
The Effects of Tamoxifen on Tolerogenic Cells in Cancer


 ,
,  , , ,
, , ,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
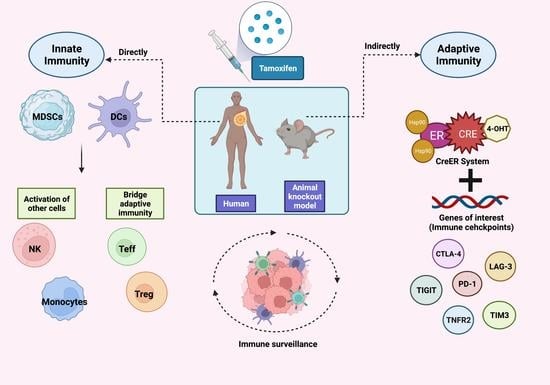
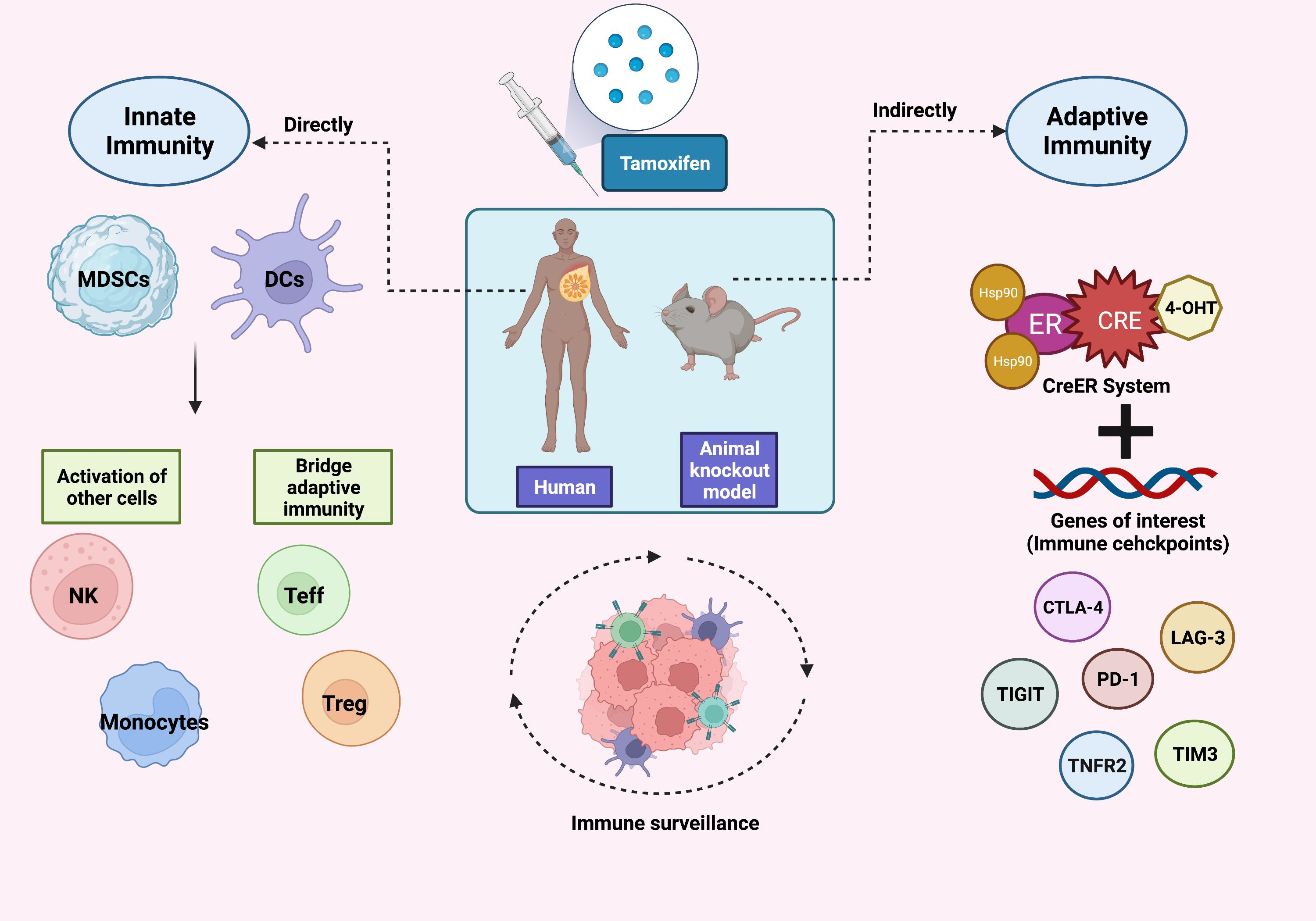
1. Introduction
2. Immune Tolerance and Cancer
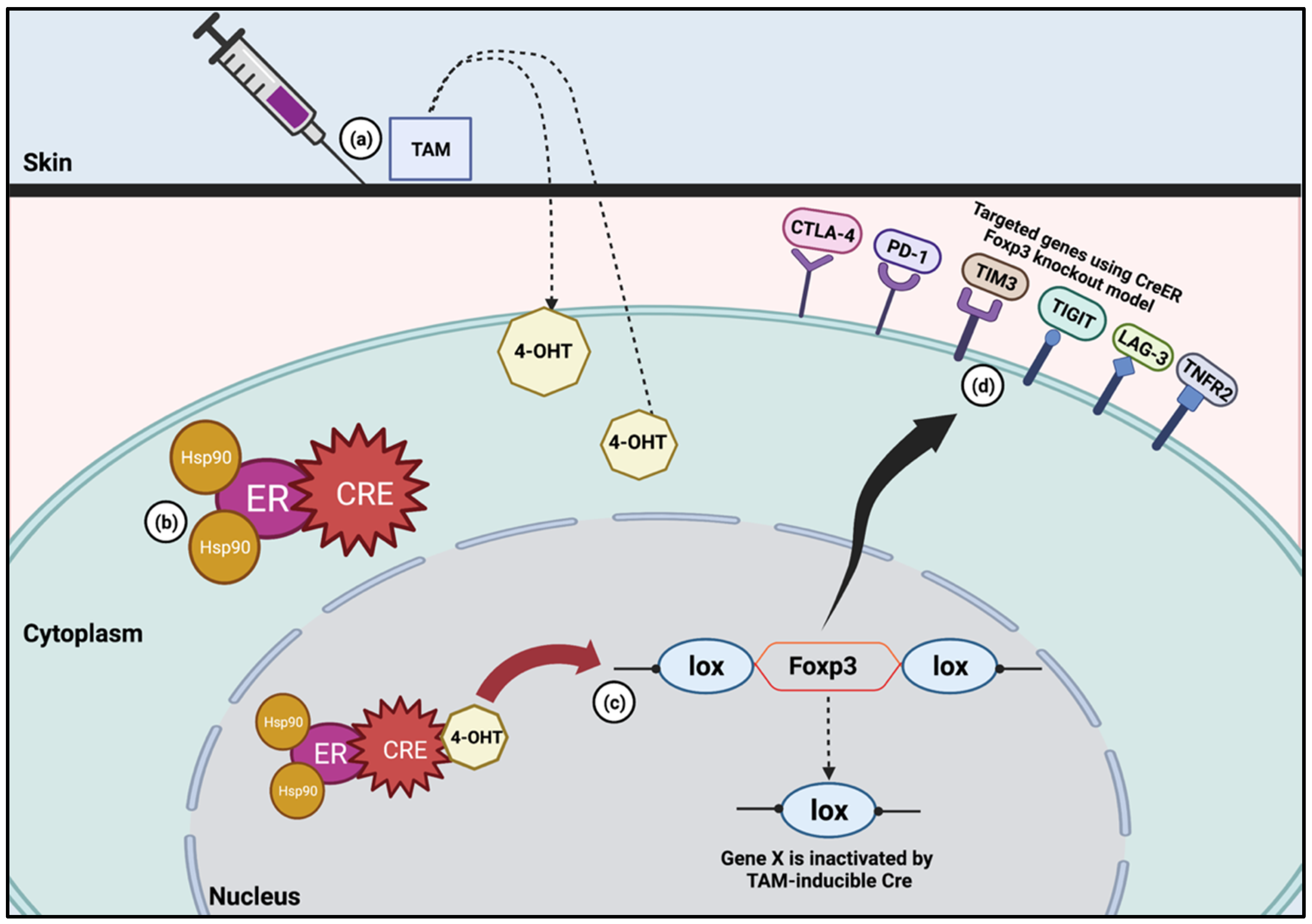
3. Utilization of TAM for Treg Regulation
3.1. Foxp3
3.2. CTLA-4
3.3. PD-1
3.4. Other Immune Checkpoints
- LAG-3
- TIM3
- TIGIT
- TNFR2
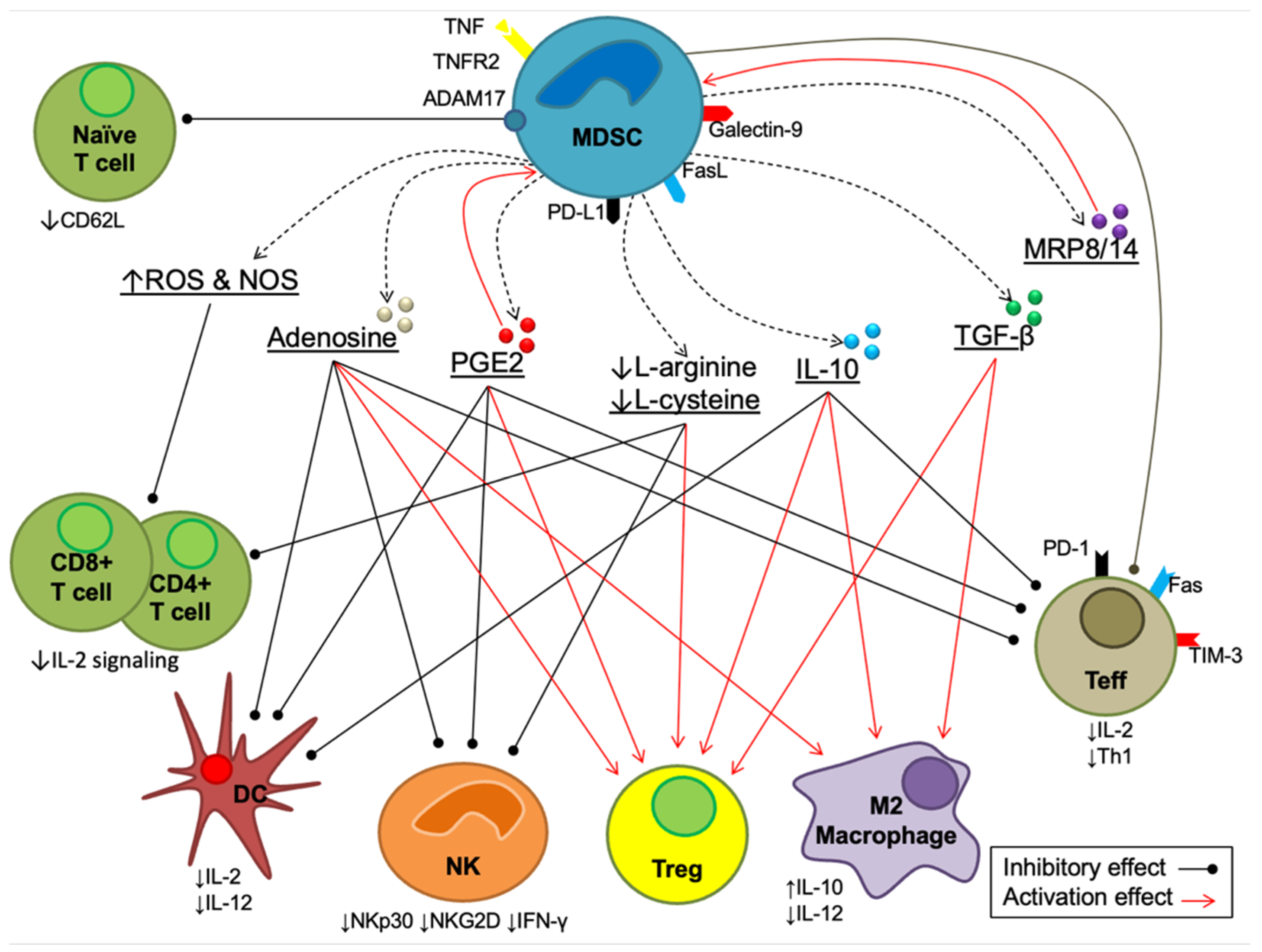
4. Myeloid-Derived Suppressor Cells (MDSCs)
Effects of Tamoxifen on MDSCs
5. Dendritic Cells (DCs)
Effects of Tamoxifen on Dendritic Cells
6. Future Directions for TAM in Investigating Tolerogenic Cells
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jordan, V.C. Tamoxifen (ICI46, 474) as a targeted therapy to treat and prevent breast cancer. Br. J. Pharmacol. 2006, 147, S269–S276. [Google Scholar] [CrossRef]
- Cole, M.P.; Jones, C.T.; Todd, I.D.H. A New Anti-oestrogenic Agent in Late Breast Cancer: An Early Clinical Appraisal of ICI46474. Br. J. Cancer 1971, 25, 270–275. [Google Scholar] [CrossRef]
- Peto, R. Effects of Adjuvant Tamoxifen and of Cytotoxic Therapy on Mortality in Early Breast Cancer. An Overview of 61 Randomised Trials among 28,896 Women. Horm. Res. 1989, 32, 165. [Google Scholar] [CrossRef]
- Ide, V.; Vanderschueren, D.; Antonio, L. Treatment of Men with Central Hypogonadism: Alternatives for Testosterone Replacement Therapy. Int. J. Mol. Sci. 2020, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Breastcancer.org. Tamoxifen; Tamoxifen (Brand Names: Nolvadex, Soltamox) is a Selective Estrogen. 2022. Available online: https://www.breastcancer.org/treatment/hormonal-therapy/tamoxifen (accessed on 5 April 2022).
- Hird, R.B.; Chang, A.; Cimmino, V.; Diehl, K.; Sabel, M.; Kleer, C.; Helvie, M.; Schott, A.; Young, J.; Hayes, D.; et al. Impact of estrogen receptor expression and other clinicopathologic features on tamoxifen use in ductal carcinoma in situ. Cancer 2006, 106, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Research, N.C. Immune Checkpoint Inhibitor. 2020. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/immune-checkpoint-inhibitor (accessed on 11 August 2022).
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.-J. Strategies for subtypes—Dealing with the diversity of breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Yu, F.; Bender, W. The mechanism of tamoxifen in breast cancer prevention. In Breast Cancer Research; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Osborne, C.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Behjati, S.; Frank, M. The Effects of Tamoxifen on Immunity. Curr. Med. Chem. 2009, 16, 3076–3080. [Google Scholar] [CrossRef]
- Corriden, R.; Hollands, A.; Olson, J.; Derieux, J.; Lopez, J.; Chang, J.T.; Gonzalez, D.J.; Nizet, V. Tamoxifen augments the innate immune function of neutrophils through modulation of intracellular ceramide. Nat. Commun. 2015, 6, 8369. [Google Scholar] [CrossRef]
- Huang, H.; Zhou, J.; Chen, H.; Li, J.; Zhang, C.; Jiang, X.; Ni, C. The immunomodulatory effects of endocrine therapy in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 19. [Google Scholar] [CrossRef]
- Lv, T.; Zhang, Z.; Yu, H.; Ren, S.; Wang, J.; Li, S.; Sun, L. Tamoxifen Exerts Anticancer Effects on Pituitary Adenoma Progression via Inducing Cell Apoptosis and Inhibiting Cell Migration. Int. J. Mol. Sci. 2022, 23, 2664. [Google Scholar] [CrossRef]
- Wüst, R.C.; Houtkooper, R.H.; Auwerx, J. Confounding factors from inducible systems for spatiotemporal gene expression regulation. J. Cell Biol. 2020, 219, e202003031. [Google Scholar] [CrossRef]
- Markowitz, J.; Wesolowski, R.; Papenfuss, T.; Brooks, T.R.; Carson, W.E., III. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res. Treat. 2013, 140, 13–21. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Nalbandian, G.; Paharkova-Vatchkova, V.; Mao, A.; Nale, S.; Kovats, S. The Selective Estrogen Receptor Modulators, Tamoxifen and Raloxifene, Impair Dendritic Cell Differentiation and Activation. J. Immunol. 2005, 175, 2666–2675. [Google Scholar] [CrossRef]
- David Male, J.B.; Roth, D.B.; Roitt, I.M. Immunology, 8th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2013. [Google Scholar]
- Waldmann, H. Mechanisms of immunological tolerance. Clin. Biochem. 2016, 49, 324–328. [Google Scholar] [CrossRef]
- Mackay, I.R. Tolerance and autoimmunity. BMJ 2000, 321, 93–96. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Basic Immunology: Functions and Disorders of Immune System, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Lee, W.; Lee, G.R. Transcriptional regulation and development of regulatory T cells. Exp. Mol. Med. 2018, 50, e456. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge Cancer Immunotherapy and Breaking Immune Tolerance. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef]
- Liu, R.; Liu, C.; Chen, D.; Yang, W.-H.; Liu, X.; Liu, C.-G.; Dugas, C.M.; Tang, F.; Zheng, P.; Liu, Y.; et al. FOXP3 Controls an miR-146/NF-κB Negative Feedback Loop That Inhibits Apoptosis in Breast Cancer CellsFOXP3–miRNA-146–NF-κB Axis Controls Tumor Growth. Cancer Res. 2015, 75, 1703–1713. [Google Scholar] [CrossRef]
- Marangoni, F.; Zhakyp, A.; Corsini, M.; Geels, S.N.; Carrizosa, E.; Thelen, M.; Mani, V.; Prüßmann, J.N.; Warner, R.D.; Ozga, A.J.; et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell 2021, 184, 3998–4015.e19. [Google Scholar] [CrossRef]
- Smigiel, K.S.; Srivastava, S.; Stolley, J.M.; Campbell, D.J. Regulatory T-cell homeostasis: Steady-state maintenance and modulation during inflammation. Immunol. Rev. 2014, 259, 40–59. [Google Scholar] [CrossRef]
- Shi, H.; Chi, H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front. Immunol. 2019, 10, 2716. [Google Scholar] [CrossRef]
- Fridman, W.H.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Hatzioannou, A.; Boumpas, A.; Papadopoulou, M.; Papafragkos, I.; Varveri, A.; Alissafi, T.; Verginis, P. Regulatory T Cells in Autoimmunity and Cancer: A Duplicitous Lifestyle. Front. Immunol. 2021, 12, 731947. [Google Scholar] [CrossRef]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- Walker, M.R.; Kasprowicz, D.J.; Gersuk, V.H.; Bénard, A.; Van Landeghen, M.; Buckner, J.H.; Ziegler, S.F. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25– T cells. J. Clin. Investig. 2003, 112, 1437–1443. [Google Scholar] [CrossRef]
- Ziegler, S.F. FOXP3: Of mice and men. Annu. Rev. Immunol. 2006, 24, 209–226. [Google Scholar] [CrossRef]
- Gavin, M.A.; Torgerson, T.R.; Houston, E.; Deroos, P.; Ho, W.Y.; Stray-Pedersen, A.; Ocheltree, E.L.; Greenberg, P.D.; Ochs, H.D.; Rudensky, A.Y. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. USA 2006, 103, 6659–6664. [Google Scholar] [CrossRef]
- Rudensky, A.Y. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef]
- Zheng, Y.; Rudensky, A.Y. Foxp3 in control of the regulatory T cell lineage. Nat. Immunol. 2007, 8, 457–462. [Google Scholar] [CrossRef]
- Szylberg, Ł.; Karbownik, D.; Marszałek, A. The Role of FOXP3 in Human Cancers. Anticancer Res. 2016, 36, 3789–3794. [Google Scholar]
- Douglass, S.; Meeson, A.P.; Overbeck-Zubrzycka, D.; Brain, J.G.; Bennett, M.R.; Lamb, C.; Lennard, T.W.J.; Browell, D.; Ali, S.; Kirby, J.A. Breast cancer metastasis: Demonstration that FOXP3 regulates CXCR4 expression and the response to CXCL12. J. Pathol. 2014, 234, 74–85. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Hao, Q.; Wang, S.; Li, H.; Li, J.; Gao, Y.; Li, M.; Li, W.; Xue, X.; et al. FOXP3 suppresses breast cancer metastasis through downregulation of CD44. Int. J. Cancer 2015, 137, 1279–1290. [Google Scholar] [CrossRef]
- Mihály, Z.; Kormos, M.; Lánczky, A.; Dank, M.; Budczies, J.; Szász, M.A.; Győrffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef]
- Tai, Y.; Sakamoto, K.; Takano, A.; Haga, K.; Harada, Y. Dysregulation of humoral immunity in Foxp3 conditional-knockout mice. Biochem. Biophys. Res. Commun. 2019, 513, 787–793. [Google Scholar] [CrossRef]
- Hori, S. Regulatory T cell plasticity: Beyond the controversies. Trends Immunol. 2011, 32, 295–300. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for regulatory T cell suppressive function. Nature 2019, 565, 495. [Google Scholar] [CrossRef]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef]
- Banerjee, H.; Nieves-Rosado, H.; Kulkarni, A.; Murter, B.; McGrath, K.V.; Chandran, U.R.; Chang, A.; Szymczak-Workman, A.L.; Vujanovic, L.; Delgoffe, G.M.; et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. 2021, 36, 109699. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef]
- Alvisi, G.; Brummelman, J.; Puccio, S.; Mazza, E.M.C.; Tomada, E.P.; Losurdo, A.; Zanon, V.; Peano, C.; Colombo, F.S.; Scarpa, A.; et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Investig. 2020, 130, 3137–3150. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- National Cancer Institute. CTLA-4. 2020. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/ctla-4 (accessed on 2 July 2022).
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Greaves, P.; Gribben, J.G. The role of B7 family molecules in hematologic malignancy. Blood 2013, 121, 734–744. [Google Scholar] [CrossRef]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Abbas, A.; Shiv Pillai, A.L. Cellular and Molecular Immunology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Phillips, K.A.; Ribi, K.; Fisher, R. Do aromatase inhibitors have adverse effects on cognitive function? Breast Cancer Res. 2011, 13, 203. [Google Scholar] [CrossRef]
- Laba, S.; Mallett, G.; Amarnath, S. The depths of PD-1 function within the tumor microenvironment beyond CD8+ T cells. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The role of PD-1/PD-L1 axis in treg development and function: Implications for cancer immunotherapy. OncoTargets Ther. 2019, 12, 8437. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2020, 218, e20182232. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Toor, S.M.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenetics 2018, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Al-Hatamleh, M.A.I.; Ahmad, S.; Boer, J.; Lim, J.; Chen, X.; Plebanski, M.; Mohamud, R. A Perspective Review on the Role of Nanomedicine in the Modulation of TNF-TNFR2 Axis in Breast Cancer Immunotherapy. J. Oncol. 2019, 2019, 6313242. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) Modulates the Ability of CD4 T-cells to Be Suppressed In Vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s enigmatic mechanism of action. Front. Immunol. 2021, 11, 615317. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in Regulatory T Cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Zhang, Q.; Chikina, M.; Szymczak-Workman, A.L.; Horne, W.; Kolls, J.K.; Vignali, K.M.; Normolle, D.; Bettini, M.; Workman, C.J.; Vignali, D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017, 2, eaah4569. [Google Scholar] [CrossRef]
- Lecocq, Q.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. The next-generation immune checkpoint LAG-3 and its therapeutic potential in oncology: Third time’sa charm. Int. J. Mol. Sci. 2020, 22, 75. [Google Scholar] [CrossRef]
- Andrews, L.P.; Somasundaram, A.; Moskovitz, J.M.; Szymczak-Workman, A.L.; Liu, C.; Cillo, A.R.; Lin, H.; Normolle, D.P.; Moynihan, K.D.; Taniuchi, I.; et al. Resistance to PD1 blockade in the absence of metalloprotease-mediated LAG3 shedding. Sci. Immunol. 2020, 5, eabc2728. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Kane, L.P. T Cell Ig and Mucin Domain Proteins and Immunity. J. Immunol. 2010, 184, 2743–2749. [Google Scholar] [CrossRef]
- Sakuishi, K.; Ngiow, S.F.; Sullivan, J.M.; Teng, M.W.L.; Kuchroo, V.K.; Smyth, M.J.; Anderson, A.C. TIM3+ FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology 2013, 2, e23849. [Google Scholar] [CrossRef]
- Liu, Z.; McMichael, E.L.; Shayan, G.; Li, J.; Chen, K.; Srivastava, R.; Kane, L.P.; Lu, B.; Ferris, R.L. Novel Effector Phenotype of Tim-3+ Regulatory T Cells Leads to Enhanced Suppressive Function in Head and Neck Cancer Patients The Role of Tim-3 on Regulatory T Cells in HNSCC Patients. Clin. Cancer Res. 2018, 24, 4529–4538. [Google Scholar] [CrossRef]
- Gautron, A.-S.; Dominguez-Villar, M.; de Marcken, M.; Hafler, D.A. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur. J. Immunol. 2014, 44, 2703–2711. [Google Scholar] [CrossRef]
- Birla, P. Investigating Functional Synergy between PD-1 and TIGIT in Anti-Tumor Immunity and Autoimmunity. Master’s Thesis, Harvard University, Cambridge, MA, USA, 2019. [Google Scholar]
- Al-Hatamleh, M.A.I.; Engku Nur Syafirah, E.A.R.; Boer, J.C.; Ferji, K.; Six, J.-L.; Chen, X.; Elkord, E.; Plebanski, M.; Mohamud, R. Synergistic Effects of Nanomedicine Targeting TNFR2 and DNA Demethylation Inhibitor—An Opportunity for Cancer Treatment. Cells 2020, 9, 33. [Google Scholar] [CrossRef]
- Sheng, Y.; Li, F.; Qin, Z. TNF Receptor 2 Makes Tumor Necrosis Factor a Friend of Tumors. Front. Immunol. 2018, 9, 1170. [Google Scholar] [CrossRef]
- Li, M.; Zhang, X.; Bai, X.; Liang, T. Targeting TNFR2: A Novel Breakthrough in the Treatment of Cancer. Front. Oncol. 2022, 12, 862154. [Google Scholar] [CrossRef]
- Chen, X.; Plebanski, M. Editorial: The Role of TNF-TNFR2 Signal in Immunosuppressive Cells and Its Therapeutic Implications. Front. Immunol. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo: Presented in part at the Keystone Symposium “Cellular and Molecular Biology of Dendritic Cells”, Santa Fe, NM, March 3–9, 1998, and at the annual meeting of the American Association for Cancer Research, March 28–April 1, 1998. Blood 1998, 92, 4150–4166. [Google Scholar]
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737. [Google Scholar]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2018, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, V.; Morandi, F.; Bianchi, G.; Pezzolo, A.; Prigione, I.; Raffaghello, L. Immunosuppressive microenvironment in neuroblastoma. Front. Oncol. 2013, 3, 167. [Google Scholar] [CrossRef] [PubMed]
- Vasievich, E.A.; Huang, L. The Suppressive Tumor Microenvironment: A Challenge in Cancer Immunotherapy. Mol. Pharm. 2011, 8, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Ren, J.P.; Wu, X.Y.; Morrison, Z.D.; El Gazzar, M.; Ning, S.; Moorman, J.P.; Yao, Z.Q. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS 2016, 30, 1521–1531. [Google Scholar] [CrossRef]
- Yang, F.; Li, Y.; Zhang, Q.; Tan, L.; Peng, L.; Zhao, Y. The Effect of Immunosuppressive Drugs on MDSCs in Transplantation. J. Immunol. Res. 2018, 2018, 5414808. [Google Scholar] [CrossRef]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef]
- Nausch, N.; Galani, I.E.; Schlecker, E.; Cerwenka, A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood 2008, 112, 4080–4089. [Google Scholar] [CrossRef]
- Mao, A.; Paharkova-Vatchkova, V.; Hardy, J.; Miller, M.M.; Kovats, S. Estrogen Selectively Promotes the Differentiation of Dendritic Cells with Characteristics of Langerhans Cells. J. Immunol. 2005, 175, 5146–5151. [Google Scholar] [CrossRef]
- Tomić, S.; Joksimović, B.; Bekić, M.; Vasiljević, M.; Milanović, M.; Čolić, M.; Vučević, D. Prostaglanin-E2 Potentiates the Suppressive Functions of Human Mononuclear Myeloid-Derived Suppressor Cells and Increases Their Capacity to Expand IL-10-Producing Regulatory T Cell Subsets. Front. Immunol. 2019, 10, 475. [Google Scholar] [CrossRef]
- Srikrishna, G. S100A8 and S100A9: New Insights into Their Roles in Malignancy. J. Innate Immun. 2011, 4, 31–40. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Al-Hatamleh, M.A.I.; Baig, A.A.; Simbak, N.B.; Nadeem, M.I.; Khan, S.U.; Ariff, T.M. Molecular Modulation of Stress Induced to Abnormal Haematological Indices in Medical Students, Malaysian Perspective. Pak. J. Biol. Sci. 2017, 20, 478–488. [Google Scholar] [CrossRef]
- Al-Hatamleh, M.A.İ.; Ismail, İ.; Al-Shajrawi, O.M.; Ariff, T.M. Effect of stress on alteration of haematological parameters: A preliminary study on preclinical medical students in Malaysia. J. Cell. Neurosci. Oxidative Stress 2020, 11, 852–860. [Google Scholar] [CrossRef]
- Kesarwani, P.; Murali, A.K.; Al-Khami, A.A.; Mehrotra, S. Redox Regulation of T-Cell Function: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2013, 18, 1497–1534. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Levring, T.B.; Hansen, A.K.; Nielsen, B.L.; Kongsbak, M.; von Essen, M.R.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci. Rep. 2012, 2, 266. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef]
- Cha, H.J.; He, C.; Zhao, H.; Dong, Y.; An, I.-S.; An, S. Intercellular and intracellular functions of ceramides and their metabolites in skin (Review). Int. J. Mol. Med. 2016, 38, 16–22. [Google Scholar] [CrossRef]
- Larsson, A.-M.; Roxå, A.; Leandersson, K.; Bergenfelz, C. Impact of systemic therapy on circulating leukocyte populations in patients with metastatic breast cancer. Sci. Rep. 2019, 9, 13451. [Google Scholar] [CrossRef]
- Millrud, C.R.; Mehmeti, M.; Leandersson, K. Docetaxel promotes the generation of anti-tumorigenic human macrophages. Exp. Cell Res. 2018, 362, 525–531. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Coussens, L.M. Inflammation and breast cancer. Balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007, 9, 212. [Google Scholar] [CrossRef]
- Gong, J.; Avigan, D.; Chen, D.; Wu, Z.; Koido, S.; Kashiwaba, M.; Kufe, D. Activation of antitumor cytotoxic T lymphocytes by fusions of human dendritic cells and breast carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Pedersen, A.E.; Johnsen, H.E.; Nielsen, D.; Kamby, C.; Gaarsdal, E.; Nikolajsen, K.; Claesson, M.H. Vaccination with p53-peptide?pulsed dendritic cells, of patients with advanced breast cancer: Report from a phase I study. Cancer Immunol. Immunother. 2004, 53, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, S.; Gennaro, M.; Vaccari, M.; Ferraris, C.; Nicola, S.; Riva, A.; Clerici, M.; Greco, M.; Villa, M.L. Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br. J. Cancer 2003, 89, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13–secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-α Production by Plasmacytoid Dendritic Cells Favors Regulatory T-cell Expansion That May Contribute to Breast Cancer Progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef]
- Komi, J.; Lassila, O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood 2000, 95, 2875–2882. [Google Scholar] [CrossRef]
- Kranich, J.; Krautler, N.J. How Follicular Dendritic Cells Shape the B-Cell Antigenome. Front. Immunol. 2016, 7, 225. [Google Scholar] [CrossRef]
- Sapino, A.; Cassoni, P.; Ferrero, E.; Bongiovanni, M.; Righi, L.; Fortunati, N.; Crafa, P.; Chiarle, R.; Bussolati, G. Estrogen Receptor α Is a Novel Marker Expressed by Follicular Dendritic Cells in Lymph Nodes and Tumor-Associated Lymphoid Infiltrates. Am. J. Pathol. 2003, 163, 1313–1320. [Google Scholar] [CrossRef]
- Kurze, A.-K.; Buhs, S.; Eggert, D.; Oliveira-Ferrer, L.; Müller, V.; Niendorf, A.; Wagener, C.; Nollau, P. Immature O-glycans recognized by the macrophage glycoreceptor CLEC10A (MGL) are induced by 4-hydroxy-tamoxifen, oxidative stress and DNA-damage in breast cancer cells. Cell Commun. Signal. 2019, 17, 107. [Google Scholar] [CrossRef]
- American Cancer Society. Tamoxifen and Raloxifene for Lowering Breast Cancer Risk; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- de Cremoux, P.; Diéras, V.; Poupon, M.-F.; Magdelénat, H.; Sigal-Zafrani, B.; Fourquet, A.; Pierga, J.-Y. Tamoxifen and aromatase inhibitors in the treatment of breast cancer in menopausal women: Pharmacological and clinical aspects. Bull. Cancer 2004, 91, 917–927. [Google Scholar]
- Schneider, R.; Barakat, A.; Pippen, J.E.; Osborne, C. Aromatase inhibitors in the treatment of breast cancer in post-menopausal female patients: An update. Breast Cancer Targets Ther. 2011, 3, 113–125. [Google Scholar] [CrossRef]
- Paharkova-Vatchkova, V.; Maldonado, R.; Kovats, S. Estrogen Preferentially Promotes the Differentiation of CD11c+CD11bintermediateDendritic Cells from Bone Marrow Precursors. J. Immunol. 2004, 172, 1426–1436. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Molecular Subtypes | Immunohistochemical Characterization |
|---|---|
| Luminal A | ER+ and (or) PR+, HER-2− and Ki-67 < 14% |
| Luminal B | ER+ and (or) PR+, HER-2− and Ki-67 ≥ 14% ER+ and (or) PR+, HER-2+, any level of Ki-67 |
| HER-2 Overexpression | ER−, PR−, HER+, any level of Ki-67 |
| Triple-Negative Type | ER−, PR−, HER−, and Ki-67 any level |
| Central Tolerance | Peripheral Tolerance | |
|---|---|---|
| Features | Inactivation of cells required for initiation of an immune response. | Inhibition of expression to the immune response. |
| Site of tolerance induction | Generative lymphoid organs. | Peripheral lymphoid tissues. |
| Site of involvement | Afferent limb of the immune response, which is concerned with sensitization and cell proliferation. | Afferent limb of immune response, which is concerned with the generation of effector cells. |
| B cell participation | Immature B cells. | Mature B cells. |
| T cell participation | Immature thymocytes. | Mature T cells. |
| Mechanisms of tolerance | Clonal deletion (apoptotic cell death, negative selection). | Clonal deletion (apoptotic cell death); clonal anergy (functional inactivation without cell death); clonal ignorance (failure to recognize or recognition of antigens without costimulation); suppression of lymphocyte activation and effector functions by regulatory lymphocytes. |
| Function | Eliminates potentially self-reactive lymphocytes. | Maintains unresponsiveness to self-antigens. |
| Immune System Involve | Specific Cells and Other Immune Checkpoints | References | |
|---|---|---|---|
| Adaptive | Foxp3+ Treg |
| [43,54] |
| CTLA4 |
| [30] | |
| PD-1 |
| [68] | |
| LAG-3 |
| [77] | |
| TIM3 |
| [53] | |
| Immune System Involve | Specific Cells and Other Immune Checkpoints | References | |
|---|---|---|---|
| Innate | MDSCs |
| [12,115] |
| DCs |
| [18,103,124,125,127,131] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Idris, R.A.; Mussa, A.; Ahmad, S.; Al-Hatamleh, M.A.I.; Hassan, R.; Tengku Din, T.A.D.A.A.; Wan Abdul Rahman, W.F.; Lazim, N.M.; Boer, J.C.; Plebanski, M.; et al. The Effects of Tamoxifen on Tolerogenic Cells in Cancer. Biology 2022, 11, 1225. https://doi.org/10.3390/biology11081225
Mohd Idris RA, Mussa A, Ahmad S, Al-Hatamleh MAI, Hassan R, Tengku Din TADAA, Wan Abdul Rahman WF, Lazim NM, Boer JC, Plebanski M, et al. The Effects of Tamoxifen on Tolerogenic Cells in Cancer. Biology. 2022; 11(8):1225. https://doi.org/10.3390/biology11081225
Chicago/Turabian StyleMohd Idris, Ros Akmal, Ali Mussa, Suhana Ahmad, Mohammad A. I. Al-Hatamleh, Rosline Hassan, Tengku Ahmad Damitri Al Astani Tengku Din, Wan Faiziah Wan Abdul Rahman, Norhafiza Mat Lazim, Jennifer C. Boer, Magdalena Plebanski, and et al. 2022. "The Effects of Tamoxifen on Tolerogenic Cells in Cancer" Biology 11, no. 8: 1225. https://doi.org/10.3390/biology11081225