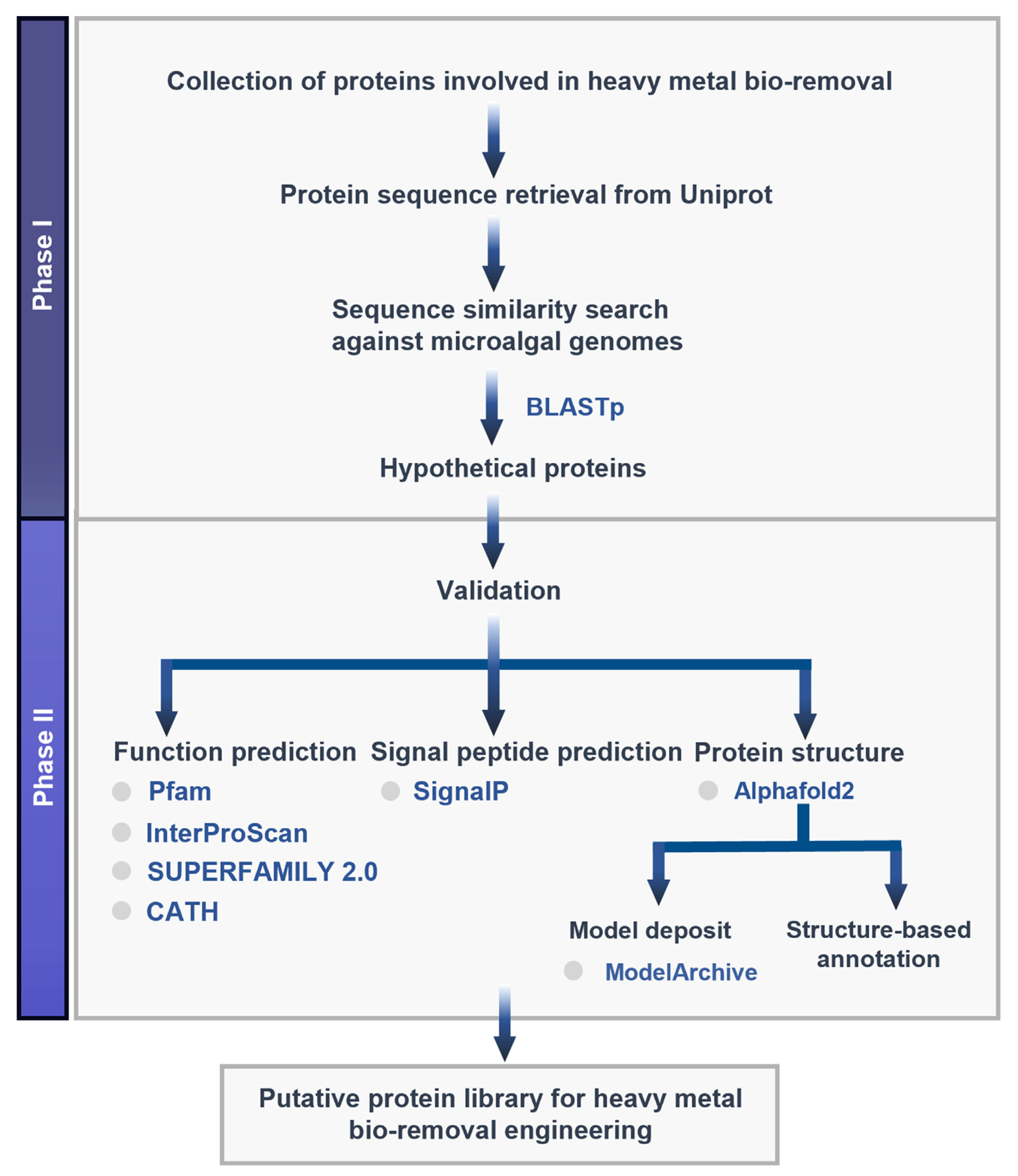
2. Materials and Methods
2.1. Target Protein Identification and Sequence Retrieval
2.2. Sequence Similarity Search
2.3. Function Prediction
2.4. Signal Peptide Prediction
2.5. Structure Modeling
2.6. Structure-Based Functional Annotation
3. Results
3.1. Target Protein Identification
3.2. Sequence Similarity Search
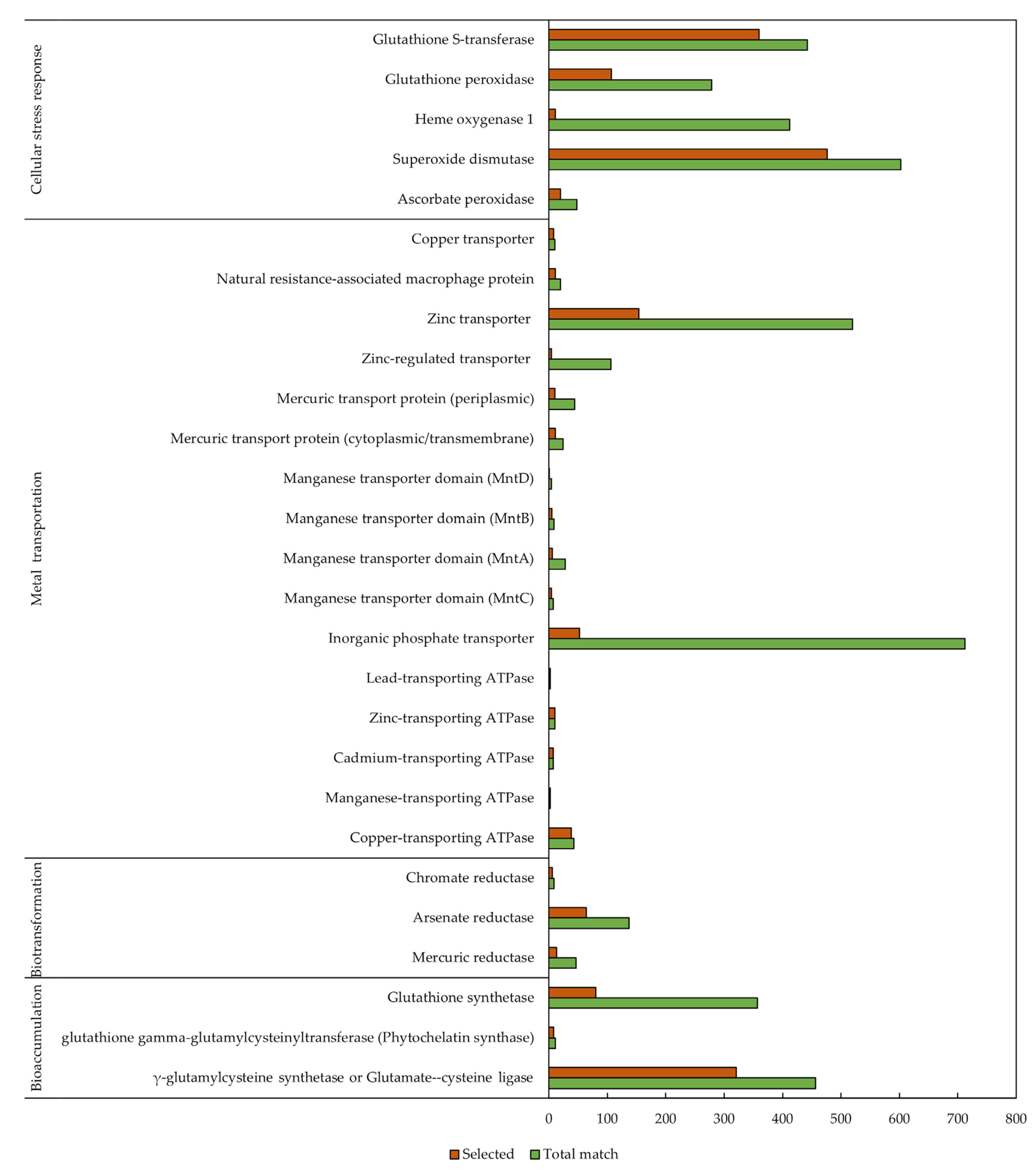
3.3. Protein Function Prediction
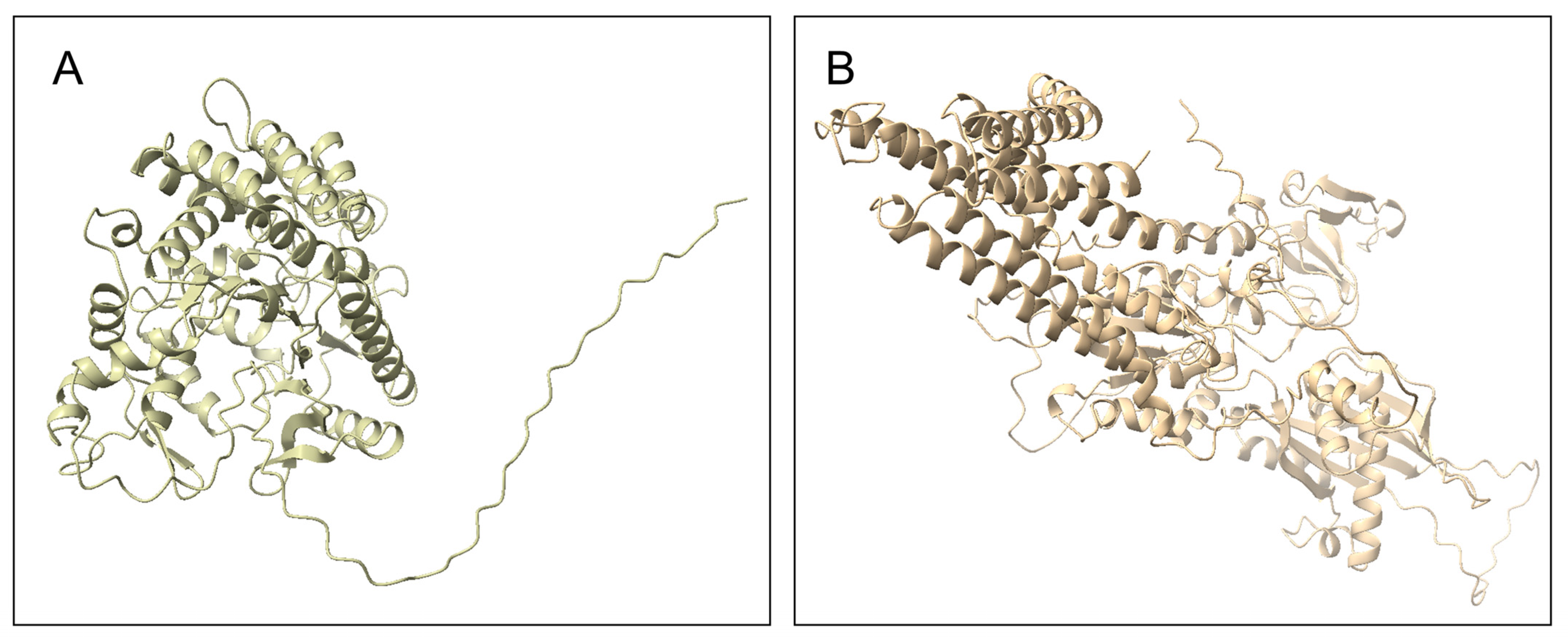
3.4. Homology Modeling
3.5. Structure-Based Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, Mechanism and Health Effects of Some Heavy Metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy Metal Pollution in the Environment and Their Toxicological Effects on Humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Namuhani, N.; Kimumwe, C. Soil Contamination with Heavy Metals around Jinja Steel Rolling Mills in Jinja Municipality, Uganda. J. Health Pollut. 2015, 5, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Huda, N.; Baumber, A.; Hossain, R.; Sahajwalla, V. Waste Battery Disposal and Recycling Behavior: A Study on the Australian Perspective. Environ. Sci. Pollut. Res. 2022. [Google Scholar] [CrossRef]
- Krekeler, M.P.S.; Barrett, H.A.; Davis, R.; Burnette, C.; Doran, T.; Ferraro, A.; Meyer, A. An Investigation of Mass and Brand Diversity in a Spent Battery Recycling Collection with an Emphasis on Spent Alkaline Batteries: Implications for Waste Management and Future Policy Concerns. J. Power Sources 2012, 203, 222–226. [Google Scholar] [CrossRef]
- Suresh Kumar, K.; Dahms, H.U.; Won, E.J.; Lee, J.S.; Shin, K.H. Microalgae—A Promising Tool for Heavy Metal Remediation. Ecotoxicol. Environ. Saf. 2015, 113, 329–352. [Google Scholar] [CrossRef]
- Sattayawat, P.; Yunus, I.S.; Noirungsee, N.; Mukjang, N.; Pathom-Aree, W.; Pekkoh, J.; Pumas, C. Synthetic Biology-Based Approaches for Microalgal Bio-Removal of Heavy Metals from Wastewater Effluents. Front. Environ. Sci. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Wei, Y.Y.; Zheng, Q.; Liu, Z.P.; Yang, Z.M. Regulation of Tolerance of Chlamydomonas Reinhardtii to Heavy Metal Toxicity by Heme Oxygenase-1 and Carbon Monoxide. Plant Cell Physiol. 2011, 52, 1665–1675. [Google Scholar] [CrossRef]
- Siripornadulsil, S.; Traina, S.; Verma, D.P.S.; Sayre, R.T. Molecular Mechanisms of Proline-Mediated Tolerance to Toxic Heavy Metals in Transgenic Microalgae. Plant Cell 2002, 14, 2837–2847. [Google Scholar] [CrossRef]
- Doshi, H.; Ray, A.; Kothari, I.L.; Gami, B. Spectroscopic and Scanning Electron Microscopy Studies of Bioaccumulation of Pollutants by Algae. Curr. Microbiol. 2006, 53, 148–157. [Google Scholar] [CrossRef]
- Peña-Castro, J.M.; Martínez-Jerónimo, F.; Esparza-García, F.; Cañizares-Villanueva, R.O. Heavy Metals Removal by the Microalga Scenedesmus incrassatulus in Continuous Cultures. Bioresour. Technol. 2004, 94, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Terry, P.A.; Stone, W. Biosorption of Cadmium and Copper Contaminated Water by Scenedesmus abundans. Chemosphere 2002, 47, 249–255. [Google Scholar] [CrossRef]
- Monteiro, C.M.; Castro, P.M.L.; Malcata, X.X. Use of the Microalga Scenedesmus obliquus to Remove Cadmium Cations from Aqueous Solutions. World J. Microbiol. Biotechnol. 2009, 25, 1573–1578. [Google Scholar] [CrossRef]
- da Costa, A.C.A.; de França, F.P. The Behaviour of the Microalgae Tetraselmis chuii in Cadmium-Contaminated Solutions. Aquac. Int. 1998, 6, 57–66. [Google Scholar] [CrossRef]
- Shanab, S.; Essa, A.; Shalaby, E. Bioremoval Capacity of Three Heavy Metals by Some Microalgae Species (Egyptian Isolates). Plant Signal. Behav. 2012, 7, 392–399. [Google Scholar] [CrossRef]
- Yan, C.; Qu, Z.; Wang, J.; Cao, L.; Han, Q. Microalgal Bioremediation of Heavy Metal Pollution in Water: Recent Advances, Challenges, and Prospects. Chemosphere 2022, 286, 131870. [Google Scholar] [CrossRef]
- Huang, C.C.; Chen, M.W.; Hsieh, J.L.; Lin, W.H.; Chen, P.C.; Chien, L.F. Expression of Mercuric Reductase from Bacillus megaterium MB1 in Eukaryotic Microalga Chlorella Sp. DT: An Approach for Mercury Phytoremediation. Appl. Microbiol. Biotechnol. 2006, 72, 197–205. [Google Scholar] [CrossRef]
- Ibuot, A.; Webster, R.E.; Williams, L.E.; Pittman, J.K. Increased Metal Tolerance and Bioaccumulation of Zinc and Cadmium in Chlamydomonas reinhardtii Expressing a AtHMA4 C-Terminal Domain Protein. Biotechnol. Bioeng. 2020, 117, 2996–3005. [Google Scholar] [CrossRef]
- Ibuot, A.; Dean, A.P.; McIntosh, O.A.; Pittman, J.K. Metal Bioremediation by CrMTP4 Over-Expressing Chlamydomonas reinhardtii in Comparison to Natural Wastewater-Tolerant Microalgae Strains. Algal Res. 2017, 24, 89–96. [Google Scholar] [CrossRef]
- Han, S.H.; Hu, Z.L.; Lei, A.P. Expression and Function Analysis of the Metallothionein-like (MT-like) Gene from Festuca rubra in Chlamydomonas reinhardtii Chloroplast. Sci. China C Life Sci. 2008, 51, 1076–1081. [Google Scholar] [CrossRef]
- Kader, M.A.; Ahammed, A.; Khan, M.S.; al Ashik, S.A.; Islam, M.S.; Hossain, M.U. Hypothetical Protein Predicted to Be Tumor Suppressor: A Protein Functional Analysis. Genom. Inform. 2022, 20, e6. [Google Scholar] [CrossRef] [PubMed]
- Shahbaaz, M.; Hassan, M.I.; Ahmad, F. Functional Annotation of Conserved Hypothetical Proteins from Haemophilus influenzae Rd KW20. PLoS ONE 2013, 8, e84263. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Krogh, A. Prediction of Signal Peptides and Signal Anchors by a Hidden Markov Model SignalP View Project. In Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology, Montreal, QC, Canada, 28 June–1 July 1998; AAAI Press: Palo Alto, CA, USA; Volume 6, pp. 122–130. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kootery, K.P.; Sarojini, S. Structural and Functional Characterization of a Hypothetical Protein in the RD7 Region in Clinical Isolates of Mycobacterium tuberculosis—An in Silico Approach to Candidate Vaccines. J. Genet. Eng. Biotechnol. 2022, 20, 55. [Google Scholar] [CrossRef]
- QIAGEN. CLC Genomics Workbench User Manual. Available online: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/801/User_Manual.pdf (accessed on 2 August 2022).
- Mubashar, M.; Naveed, M.; Mustafa, A.; Ashraf, S.; Baig, K.S.; Alamri, S.; Siddiqui, M.H.; Zabochnicka-światek, M.; Szota, M.; Kalaji, H.M. Experimental Investigation of Chlorella Vulgaris and Enterobacter Sp. Mn17 for Decolorization and Removal of Heavy Metals from Textile Wastewater. Water 2020, 12, 3034. [Google Scholar] [CrossRef]
- Shivaji, S.; Dronamaraju, S.V.L. Scenedesnus Rotundus Isolated from the Petroleum Effluent Employs Alternate Mechanisms of Tolerance to Elevated Levels of Cadmium and Zinc. Sci. Rep. 2019, 9, 8485. [Google Scholar] [CrossRef]
- Da Rosa Tavares, B.A.; Paes, J.A.; Zaha, A.; Ferreira, H.B. Reannotation of Mycoplasma Hyopneumoniae Hypothetical Proteins Revealed Novel Potential Virulence Factors. Microb. Pathog. 2022, 162, 105344. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A Server for Predicting Protein Function from 3D Structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef]
- Balzano, S.; Sardo, A.; Blasio, M.; Chahine, T.B.; Dell’Anno, F.; Sansone, C.; Brunet, C. Microalgal Metallothioneins and Phytochelatins and Their Potential Use in Bioremediation. Front. Microbiol. 2020, 11, 517. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Z.; Luo, L.; Zhao, X.; Lu, Z.; Luo, T.; Li, M.; Zhang, Y. An Aldo-Keto Reductase, Bbakr1, Is Involved in Stress Response and Detoxification of Heavy Metal Chromium but Not Required for Virulence in the Insect Fungal Pathogen Beauveria bassiana. Fungal Genet. Biol. 2018, 111, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, V. Mercury Detoxification by Absorption, Mercuric Ion Reductase, and Exopolysaccharides: A Comprehensive Study. Environ. Sci. Pollut. Res. 2020, 27, 27181–27201. [Google Scholar] [CrossRef] [PubMed]
- Danouche, M.; el Ghachtouli, N.; el Arroussi, H. Phycoremediation Mechanisms of Heavy Metals Using Living Green Microalgae: Physicochemical and Molecular Approaches for Enhancing Selectivity and Removal Capacity. Heliyon 2021, 7, e07609. [Google Scholar] [CrossRef]
- Blaby-Haas, C.E.; Merchant, S.S. The Ins and Outs of Algal Metal Transport. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 1531–1552. [Google Scholar] [CrossRef]
- Danouche, M.; el Ghatchouli, N.; Arroussi, H. Overview of the Management of Heavy Metals Toxicity by Microalgae. J. Appl. Phycol. 2022, 34, 475–488. [Google Scholar] [CrossRef]
- Kotrba, P.; Ruml, T. Surface Display of Metal Fixation Motifs of Bacterial Pi-Type ATPases Specifically Promotes Biosorption of Pb2+ by Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2010, 76, 2615–2622. [Google Scholar] [CrossRef]
- Ma, X.; Chen, Y.; Liu, F.; Zhang, S.; Wei, Q. Enhanced Tolerance and Resistance Characteristics of Scenedesmus obliquus FACHB-12 with K3 Carrier in Cadmium Polluted Water. Algal Res. 2021, 55, 102267. [Google Scholar] [CrossRef]
- Bøttger, P.; Pedersen, L. Evolutionary and Experimental Analyses of Inorganic Phosphate Transporter PiT Family Reveals Two Related Signature Sequences Harboring Highly Conserved Aspartic Acids Critical for Sodium-Dependent Phosphate Transport Function of Human PiT2. FEBS J. 2005, 272, 3060–3074. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, A.P.; Stahlhacke, J.; Oates, M.E.; Smithers, B.; Gough, J. The SUPERFAMILY 2.0 Database: A Significant Proteome Update and a New Webserver. Nucleic Acids Res. 2019, 47, D490–D494. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sitsel, O.; Meloni, G.; Autzen, H.E.; Andersson, M.; Klymchuk, T.; Nielsen, A.M.; Rees, D.C.; Nissen, P.; Gourdon, P. Structure and Mechanism of Zn2+-Transporting P-Type ATPases. Nature 2014, 514, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Garcion, C.; Béven, L.; Foissac, X. Comparison of Current Methods for Signal Peptide Prediction in Phytoplasmas. Front. Microbiol. 2021, 12, 661524. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T. Protein Modeling: What Happened to the “Protein Structure Gap”? Structure 2013, 21, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Mochida, K.; Suzuki, K. Diverse Biosynthetic Pathways and Protective Functions against Environmental Stress of Antioxidants in Microalgae. Plants 2021, 10, 1250. [Google Scholar] [CrossRef]
- Shi, L.D.; Chen, Y.S.; Du, J.J.; Hu, Y.Q.; Shapleigh, J.P.; Zhao, H.P. Metagenomic Evidence for a Methylocystis Species Capable of Bioremediation of Diverse Heavy Metals. Front. Microbiol. 2019, 10, 3297. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Zhang, X.; Xu, H.; Lescop, E.; Xia, B.; Jin, C. Conformational Fluctuations Coupled to the Thiol-Disulfide Transfer between Thioredoxin and Arsenate Reductase in Bacillus subtilis. J. Biol. Chem. 2007, 282, 11078–11083. [Google Scholar] [CrossRef]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Preben Morth, J.; Palmgren, M.G.; Nissen, P. Crystal Structure of the Plasma Membrane Proton Pump. Nature 2007, 450, 1111–1114. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biology, Structure and Mechanism of P-Type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Migocka, M. Copper-Transporting ATPases: The Evolutionarily Conserved Machineries for Balancing Copper in Living Systems. IUBMB Life 2015, 67, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Norambuena, L.; Maturana, D.; Toro, M.; Vergara, C.; Orellana, A.; Zurita-Silva, A.; Ordenes, V.R. AtHMA1 Is a Thapsigargin-Sensitive Ca2+/Heavy Metal Pump. J. Biol. Chem. 2008, 283, 9633–9641. [Google Scholar] [CrossRef] [PubMed]
- Sael, L.; Chitale, M.; Kihara, D. Structure- and Sequence-Based Function Prediction for Non-Homologous Proteins. J. Struct. Funct. Genom. 2012, 13, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Meharenna, Y.T.; Oertel, P.; Bhaskar, B.; Poulos, T.L. Engineering Ascorbate Peroxidase Activity into Cytochrome c Peroxidase. Biochemistry 2008, 47, 10324–10332. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Rahman, I.A.; Islam, T.; Ghosh, A. Genome-Wide Identification and Expression Analysis of Glutathione S-Transferase Gene Family in Tomato: Gaining an Insight to Their Physiological and Stress-Specific Roles. PLoS ONE 2017, 12, e0187504. [Google Scholar] [CrossRef]
- Naseri, G.; Koffas, M.A.G. Application of Combinatorial Optimization Strategies in Synthetic Biology. Nat. Commun. 2020, 11, 2446. [Google Scholar] [CrossRef]
- Li, N.; Qin, L.; Jin, M.; Zhang, L.; Geng, W.; Xiao, X. Extracellular Adsorption, Intracellular Accumulation and Tolerance Mechanisms of Cyclotella Sp. to Cr(VI) Stress. Chemosphere 2021, 270, 128662. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microalga | Reported Mechanism | Reference |
|---|---|---|
| Chlorella (taxid 3071) | Biosorption, bioaccumulation, biotransformation | [27] |
| Scenedesmus (taxid 3087) | Bioaccumulation, biotransformation, cellular stress response | [28] |
| Protein | Microalgal Genome | Hypothetical Protein | Hypothetical Protein after Primary Function Prediction |
|---|---|---|---|
| Bioaccumulation | |||
| γ-Glutamylcysteine synthetase or Glutamate—cysteine ligase | Chlorella | 4 | 2 |
| Scenedesmus | 1 | 0 | |
| Phytochelatin synthase | Chlorella | 2 | 2 |
| Scenedesmus | 0 | 0 | |
| Glutathione synthetase | Chlorella | 2 | 2 |
| Scenedesmus | 0 | 0 | |
| Inorganic phosphate transporter | Chlorella | 10 | 7 |
| Scenedesmus | 0 | 0 | |
| Biotransformation | |||
| Mercuric reductase | Chlorella | 10 | 0 |
| Scenedesmus | 5 | 0 | |
| Arsenate reductase | Chlorella | 6 | 0 |
| Scenedesmus | 1 | 0 | |
| Chromate reductase | Chlorella | 0 | 0 |
| Scenedesmus | 0 | 0 | |
| Metal transportation | |||
| Copper-transporting ATPase | Chlorella | 26 | 9 |
| Scenedesmus | 8 | 2 | |
| Manganese-transporting ATPase | Chlorella | 9 | 0 |
| Scenedesmus | 1 | 0 | |
| Cadmium-transporting ATPase | Chlorella | 22 | 0 |
| Scenedesmus | 4 | 0 | |
| Zinc-transporting ATPase | Chlorella | 25 | 0 |
| Scenedesmus | 6 | 0 | |
| Lead-transporting ATPase | Chlorella | 11 | 0 |
| Scenedesmus | 1 | 0 | |
| Manganese transporter domain (MntA) | Chlorella | 43 | 1 |
| Scenedesmus | 10 | 1 | |
| Manganese transporter domain (MntB) | Chlorella | 27 | 1 |
| Scenedesmus | 6 | 1 | |
| Manganese transporter domain (MntC) | Chlorella | 0 | 0 |
| Scenedesmus | 0 | 0 | |
| Manganese transporter domain (MntD) | Chlorella Scenedesmus | 0 0 | 0 0 |
| Mercuric transport protein (cytoplasmic/transmembrane) | Scenedesmus | 0 | 0 |
| Scenedesmus | 0 | 0 | |
| Mercuric transport protein (periplasmic) | Chlorella | 0 | 0 |
| Scenedesmus | 0 | 0 | |
| Zinc-regulated transporter (ZRT) | Chlorella | 5 | 1 |
| Scenedesmus | 1 | 0 | |
| Zinc transporter (ZIP) | Chlorella | 13 | 1 |
| Scenedesmus | 4 | 1 | |
| Natural resistance-associated macrophage protein | Chlorella | 2 | 2 |
| Scenedesmus | 0 | 0 | |
| Copper transporter | Chlorella | 0 | 0 |
| Scenedesmus | 0 | 0 | |
| Cellular tolerance | |||
| Ascorbate peroxidase | Chlorella | 9 | 8 |
| Scenedesmus | 0 | 0 | |
| Superoxide dismutase | Chlorella | 10 | 9 |
| Scenedesmus | 4 | 4 | |
| Heme oxygenase | Chlorella | 5 | 4 |
| Scenedesmus | 2 | 2 | |
| Glutathione peroxidase | Chlorella | 15 | 10 |
| Scenedesmus | 0 | 0 | |
| Glutathione S-transferase | Chlorella | 40 | 20 |
| Scenedesmus | 11 | 2 | |
| Genome | Accession No. | Signal Peptide * | Putative Function |
|---|---|---|---|
| Bioaccumulation | |||
| Chlorella desiccata (nom. nud.) | KAG7671258.1 | N | Glutamate-cysteine ligase |
| Chlorella variabilis | XP_005844806.1 | N | Glutamate-cysteine ligase |
| Chlorella desiccata (nom. nud.) | KAG7668718.1 | N | Phytochelatin synthase |
| Chlorella variabilis | XP_005845668.1 | N | Phytochelatin synthase |
| Chlorella desiccata (nom. nud.) | KAG7673317.1 | N | Glutathione synthetase |
| Chlorella variabilis | XP_005847003.1 | N | Glutathione synthetase |
| Metal transportation | |||
| Chlorella variabilis | XP_005845243.1 | Y | Heavy metal transporting ATPase |
| Chlorella variabilis | XP_005851032.1 | Y | Heavy metal transporting ATPase |
| Scenedesmus sp. NREL 46B-D10 | KAF6264708.1 | Y | Manganese transporter domain (MntA) ** |
| Chlorella desiccata (nom. nud.) | KAG7670010.1 | Y | Manganese transporter domain (MntB) |
| Chlorella variabilis | XP_005845281.1 | Y | Manganese transporter domain (MntB) ** |
| Chlorella variabilis | XP_005844148.1 | Y | Zinc-regulated transporter (ZRT)/Zinc transporter (ZIP) ** |
| Chlorella variabilis | XP_005846850.1 | Y | Zinc-regulated transporter (ZRT)/Zinc transporter (ZIP) ** |
| Chlorella desiccata (nom. nud.) | KAG7667456.1 | Y | Zinc-regulated transporter (ZRT)/Zinc transporter (ZIP) |
| Chlorella desiccata (nom. nud.) | KAG7675010.1 | N | Natural resistance-associated macrophage protein |
| Chlorella variabilis | XP_005847346.1 | N | Natural resistance-associated macrophage protein |
| Cellular tolerance | |||
| Chlorella variabilis | XP_005842918.1 | N | Ascorbate peroxidase |
| Chlorella variabilis | XP_005847371.1 | N | Ascorbate peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7671272.1 | N | Ascorbate peroxidase ** |
| Chlorella desiccata (nom. nud.) | KAG7672626.1 | N | Ascorbate peroxidase |
| Chlorella variabilis | XP_005842951.1 | N | Ascorbate peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7671850.1 | N | Ascorbate peroxidase |
| Chlorella variabilis | XP_005851196.1 | N | Ascorbate peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7671273.1 | N | Ascorbate peroxidase |
| Chlorella variabilis | XP_005852313.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005852314.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005850331.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005850533.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005850825.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005851580.1 | N | Superoxide dismutase |
| Chlorella desiccata (nom. nud.) | KAG7672127.1 | N | Superoxide dismutase |
| Chlorella desiccata (nom. nud.) | KAG7672915.1 | N | Superoxide dismutase |
| Chlorella desiccata (nom. nud.) | KAG7673432.1 | N | Superoxide dismutase |
| Scenedesmus sp. NREL 46B-D3 | KAF6253844.1 | N | Superoxide dismutase |
| Scenedesmus sp. PABB004 | KAF8054759.1 | N | Superoxide dismutase |
| Scenedesmus sp. PABB004 | KAF8070899.1 | N | Superoxide dismutase |
| Scenedesmus sp. PABB004 | KAF8072345.1 | N | Superoxide dismutase |
| Chlorella variabilis | XP_005851913.1 | N | Heme oxygenase 1 |
| Chlorella desiccata (nom. nud.) | KAG7671693.1 | N | Heme oxygenase 1 |
| Chlorella variabilis | XP_005845884.1 | N | Heme oxygenase 1 |
| Chlorella variabilis | XP_005842792.1 | N | Heme oxygenase 1 |
| Scenedesmus sp. NREL 46B-D3 | KAF6256065.1 | N | Heme oxygenase 1 |
| Scenedesmus sp. PABB004 | KAF8061310.1 | N | Heme oxygenase 1 |
| Chlorella variabilis | XP_005852198.1 | N | Glutathione peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7666639.1 | N | Glutathione peroxidase |
| Chlorella variabilis | XP_005847444.1 | N | Glutathione peroxidase |
| Chlorella variabilis | XP_005848232.1 | N | Glutathione peroxidase |
| Chlorella variabilis | XP_005851691.1 | N | Glutathione peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7666823.1 | N | Glutathione peroxidase |
| Chlorella variabilis | XP_005850288.1 | N | Glutathione peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7675006.1 | N | Glutathione peroxidase |
| Chlorella variabilis | XP_005844151.1 | N | Glutathione peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7671063.1 | N | Glutathione peroxidase |
| Chlorella desiccata (nom. nud.) | KAG7667083.1 | N | Glutathione S-transferase ** |
| Chlorella desiccata (nom. nud.) | KAG7667402.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7667544.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7667774.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7667817.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7669352.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7669598.1 | N | Glutathione S-transferase ** |
| Chlorella desiccata (nom. nud.) | KAG7670514.1 | N | Glutathione S-transferase |
| Chlorella desiccata (nom. nud.) | KAG7675170.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005843180.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005845006.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005845127.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005845396.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005847002.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005848700.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005849485.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005849684.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005850654.1 | N | Glutathione S-transferase |
| Chlorella variabilis | XP_005852104.1 | N | Glutathione S-transferase |
| Scenedesmus sp. NREL 46B-D3 | KAF6265595.1 | N | Glutathione S-transferase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uttarotai, T.; Mukjang, N.; Chaisoung, N.; Pathom-Aree, W.; Pekkoh, J.; Pumas, C.; Sattayawat, P. Putative Protein Discovery from Microalgal Genomes as a Synthetic Biology Protein Library for Heavy Metal Bio-Removal. Biology 2022, 11, 1226. https://doi.org/10.3390/biology11081226
Uttarotai T, Mukjang N, Chaisoung N, Pathom-Aree W, Pekkoh J, Pumas C, Sattayawat P. Putative Protein Discovery from Microalgal Genomes as a Synthetic Biology Protein Library for Heavy Metal Bio-Removal. Biology. 2022; 11(8):1226. https://doi.org/10.3390/biology11081226
Chicago/Turabian StyleUttarotai, Toungporn, Nilita Mukjang, Natcha Chaisoung, Wasu Pathom-Aree, Jeeraporn Pekkoh, Chayakorn Pumas, and Pachara Sattayawat. 2022. "Putative Protein Discovery from Microalgal Genomes as a Synthetic Biology Protein Library for Heavy Metal Bio-Removal" Biology 11, no. 8: 1226. https://doi.org/10.3390/biology11081226