
Gut Microbiota and Antibiotic Treatments for the Main Non-Oncologic Hepato-Biliary-Pancreatic Disorders
 , , ,
, , ,
Abstract
:1. Introduction
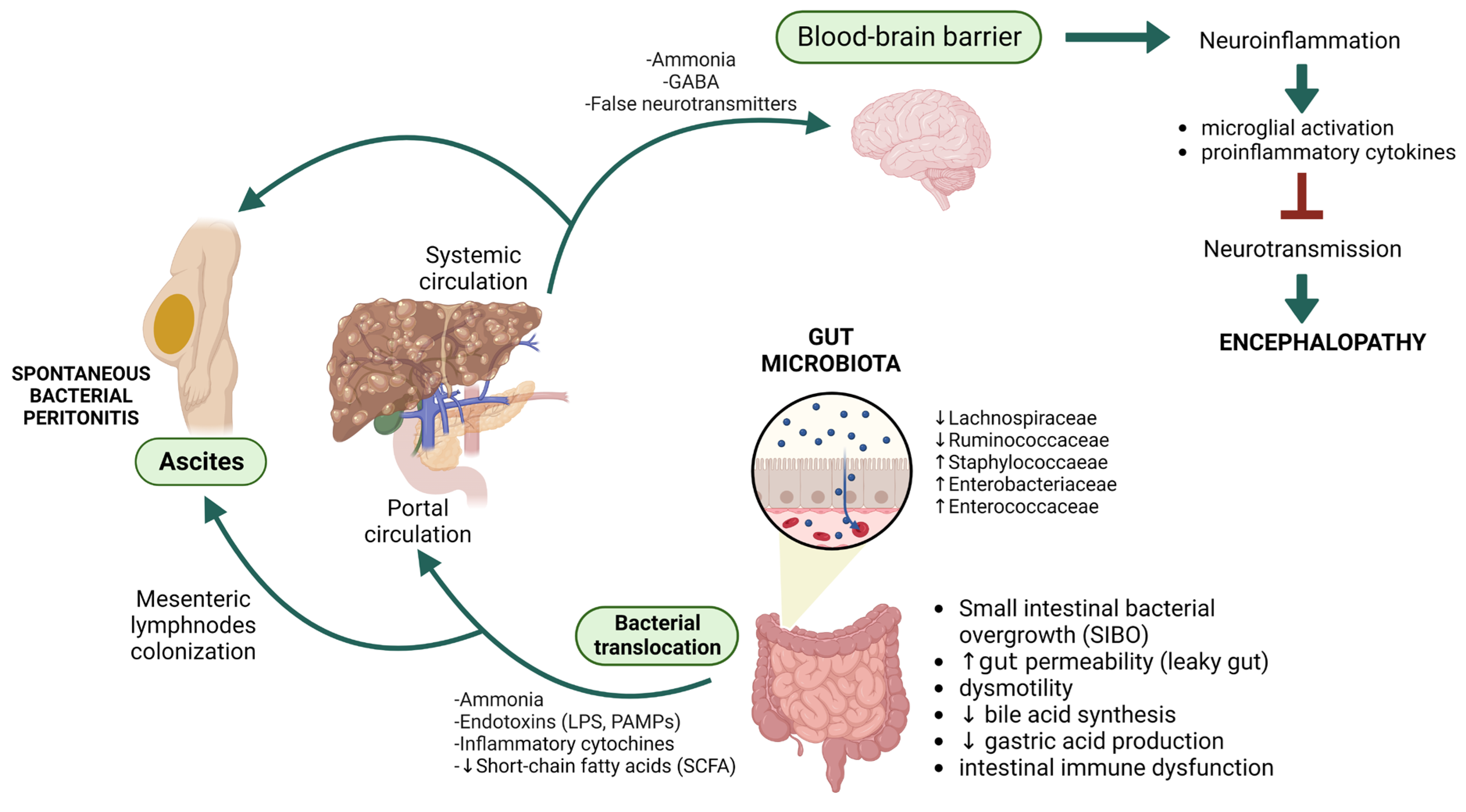
2. Liver and Gut Microbiota
2.1. Hepatic Encephalopathy
2.1.1. Gut Microbiota and Hepatic Encephalopathy: Towards the Gut–Liver–Brain Axis
2.1.2. Antibiotic Treatment for Overt Hepatic Encephalopathy
2.2. Spontaneous Bacterial Peritonitis
2.2.1. Spontaneous Bacterial Peritonitis and Intestinal Microbiota in Cirrhosis
2.2.2. Antibiotics and Spontaneous Bacterial Peritonitis
3. The Gut Microbiota in Acute Pancreatitis
Antibiotics in Acute Pancreatitis
4. Gut Microbiota and Antibiotics in Biliary Diseases
4.1. Primary Sclerosing Cholangitis

{kind=link}
| AUTHOR OF THE STUDY | STUDY DESIGN | STUDY POPULATION | AIM OF THE STUDY | PRIMARY ENDPOINTS | SECONDARY ENDPOINTS | RESULTS (% Change from Baseline Post-Therapy) | |
|---|---|---|---|---|---|---|---|
| Tabibian et al. (2017) [157] | 12-week open-label pilot study | 16 patients with PSC. 13 M and 3 F, median age 40 years old, 81% with IBD | Efficacy and safety of oral rifaximin 550 mg twice daily | Serum ALP at 12 weeks | Serum bilirubin, γ GT, PSC MRS at 12 weeks |
ALP (+3.00—p = 0.47)
MRS (+0.15—p = 0.21) | |
| Silveira et al. (2009) [158] | 1-year pilot study | 16 patients with PSC, gender non specified, median age 50 years old, 88% with IBD | Safety and efficacy of Minocycline 100 mg orally twice daily | Serum ALP at 1 year | PSC MRS at 1 year | ALP (−65—p = 0.04) MRS (−0.53—p = 0.05) | |
| Färkkilä et al. (2004) [159] | 36-year multicenter, randomized, double-blind, placebo-controlled trial | 80 patients with PSC (41 placebo, 39 MTZ), 42 M and 38 F, median age 16–65 years old, 81% with IBD | Effect of Metronidazole 800 mg compared with placebo on the progression of PSC | Serum ALP at 36 months | PSC MRS at 36 months | Metronidazole: ALP (−337—p = 0.05) MRS (−0.32—p = 0.05) | Placebo: ALP (−214—p < 0.01) MRS (−0.06—p < 0.01) |
| Rahimpour et al. (2016) [161] | Triple blinded, randomized, placebo-controlled trial | 29 patients with PSC (11 placebo, 18 vancomycin), 17 M and 12 F, median age 36 years old, 75% with IBD | Safety and efficacy of oral Vancomycin (125 mg, four times a day) | ALP levels and the PSC MRS at 12 weeks | Serum level of ESR, AST, ALT, bilirubin, WBC, PLT, γ GT and symptoms at 12 weeks | ALP (−519.68—p = 0.11) Bilirubin (−1.35—p = 0.41) MRS (−0.59—p = 0.03) | |
| Tabibian et al. (2013) [160] | 12-week randomized clinical trial | 35 patients with PSC: 8 Vancomycin 125 mg/24 h 9 Vancomycin 250 mg/6 h 9 Metronidazole 250 mg/8 h 9 Metronidazole 500 mg/8 h (21 males and 14 females, median age 40 years old, 71% with IBD) | Safety and efficacy of oral Vancomycin and Metronidazole in patients with PSC | Serum ALP at 12 weeks | Serum bilirubin, PSC MRS, pruritus, adverse effects at 12 weeks | Vancomycin low-dose ALP (−188—p = 0.03) Bilirubin (−0.3—p = 0.06) MRS (−0.55—p = 0.03) Metronidazole low-dose ALP (+46—p = 0.47) Bilirubin (−0.2—p = 0.03) MRS (−0.16—p = 0.03) | Vancomycin high-dose ALP (−136—p = 0.02) Bilirubin (0—p = 0.48) MRS (−0.03—p = 0.98) Metronidazole high-dose ALP (−138—p = 0.22) Bilirubin (0.1—p = 0.78) MRS (−0.28—p = 0.16) |
| Ali et al. (2020) [162] | Open-label clinical trial | 59 patients with PSC, 38 M and 21 F, median age 13.5 years old, 95% with IBD | Safety and efficacy of oral Vancomycin in patients with PSC | Decrease of ALP, γ GT and ALT from baseline | Not specified | ALP 81.3% γ GT 96% ALT 94.9% | |
| Deneau et al. (2018) [165] | Retrospective Study (data from Pediatric Consortium) | 264 patients with PSC: 88 oral vancomicine (66 M and 22 F, median age 14 years old, 86% with IBD), 88 UCDA (72 M and 16 F, median age 12 years old, 85% with IBD), 88 observation (69 M and 19 F, median age 14 years old, 86% with IBD) | Safety and efficacy of oral Vancomycin and UCDA in patients with PSC | Serum γ GT < 50 U/L or ≥75% less than the pretreatment serum γ GT at 1 year | Improvement of liver fibrosis staging | Oral Vancomicine γ GT 53% (p = 0.918) Fibrosis 20% (p = 0.193) UCDA γ GT 49% (p = 0.918) Fibrosis 13% (p = 0.193) Observation γ GT 52% (p = 0.918) Fibrosis 18% (p = 0.193) | |
| Davies et al. (2008) [166] | Retrospective Study (data from Pediatric Consortium) | 14 patients with PSC M/F 2.3:1 median age 12 years old 100% with IBD | Safety and efficacy of oral Vancomycin in patients with PSC | Serum γ GT < 50 U/L or ≥75% less than the pretreatment serum γ GT at 1 year | Improvement of liver fibrosis staging | Oral Vancomicine γ GT 53% (p = 0.918) Fibrosis 20% (p = 0.193) UCDA γ GT 49% (p = 0.918) Fibrosis 13% (p = 0.193) Observation γ GT 52% (p = 0.918) Fibrosis 18% (p = 0.193) | |
4.2. Primary Biliary Cholangitis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leal-Lopes, C.; Velloso, F.J.; Campopiano, J.C.; Sogayar, M.C.; Correa, R.G. Roles of Commensal Microbiota in Pancreas Homeostasis and Pancreatic Pathologies. J. Diabetes Res. 2015, 2015, 284680. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, B. Role of the gut microbiota in human nutrition and metabolism: Role of the Gut Microbiota. J. Gastroenterol. Hepatol. 2013, 28, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Burcelin, R.; Tremaroli, V. Liver tissue microbiome in NAFLD: Next step in understanding the gut–liver axis? Gut 2020, 69, 1373–1374. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Tuomisto, S.; Pessi, T.; Collin, P.; Vuento, R.; Aittoniemi, J.; Karhunen, P.J. Changes in gut bacterial populations and their translocation into liver and ascites in alcoholic liver cirrhotics. BMC Gastroenterol. 2014, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Wu, Z.; Xu, W.; Yang, J.; Chen, Y.; Li, L. Intestinal Microbiota Was Assessed in Cirrhotic Patients with Hepatitis B Virus Infection: Intestinal Microbiota of HBV Cirrhotic Patients. Microb. Ecol. 2011, 61, 693–703. [Google Scholar] [CrossRef]
- Wei, X.; Yan, X.; Zou, D.; Yang, Z.; Wang, X.; Liu, W.; Wang, S.; Li, X.; Han, J.; Huang, L.; et al. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. 2013, 13, 175. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhai, H.; Geng, J.; Yu, R.; Ren, H.; Fan, H.; Shi, P. Large-Scale Survey of Gut Microbiota Associated With MHE Via 16S rRNA-Based Pyrosequencing. Am. J. Gastroenterol. 2013, 108, 1601–1611. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-S.; Yang, S.-S.; Kao, C.-H.; Yeh, H.-Z.; Chen, G.-H. Small Intestinal Bacterial Overgrowth versus Antimicrobial Capacity in Patients with Spontaneous Bacterial Peritonitis. Scand. J. Gastroenterol. 2001, 36, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Haderer, M.; Neubert, P.; Rinner, E.; Scholtis, A.; Broncy, L.; Gschwendtner, H.; Kandulski, A.; Pavel, V.; Mehrl, A.; Brochhausen, C.; et al. Novel pathomechanism for spontaneous bacterial peritonitis: Disruption of cell junctions by cellular and bacterial proteases. Gut 2022, 71, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Patidar, K.R.; Bajaj, J.S. Covert and Overt Hepatic Encephalopathy: Diagnosis and Management. Clin. Gastroenterol. Hepatol. 2015, 13, 2048–2061. [Google Scholar] [CrossRef] [Green Version]
- Karanfilian, B.V.; Park, T.; Senatore, F.; Rustgi, V.K. Minimal Hepatic Encephalopathy. Clin. Liver Dis. 2020, 24, 209–218. [Google Scholar] [CrossRef]
- Elsaid, M.I.; Rustgi, V.K. Epidemiology of Hepatic Encephalopathy. Clin. Liver Dis. 2020, 24, 157–174. [Google Scholar] [CrossRef]
- Allampati, S.; Duarte-Rojo, A.; Thacker, L.R.; Patidar, K.R.; White, M.B.; Klair, J.S.; John, B.; Heuman, D.M.; Wade, J.B.; Flud, C.; et al. Diagnosis of Minimal Hepatic Encephalopathy Using Stroop EncephalApp: A Multicenter US-Based, Norm-Based Study. Am. J. Gastroenterol. 2016, 111, 78–86. [Google Scholar] [CrossRef]
- Cordoba, J.; Ventura-Cots, M.; Simón-Talero, M.; Amorós, À.; Pavesi, M.; Vilstrup, H.; Angeli, P.; Domenicali, M.; Ginés, P.; Bernardi, M.; et al. Characteristics, risk factors, and mortality of cirrhotic patients hospitalized for hepatic encephalopathy with and without acute-on-chronic liver failure (ACLF). J. Hepatol. 2014, 60, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Shanahan, E.; Macdonald, G.; Fletcher, L.; Ghasemi, P.; Morrison, M.; Jones, M.; Holtmann, G. Systematic Review and Meta-Analysis: Prevalence of Small Intestinal Bacterial Overgrowth in Chronic Liver Disease. Semin. Liver Dis. 2017, 37, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dhiman, R.K.; Kumari, S.; Rana, S.; Agarwal, R.; Duseja, A.; Chawla, Y. Role of small intestinal bacterial overgrowth and delayed gastrointestinal transit time in cirrhotic patients with minimal hepatic encephalopathy. J. Hepatol. 2010, 53, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Sakamaki, A.; Takahashi, K.; Naruse, T.; Sato, C.; Kawata, Y.; Tominaga, K.; Abe, H.; Sato, H.; Tsuchiya, A.; et al. Hydrogen-producing small intestinal bacterial overgrowth is associated with hepatic encephalopathy and liver function. PLoS ONE 2022, 17, e0264459. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M.; Panasevich, M.R.; Peppler, W.T.; et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Liver Physiol. 2012, 302, G168–G175. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Liver Physiol. 2012, 303, G675–G685. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Hu, F.-R.; Xin, R.-J.; Yao, L.; Hu, S.-J.; Bai, F.-H. Altered gut microbiota is associated with sleep disturbances in patients with minimal hepatic encephalopathy caused by hepatitis B-related liver cirrhosis. Expert Rev. Gastroenterol. Hepatol. 2022, 16, 797–807. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Ladinsky, M.S.; Yu, K.B.; Sanders, J.G.; Yoo, B.B.; Chou, W.-C.; Conner, M.E.; Earl, A.M.; Knight, R.; Bjorkman, P.J.; et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science 2018, 360, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Zong, X.; Fu, J.; Xu, B.; Wang, Y.; Jin, M. Interplay between gut microbiota and antimicrobial peptides. Anim. Nutr. 2020, 6, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Alaish, S.M.; Smith, A.D.; Timmons, J.; Greenspon, J.; Eyvazzadeh, D.; Murphy, E.; Shea-Donahue, T.; Cirimotich, S.; Mongodin, E.; Zhao, A.; et al. Gut microbiota, tight junction protein expression, intestinal resistance, bacterial translocation and mortality following cholestasis depend on the genetic background of the host. Gut Microbes 2013, 4, 292–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, N.; Kanai, T. Role of Toll-Like Receptors in Immune Activation and Tolerance in the Liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.J.; Adams, D.H. Gut–liver immunity. J. Hepatol. 2016, 64, 1187–1189. [Google Scholar] [CrossRef] [Green Version]
- Mangas-Losada, A.; García-García, R.; Urios, A.; Escudero-García, D.; Tosca, J.; Giner-Durán, R.; Serra, M.A.; Montoliu, C.; Felipo, V. Minimal hepatic encephalopathy is associated with expansion and activation of CD4+CD28−, Th22 and Tfh and B lymphocytes. Sci. Rep. 2017, 7, 6683. [Google Scholar] [CrossRef] [PubMed]
- Shawcross, D.; Wright, G.; Olde Damink, S.W.M.; Jalan, R. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 125–138. [Google Scholar] [CrossRef]
- Lukiw, W.J. Gastrointestinal (GI) Tract Microbiome-Derived Neurotoxins—Potent Neuro-Inflammatory Signals From the GI Tract via the Systemic Circulation Into the Brain. Front. Cell. Infect. Microbiol. 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. The liver–brain axis in liver failure: Neuroinflammation and encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 522–528. [Google Scholar] [CrossRef]
- Balzano, T.; Dadsetan, S.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Gil-Perotin, S.; Cubas-Nuñez, L.; Casanova, B.; Castro-Quintas, A.; et al. Chronic hyperammonemia induces peripheral inflammation that leads to cognitive impairment in rats: Reversed by anti-TNF-α treatment. J. Hepatol. 2020, 73, 582–592. [Google Scholar] [CrossRef]
- Kang, D.J.; Betrapally, N.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.P.B.; Gillevet, P.M.P.M.; Sanyal, A.J.A.J.; Heuman, D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Kang, J.D.; Sartor, R.B.; Sikaroodi, M.; Fagan, A.; Gavis, E.A.; Zhou, H.; Hylemon, P.B.; Herzog, J.W.; Li, X.; et al. Neuroinflammation in Murine Cirrhosis Is Dependent on the Gut Microbiome and Is Attenuated by Fecal Transplant. Hepatology 2020, 71, 611–626. [Google Scholar] [CrossRef]
- Collins, C.M.; D’Orazio, S.E.F. Bacterial ureases: Structure, regulation of expression and role in pathogenesis. Mol. Microbiol. 1993, 9, 907–913. [Google Scholar] [CrossRef]
- Liotta, E.M.; Kimberly, W.T. Cerebral edema and liver disease: Classic perspectives and contemporary hypotheses on mechanism. Neurosci. Lett. 2020, 721, 134818. [Google Scholar] [CrossRef]
- Jayakumar, A.; Rao, K.R.; Murthy, C.; Norenberg, M. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem. Int. 2006, 48, 623–628. [Google Scholar] [CrossRef]
- Rodrigo, R.; Cauli, O.; Gomez–Pinedo, U.; Agusti, A.; Hernandez–Rabaza, V.; García-Verdugo, J.M.; Felipo, V. Hyperammonemia Induces Neuroinflammation That Contributes to Cognitive Impairment in Rats With Hepatic Encephalopathy. Gastroenterology 2010, 139, 675–684. [Google Scholar] [CrossRef]
- Hernández-Rabaza, V.; Cabrera-Pastor, A.; Taoro-González, L.; Malaguarnera, M.; Agustí, A.; Llansola, M.; Felipo, V. Hyperammonemia induces glial activation, neuroinflammation and alters neurotransmitter receptors in hippocampus, impairing spatial learning: Reversal by sulforaphane. J. Neuroinflamm. 2016, 13, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Sanyal, A.J.; Hylemon, P.B.; Sterling, R.K.; Stravitz, R.T.; Fuchs, M.; Ridlon, J.M.; Daita, K.; Monteith, P.; et al. Modulation of the Metabiome by Rifaximin in Patients with Cirrhosis and Minimal Hepatic Encephalopathy. PLoS ONE 2013, 8, e60042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, P.; Francés, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2012, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain, Behav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Takechi, M.; Kiyonari, H.; Shioi, G.; Tamura, A.; Tsukita, S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015, 64, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.; Davies, N.A.; Shawcross, D.L.; Hodges, S.J.; Zwingmann, C.; Brooks, H.F.; Mani, A.R.; Harry, D.; Stadlbauer, V.; Zou, Z.; et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007, 45, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Bajaj, J.S.; Bin Wang, J.; Shang, J.; Zhou, X.M.; Guo, X.L.; Zhu, X.; Na Meng, L.; Jiang, H.X.; Mi, Y.Q.; et al. Lactulose improves cognition, quality of life, and gut microbiota in minimal hepatic encephalopathy: A multicenter, randomized controlled trial. J. Dig. Dis. 2019, 20, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, M.; Kieling, C.; Wolff, F.H.; Medeiros, L.R.; Cheinquer, H. Antibiotics for Hepatic Encephalopathy. In Cochrane Database of Systematic Reviews; The Cochrane Collaboration, Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2007; p. CD006314. [Google Scholar]
- Alexander, T.; Thomas, K.; Cherian, A.M. Kanakasabapathy, null Effect of Three Antibacterial Drugs in Lowering Blood & Stool Ammonia Production in Hepatic Encephalopathy. Indian J. Med. Res. 1992, 96, 292–296. [Google Scholar] [PubMed]
- Forbes, A.; Murray-Lyon, I. Vancomycin in resistant hepatic encephalopathy. Gut 1990, 31, 1424. [Google Scholar] [CrossRef] [Green Version]
- Glal, K.A.M.; Abd-Elsalam, S.M.; Mostafa, T.M. Nitazoxanide versus rifaximin in preventing the recurrence of hepatic encephalopathy: A randomized double-blind controlled trial. J. Hepato-Biliary-Pancreat. Sci. 2021, 28, 812–824. [Google Scholar] [CrossRef]
- Rajpurohit, S.; Musunuri, B.; Shailesh; Mohan, P.B.; Shetty, S. Novel Drugs for the Management of Hepatic Encephalopathy: Still a Long Journey to Travel. J. Clin. Exp. Hepatol. 2022, 12, 1200–1214. [Google Scholar] [CrossRef] [PubMed]
- Scarpignato, C.; Pelosini, I. Rifaximin, a Poorly Absorbed Antibiotic: Pharmacology and Clinical Potential. Chemotherapy 2005, 51, 36–66. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Zocco, M.A.; D’Aversa, F.; Pompili, M.; Gasbarrini, A. Eubiotic Properties of Rifaximin: Disruption of the Traditional Concepts in Gut Microbiota Modulation. World J. Gastroenterol. 2017, 23, 4491. [Google Scholar] [CrossRef]
- Bass, N.M.; Mullen, K.D.; Sanyal, A.; Poordad, F.; Neff, G.; Leevy, C.B.; Sigal, S.; Sheikh, M.Y.; Beavers, K.; Frederick, T.; et al. Rifaximin Treatment in Hepatic Encephalopathy. N. Engl. J. Med. 2010, 362, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Eltawil, K.M. Rifaximin vs. conventional oral therapy for hepatic encephalopathy: A meta-analysis. World J. Gastroenterol. 2012, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.C.; Lee, S.; McPhail, M.J.; Da Silva, K.; Guilly, S.; Zamalloa, A.; Witherden, E.; Støy, S.; Vijay, G.K.M.; Pons, N.; et al. Rifaximin-α reduces gut-derived inflammation and mucin degradation in cirrhosis and encephalopathy: RIFSYS randomised controlled trial. J. Hepatol. 2022, 76, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Fan, H.; Tang, X.; Chen, Y.; Xun, L.; Li, Y.; Song, Z.; Zhai, H. Effect of different treatments and alcohol addiction on gut microbiota in minimal hepatic encephalopathy patients. Exp. Ther. Med. 2017, 14, 4887–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S. Review article: Potential mechanisms of action of rifaximin in the management of hepatic encephalopathy and other complications of cirrhosis. Aliment. Pharmacol. Ther. 2016, 43, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Kaji, K.; Takaya, H.; Saikawa, S.; Furukawa, M.; Sato, S.; Kawaratani, H.; Kitade, M.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Rifaximin ameliorates hepatic encephalopathy and endotoxemia without affecting the gut microbiome diversity. World J. Gastroenterol. 2017, 23, 8355–8366. [Google Scholar] [CrossRef]
- Kaji, K.; Saikawa, S.; Takaya, H.; Fujinaga, Y.; Furukawa, M.; Kitagawa, K.; Ozutsumi, T.; Kaya, D.; Tsuji, Y.; Sawada, Y.; et al. Rifaximin Alleviates Endotoxemia with Decreased Serum Levels of Soluble CD163 and Mannose Receptor and Partial Modification of Gut Microbiota in Cirrhotic Patients. Antibiotics 2020, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Schütte, K.; Vilchez-Vargas, R.; Vasapolli, R.; Malfertheiner, P. Long-Term Effect of Rifaximin with and without Lactulose on the Active Bacterial Assemblages in the Proximal Small Bowel and Faeces in Patients with Minimal Hepatic Encephalopathy. Dig. Dis. 2019, 37, 161–169. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Salzman, N.; Acharya, C.; Takei, H.; Kakiyama, G.; Fagan, A.; White, M.B.; Gavis, E.A.; Holtz, M.L.; Hayward, M.; et al. Microbial functional change is linked with clinical outcomes after capsular fecal transplant in cirrhosis. J. Clin. Investig. 2019, 4, e133410. [Google Scholar] [CrossRef] [Green Version]
- Madsen, M.; Kimer, N.; Bendtsen, F.; Petersen, A.M. Fecal microbiota transplantation in hepatic encephalopathy: A systematic review. Scand. J. Gastroenterol. 2021, 56, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Shamsaddini, A.; Fagan, A.; Sterling, R.K.; Gavis, E.; Khoruts, A.; Fuchs, M.; Lee, H.; Sikaroodi, M.; Gillevet, P.M. Fecal Microbiota Transplant in Cirrhosis Reduces Gut Microbial Antibiotic Resistance Genes: Analysis of Two Trials. Hepatol. Commun. 2021, 5, 258–271. [Google Scholar] [CrossRef]
- Marciano, S.; Díaz, J.M.; Dirchwolf, M.; Gadano, A. Spontaneous bacterial peritonitis in patients with cirrhosis: Incidence, outcomes, and treatment strategies. Hepatic Med. Évid. Res. 2019, 11, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Llach, J.; Rimola, A.; Navasa, M.; Ginès, P.; Salmerón, J.M.; Ginès, A.; Arroyo, V.; Rodés, J. Incidence and predictive factors of first episode of spontaneous bacterial peritonitis in cirrhosis with ascites: Relevance of ascitic fluid protein concentration. Hepatology 1992, 16, 724–727. [Google Scholar] [CrossRef]
- Saab, S.; Hernandez, J.C.; Chi, A.C.; Tong, M.J. Oral Antibiotic Prophylaxis Reduces Spontaneous Bacterial Peritonitis Occurrence and Improves Short-Term Survival in Cirrhosis: A Meta-Analysis. Am. J. Gastroenterol. 2009, 104, 993–1001. [Google Scholar] [CrossRef]
- Nischalke, H.; Berger, C.; Aldenhoff, K.; Thyssen, L.; Gentemann, M.; Grünhage, F.; Lammert, F.; Nattermann, J.; Sauerbruch, T.; Spengler, U.; et al. Toll-like receptor (TLR) 2 promoter and intron 2 polymorphisms are associated with increased risk for spontaneous bacterial peritonitis in liver cirrhosis. J. Hepatol. 2011, 55, 1010–1016. [Google Scholar] [CrossRef]
- Reiberger, T.; Ferlitsch, A.; Payer, B.A.; Mandorfer, M.; Heinisch, B.B.; Hayden, H.; Lammert, F.; Trauner, M.; Peck-Radosavljevic, M.; Vogelsang, H. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J. Hepatol. 2013, 58, 911–921. [Google Scholar] [CrossRef]
- Rogers, G.B.; Russell, L.E.; Preston, P.G.; Marsh, P.; Collins, J.E.; Saunders, J.; Sutton, J.; Fine, D.; Bruce, K.D.; Wright, M. Characterisation of bacteria in ascites—Reporting the potential of culture-independent, molecular analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Van Der Gast, C.J.; Bruce, K.D.; Marsh, P.; Collins, J.E.; Sutton, J.; Wright, M. Ascitic Microbiota Composition Is Correlated with Clinical Severity in Cirrhosis with Portal Hypertension. PLoS ONE 2013, 8, e74884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginés, P.; Rimola, A.; Planas, R.; Vargas, V.; Marco, F.; Almela, M.; Forne, M.; Miranda, M.L.; Llach, J.; Salmerón, J.M.; et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: Results of a double-blind, placebo-controlled trial. Hepatology 1990, 12, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Runyon, B.A. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013, 57, 1651–1653. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Tapia, N.C.; Barrientos-Gutierrez, T.; Tellez-Avila, F.; Soares-Weiser, K.; Mendez-Sanchez, N.; Gluud, C.; Uribe, M. Meta-analysis: Antibiotic prophylaxis for cirrhotic patients with upper gastrointestinal bleeding—An updated Cochrane review: Meta-Analysis: Antibiotics and Cirrhotic Gastrointestinal Bleeding. Aliment. Pharmacol. Ther. 2011, 34, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Angeli, P.; Bernardi, M.; Villanueva, C.; Francoz, C.; Mookerjee, R.P.; Trebicka, J.; Krag, A.; Laleman, W.; Gines, P. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runyon, B.A.; AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: An update. Hepatology 2009, 49, 2087–2107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lv, H.; Lv, J.; Shi, Y.; Huang, H.; Chen, L.; Shi, D. Alterations of gut microbiota in cirrhotic patients with spontaneous bacterial peritonitis: A distinctive diagnostic feature. Front. Cell. Infect. Microbiol. 2022, 12, 999418. [Google Scholar] [CrossRef]
- Fernández, J.; Tandon, P.; Mensa, J.; Garcia-Tsao, G. Antibiotic prophylaxis in cirrhosis: Good and bad. Hepatology 2016, 63, 2019–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalambokis, G.N. Rifaximin for the prevention of spontaneous bacterial peritonitis. World J. Gastroenterol. 2012, 18, 1700–1702. [Google Scholar] [CrossRef]
- Dănulescu, R.M.; Ciobică, A.; Stanciu, C.; Trifan, A. The role of rifaximine in the prevention of the spontaneous bacterial peritonitis. Rev. Med.-Chir. 2013, 117, 315–320. [Google Scholar]
- Hanouneh, M.A.; Hanouneh, I.A.; Hashash, J.G.; Law, R.; Esfeh, J.M.; Lopez, R.; Hazratjee, N.; Smith, T.; Zein, N.N. The Role of Rifaximin in the Primary Prophylaxis of Spontaneous Bacterial Peritonitis in Patients With Liver Cirrhosis. J. Clin. Gastroenterol. 2012, 46, 709–715. [Google Scholar] [CrossRef]
- Mostafa, T.; Badra, G.; Abdallah, M. The efficacy and the immunomodulatory effect of rifaximin in prophylaxis of spontaneous bacterial peritonitis in cirrhotic Egyptian patients. Turk. J. Gastroenterol. 2015, 26, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, G.S.; Go, A.; Attar, B.M.; Mutneja, H.; Arora, S.; Patel, S.A. Rifaximin versus norfloxacin for prevention of spontaneous bacterial peritonitis: A systematic review. BMJ Open Gastroenterol. 2017, 4, e000154. [Google Scholar] [CrossRef]
- Goel, A.; Rahim, U.; Nguyen, L.H.; Stave, C.; Nguyen, M.H. Systematic review with meta-analysis: Rifaximin for the prophylaxis of spontaneous bacterial peritonitis. Aliment. Pharmacol. Ther. 2017, 46, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, N.; Yamada, A.; Haider, H.; Komaki, Y.; Komaki, F.; Micic, D.; Sakuraba, A. Systemic review and network meta-analysis: Prophylactic antibiotic therapy for spontaneous bacterial peritonitis. World J. Hepatol. 2020, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Menshawy, A.; Mattar, O.; Barssoum, K.; Aboel-Naga, A.M.; Salim, H.M.; Mohamed, A.M.F.; Elgebaly, A.; Abd-Elsalam, S. Safety and Efficacy of Rifaximin in Prophylaxis of Spontaneous Bacterial Peritonitis: A Systematic Review and Meta-analysis. Curr. Drug Targets 2019, 20, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Assem, M.; Elsabaawy, M.; Abdelrashed, M.; Elemam, S.; Khodeer, S.; Hamed, W.; Abdelaziz, A.; El-Azab, G. Efficacy and safety of alternating norfloxacin and rifaximin as primary prophylaxis for spontaneous bacterial peritonitis in cirrhotic ascites: A prospective randomized open-label comparative multicenter study. Hepatol. Int. 2016, 10, 377–385. [Google Scholar] [CrossRef]
- Johnson, C.D.; Besselink, M.G.; Carter, R. Acute pancreatitis. BMJ 2014, 349, g4859. [Google Scholar] [CrossRef]
- Roberts, S.E.; Morrison-Rees, S.; John, A.; Williams, J.G.; Brown, T.H.; Samuel, D.G. The incidence and aetiology of acute pancreatitis across Europe. Pancreatology 2017, 17, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Marshall, J.C.; Cook, D.J.; Christou, N.V.; Bernard, G.R.; Sprung, C.L.; Sibbald, W.J. Multiple Organ Dysfunction Score: A Reliable Descriptor of a Complex Clinical Outcome. Crit. Care Med. 1995, 23, 1638–1652. [Google Scholar] [CrossRef]
- Spanier, B.; Nio, Y.; van der Hulst, R.; Tuynman, H.; Dijkgraaf, M.; Bruno, M. Practice and Yield of Early CT Scan in Acute Pancreatitis: A Dutch Observational Multicenter Study. Pancreatology 2010, 10, 222–228. [Google Scholar] [CrossRef]
- Balthazar, E.J.; Robinson, D.L.; Megibow, A.; Ranson, J.H. Acute pancreatitis: Value of CT in establishing prognosis. Radiology 1990, 174, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Werge, M.; Novovic, S.; Schmidt, P.N.; Gluud, L.L. Infection increases mortality in necrotizing pancreatitis: A systematic review and meta-analysis. Pancreatology 2016, 16, 698–707. [Google Scholar] [CrossRef]
- Guo, Q.; Li, A.; Xia, Q.; Liu, X.; Tian, B.; Mai, G.; Huang, Z.; Chen, G.; Tang, W.; Jin, X.; et al. The Role of Organ Failure and Infection in Necrotizing Pancreatitis. Ann. Surg. 2014, 259, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.S.; Shanbhag, S.; Chakraborty, M.; Phillips, A.R.; Windsor, J.A. Organ Failure and Infection of Pancreatic Necrosis as Determinants of Mortality in Patients With Acute Pancreatitis. Gastroenterology 2010, 139, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Yang, C.; Tang, M.; Wang, M.; Cheng, Z.; Chen, D.; Chen, X.; Liu, K. The Role of Gut Microbiota and Genetic Susceptibility in the Pathogenesis of Pancreatitis. Gut Liver 2022, 16, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of Acute and Chronic Pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Zhang, Z.Y.; Zhang, C.H.; Wu, J.; Wang, Y.X.; Zhang, G.X. Intestinal Microbial Community Differs between Acute Pancreatitis Patients and Healthy Volunteers. Biomed. Environ. Sci. 2018, 31, 81–86. [Google Scholar]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Bacteremia in Patients With Acute Pancreatitis as Revealed by 16S Ribosomal RNA Gene-Based Techniques*. Crit. Care Med. 2013, 41, 1938–1950. [Google Scholar] [CrossRef]
- Yu, S.; Xiong, Y.; Xu, J.; Liang, X.; Fu, Y.; Liu, D.; Yu, X.; Wu, D. Identification of Dysfunctional Gut Microbiota Through Rectal Swab in Patients with Different Severity of Acute Pancreatitis. Dig. Dis. Sci. 2020, 65, 3223–3237. [Google Scholar] [CrossRef]
- Zhu, Y.; He, C.; Li, X.; Cai, Y.; Hu, J.; Liao, Y.; Zhao, J.; Xia, L.; He, W.; Liu, L.; et al. Gut microbiota dysbiosis worsens the severity of acute pancreatitis in patients and mice. J. Gastroenterol. 2019, 54, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, L.; Liu, S.; Hu, S.; Wu, L.; Liu, Y.; Yang, M.; Huang, S.; Tang, X.; Tang, T.; et al. Parabacteroides produces acetate to alleviate heparanase-exacerbated acute pancreatitis through reducing neutrophil infiltration. Microbiome 2021, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Párniczky, A.; Lantos, T.; Tóth, E.M.; Szakács, Z.; Gódi, S.; Hágendorn, R.; Illés, D.; Koncz, B.; Márta, K.; Mikó, A.; et al. Antibiotic therapy in acute pancreatitis: From global overuse to evidence based recommendations. Pancreatology 2019, 19, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Deviere, J. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013, 13, e1–e15. [Google Scholar] [CrossRef]
- De Waele, J.J. Rational Use of Antimicrobials in Patients with Severe Acute Pancreatitis. Semin. Respir. Crit. Care Med. 2011, 32, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenner, S.; Baillie, J.; DeWitt, J.; Vege, S.S. American College of Gastroenterology Guideline: Management of Acute Pancreatitis. Am. J. Gastroenterol. 2013, 108, 1400–1415. [Google Scholar] [CrossRef]
- Tran, A.; Fernando, S.M.; Rochwerg, B.; Inaba, K.; Bertens, K.A.; Engels, P.T.; Balaa, F.K.; Kubelik, D.; Matar, M.; Lenet, T.I.; et al. Prognostic factors associated with development of infected necrosis in patients with acute necrotizing or severe pancreatitis—A systematic review and meta-analysis. J. Trauma: Inj. Infect. Crit. Care 2022, 92, 940–948. [Google Scholar] [CrossRef]
- Li, W.; Ou, L.; Fu, Y.; Chen, Y.; Yin, Q.; Song, H. Risk factors for concomitant infectious pancreatic necrosis in patients with severe acute pancreatitis: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101901. [Google Scholar] [CrossRef]
- Barrie, J.; Jamdar, S.; Smith, N.; McPherson, S.J.; Siriwardena, A.K.; O’Reilly, D.A. Mis-use of antibiotics in acute pancreatitis: Insights from the United Kingdom’s National Confidential Enquiry into patient outcome and death (NCEPOD) survey of acute pancreatitis. Pancreatology 2018, 18, 721–726. [Google Scholar] [CrossRef]
- Baltatzis, M.; Mason, J.; Chandrabalan, V.; Stathakis, P.; McIntyre, B.; Jegatheeswaran, S.; Jamdar, S.; O’Reilly, D.A.; Siriwardena, A.K. Antibiotic use in acute pancreatitis: An audit of current practice in a tertiary centre. Pancreatology 2016, 16, 946–951. [Google Scholar] [CrossRef]
- Siriwardena, A.K.; Jegatheeswaran, S.; Mason, J.M.; Baltatzis, M.; Sheen, A.J.; O’Reilly, D.A.; Jamdar, S.; Deshpande, R.; Carino, N.D.L.; Satyadas, T.; et al. A procalcitonin-based algorithm to guide antibiotic use in patients with acute pancreatitis (PROCAP): A single-centre, patient-blinded, randomised controlled trial. Lancet Gastroenterol. Hepatol. 2022, 7, 913–921. [Google Scholar] [CrossRef]
- Reuken, P.A.; Albig, H.; Rödel, J.; Hocke, M.; Will, U.; Stallmach, A.; Bruns, T. Fungal Infections in Patients With Infected Pancreatic Necrosis and Pseudocysts. Pancreas 2018, 47, 92–98. [Google Scholar] [CrossRef]
- Büchler, M.; Malfertheiner, P.; Frieβ, H.; Isenmann, R.; Vanek, E.; Grimm, H.; Schlegel, P.; Friess, T.; Beger, H.G. Human pancreatic tissue concentration of bactericidal antibiotics. Gastroenterology 1992, 103, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Otto, W.; Komorzycki, K.; Krawczyk, M. Efficacy of antibiotic penetration into pancreatic necrosis. HPB 2006, 8, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, S.; Dalhoff, A. Activity of Moxifloxacin, Imipenem, and Ertapenem against Escherichia coli, Enterobacter cloacae, Enterococcus faecalis, and Bacteroides fragilis in Monocultures and Mixed Cultures in an In Vitro Pharmacokinetic/Pharmacodynamic Model Simulating Concentrations in the Human Pancreas. Antimicrob. Agents Chemother. 2012, 56, 6434–6436. [Google Scholar] [CrossRef] [Green Version]
- Leppäniemi, A.; Tolonen, M.; Tarasconi, A.; Segovia-Lohse, H.; Gamberini, E.; Kirkpatrick, A.W.; Ball, C.G.; Parry, N.; Sartelli, M.; Wolbrink, D.; et al. 2019 WSES guidelines for the management of severe acute pancreatitis. World J. Emerg. Surg. 2019, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Boxhoorn, L.; van Dijk, S.M.; van Grinsven, J.; Verdonk, R.C.; Boermeester, M.A.; Bollen, T.L.; Bouwense, S.A.; Bruno, M.J.; Cappendijk, V.C.; Dejong, C.H.; et al. Immediate versus Postponed Intervention for Infected Necrotizing Pancreatitis. N. Engl. J. Med. 2021, 385, 1372–1381. [Google Scholar] [CrossRef]
- Beger, H.; Rau, B.; Isenmann, R.; Schwarz, M.; Gansauge, F.; Poch, B. Antibiotic prophylaxis in severe acute pancreatitis. Pancreatology 2005, 5, 10–19. [Google Scholar] [CrossRef]
- Wittau, M.; Mayer, B.; Scheele, J.; Henne-Bruns, D.; Dellinger, E.P.; Isenmann, R. Systematic review and meta-analysis of antibiotic prophylaxis in severe acute pancreatitis. Scand. J. Gastroenterol. 2011, 46, 261–270. [Google Scholar] [CrossRef]
- Lim, C.L.L.; Lee, W.; Liew, Y.X.; Tang, S.S.L.; Chlebicki, M.P.; Kwa, A.L.-H. Role of Antibiotic Prophylaxis in Necrotizing Pancreatitis: A Meta-Analysis. J. Gastrointest. Surg. 2015, 19, 480–491. [Google Scholar] [CrossRef]
- Guo, D.; Dai, W.; Shen, J.; Zhang, M.; Shi, Y.; Jiang, K.; Guo, L. Assessment of Prophylactic Carbapenem Antibiotics Administration for Severe Acute Pancreatitis: An Updated Systematic Review and Meta-Analysis. Digestion 2022, 103, 183–191. [Google Scholar] [CrossRef]
- Nakaharai, K.; Morita, K.; Jo, T.; Matsui, H.; Fushimi, K.; Yasunaga, H. Early prophylactic antibiotics for severe acute pancreatitis: A population-based cohort study using a nationwide database in Japan. J. Infect. Chemother. 2018, 24, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Wani, S.; Gardner, T.B.; Falck-Ytter, Y.; Barkun, A.N.; Feuerstein, J.; Flamm, S.; Gellad, Z.; Gerson, L.; Gupta, S.; et al. American Gastroenterological Association Institute Guideline on Initial Management of Acute Pancreatitis. Gastroenterology 2018, 154, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Baltatzis, M.; Jegatheeswaran, S.; O’Reilly, D.A.; Siriwardena, A.K. Antibiotic use in acute pancreatitis: Global overview of compliance with international guidelines. Pancreatology 2016, 16, 189–193. [Google Scholar] [CrossRef]
- Powell, J.J.; Campbell, E.; Johnson, C.D.; Siriwardena, A.K. Survey of antibiotic prophylaxis in acute pancreatitis in the UK and Ireland. Br. J. Surg. 1999, 86, 320–322. [Google Scholar] [CrossRef] [PubMed]
- King, N.K.K.; Siriwardena, A.K. European Survey of Surgical Strategies for the Management of Severe Acute Pancreatitis. Am. J. Gastroenterol. 2004, 99, 719–728. [Google Scholar] [CrossRef]
- Sun, E.; Tharakan, M.; Kapoor, S.; Chakravarty, R.; Salhab, A.; Buscaglia, J.M.; Nagula, S. Poor Compliance with ACG Guidelines for Nutrition and Antibiotics in the Management of Acute Pancreatitis: A North American Survey of Gastrointestinal Specialists and Primary Care Physicians. JOP J. Pancreas 2013, 14, 221–227. [Google Scholar] [CrossRef]
- Sekimoto, M.; Shikata, S.; Takada, T.; Hirata, K.; Yoshida, M.; Hirota, M.; Kitamura, N.; Shirai, K.; Kimura, Y.; Wada, K.; et al. Changes in management of acute pancreatitis before and after the publication of evidence-based practice guidelines in 2003. J. Hepato-Biliary-Pancreat. Sci. 2010, 17, 17–23. [Google Scholar] [CrossRef]
- Mowbray, N.G.; Ben-Ismaeil, B.; Hammoda, M.; Shingler, G.; Al-Sarireh, B. The microbiology of infected pancreatic necrosis. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 456–460. [Google Scholar] [CrossRef]
- Vege, S.S.; DiMagno, M.J.; Forsmark, C.E.; Martel, M.; Barkun, A.N. Initial Medical Treatment of Acute Pancreatitis: American Gastroenterological Association Institute Technical Review. Gastroenterology 2018, 154, 1103–1139. [Google Scholar] [CrossRef] [Green Version]
- Besselink, M.G.; Van Santvoort, H.C.; Buskens, E.; Boermeester, M.A.; Van Goor, H.; Timmerman, H.M.; Nieuwenhuijs, V.B.; Bollen, T.L.; van Ramshorst, B.; Witterman, B.J.; et al. Probiotic prophylaxis in predicted severe acute pancreatitis: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 371, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Solomkin, J.S.; Mazuski, J.E.; Bradley, J.S.; Rodvold, K.A.; Goldstein, E.J.; Baron, E.J.; O’Neill, P.J.; Chow, A.W.; Dellinger, E.P.; Eachempati, S.R.; et al. Diagnosis and Management of Complicated Intra-Abdominal Infection in Adults and Children: Guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg. Infect. 2010, 11, 79–109. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Okamoto, K.; Takada, T.; Strasberg, S.M.; Asbun, H.J.; Pitt, H.A.; Gomi, H.; Solomkin, J.; Schlossberg, D.; Han, H.-S.; et al. Tokyo Guidelines 2018: Initial management of acute biliary infection and flowchart for acute cholangitis. J. Hepato-Biliary-Pancreat. Sci. 2018, 25, 31–40. [Google Scholar] [CrossRef]
- Floreani, A.; De Martin, S. Treatment of primary sclerosing cholangitis. Dig. Liver Dis. 2021, 53, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Maurice, J.B.; Thorburn, D. Guideline review: British Society of Gastroenterology/UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Front. Gastroenterol. 2021, 12, 62–66. [Google Scholar] [CrossRef]
- Chapman, M.H.; Thorburn, D.; Hirschfield, G.M.; Webster, G.G.J.; Rushbrook, S.M.; Alexander, G.; Collier, J.; Dyson, J.K.; Jones, D.E.; Patanwala, I.; et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 2019, 68, 1356–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Gong, W.; Jiang, B. Antibiotic prophylaxis for GI endoscopy. Gastrointest. Endosc. 2015, 81, 1503–1504. [Google Scholar] [CrossRef]
- Byl, B.; Devière, J.; Struelens, M.J.; Roucloux, I.; De Coninck, A.; Thys, J.-P.; Cremer, M. Antibiotic Prophylaxis for Infectious Complications after Therapeutic Endoscopic Retrograde Cholangiopancreatography: A Randomized, Double-Blind, Placebo-Controlled Study. Clin. Infect. Dis. 1995, 20, 1236–1240. [Google Scholar] [CrossRef]
- Bangarulingam, S.Y.; Gossard, A.A.; Petersen, B.T.; Ott, B.J.; Lindor, K.D. Complications of Endoscopic Retrograde Cholangiopancreatography in Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2009, 104, 855–860. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Hov, J.R.; Karlsen, T.H. The microbiota and the gut–liver axis in primary sclerosing cholangitis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 135–154. [Google Scholar] [CrossRef]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Ponziani, F.R.; Nardella, E.; Ianiro, G.; Gasbarrini, A.; Zileri Dal Verme, L. Biliary Tract Microbiota: A New Kid on the Block of Liver Diseases? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2750–2775. [Google Scholar] [CrossRef] [PubMed]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, P.; Aho, V.; Arola, J.; Boyd, S.; Jokelainen, K.; Paulin, L.; Auvinen, P.; Färkkilä, M. Bile microbiota in primary sclerosing cholangitis: Impact on disease progression and development of biliary dysplasia. PLoS ONE 2017, 12, e0182924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabibian, J.H.; Gossard, A.; El-Youssef, M.; Eaton, J.E.; Petz, J.; Jorgensen, R.; Enders, F.B.; Tabibian, A.; Lindor, K.D. Prospective Clinical Trial of Rifaximin Therapy for Patients With Primary Sclerosing Cholangitis. Am. J. Ther. 2017, 24, e56–e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, M.G.; Torok, N.J.; Gossard, A.A.; Keach, J.C.; Jorgensen, R.R.A.; Petz, R.J.L.; Lindor, K.D. Minocycline in the Treatment of Patients With Primary Sclerosing Cholangitis: Results of a Pilot Study. Am. J. Gastroenterol. 2009, 104, 83–88. [Google Scholar] [CrossRef]
- Färkkilä, M.; Karvonen, A.-L.; Nurmi, H.; Nuutinen, H.; Taavitsainen, M.; Pikkarainen, P.; Kärkkäinen, P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: A randomized placebo-controlled trial. Hepatology 2004, 40, 1379–1386. [Google Scholar] [CrossRef] [Green Version]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Daryani, N.E.; Taromlou, M.K.N.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointest. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef]
- Ali, A.H.; Damman, J.; Shah, S.B.; Davies, Y.; Hurwitz, M.; Stephen, M.; Lemos, L.M.; Carey, E.J.; Lindor, K.D.; Buness, C.W.; et al. Open-label prospective therapeutic clinical trials: Oral vancomycin in children and adults with primary sclerosing cholangitis. Scand. J. Gastroenterol. 2020, 55, 941–950. [Google Scholar] [CrossRef]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of Antibiotic Therapy in Primary Sclerosing Cholangitis with and without Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.R.; Mack, C.; Mogul, D.; Perito, E.R.; Valentino, P.L.; Amir, A.Z.; DiGuglielmo, M.; Draijer, L.G.; El-Matary, W.; Furuya, K.N.; et al. Oral Vancomycin, Ursodeoxycholic Acid, or No Therapy for Pediatric Primary Sclerosing Cholangitis: A Matched Analysis. Hepatology 2021, 73, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Davies, Y.K.; Cox, K.M.; Abdullah, B.A.; Safta, A.; Terry, A.B.; Cox, K.L. Long-term Treatment of Primary Sclerosing Cholangitis in Children With Oral Vancomycin: An Immunomodulating Antibiotic. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef]
- Cazzagon, N.; Floreani, A. Primary biliary cholangitis: Treatment. Curr. Opin. Gastroenterol. 2021, 37, 99–104. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef] [Green Version]
- European Association for the Study of the Liver. European Association for the Stud EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [Google Scholar] [CrossRef]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Pathogen infections and primary biliary cholangitis. Clin. Exp. Immunol. 2018, 195, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanos, D.P.; Vergani, D. Bacteria and Primary Biliary Cirrhosis. Clin. Rev. Allergy Immunol. 2009, 36, 30–39. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Ansari, A.A.; Mackay, I.R.; Nakanuma, Y.; Nishio, A.; Rowley, M.J.; Coppel, R.L. Primary biliary cirrhosis: An orchestrated immune response against epithelial cells. Immunol. Rev. 2000, 174, 210–225. [Google Scholar] [CrossRef]
- Kaplan, M.M. Primary Biliary Cirrhosis. N. Engl. J. Med. 1996, 335, 1570–1580. [Google Scholar] [CrossRef]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Van De Water, J.; Nantz, M.H.; et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef]
- Butler, P.; Valle, F.; Hamilton-Miller, J.M.; Brumfitt, W.; Baum, H.; Burroughs, A.K. M2 mitochondrial antibodies and urinary rough mutant bacteria in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J. Hepatol. 1993, 17, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Baum, H.; Vergani, D.; Burroughs, A.K. The Role of E. coli Infection in the Pathogenesis of Primary Biliary Cirrhosis. Dis. Markers 2010, 29, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Smyk, D.; Rigopoulou, E.I.; Zen, Y.; Abeles, R.D.; Billinis, C.; Parés, A.; Bogdanos, D.P. Role for mycobacterial infection in pathogenesis of primary biliary cirrhosis? World J. Gastroenterol. 2012, 18, 4855–4865. [Google Scholar] [CrossRef] [PubMed]
- Abdulkarim, A.S.; Petrovic, L.M.; Kim, W.R.; Angulo, P.; Lloyd, R.V.; Lindor, K.D. Primary biliary cirrhosis: An infectious disease caused by Chlamydia pneumoniae? J. Hepatol. 2004, 40, 380–384. [Google Scholar] [CrossRef]
- Floreani, A.; Biagini, M.R.; Zappalà, F.; Farinati, F.; Plebani, M.; Rugge, M.; Surrenti, C.; Naccarato, R. Chronic Atrophic Gastritis and Helicobacter Pylori Infection in Primary Biliary Cirrhosis: A Cross-Sectional Study with Match-ing. Ital. J. Gastroenterol. Hepatol. 1997, 29, 13–17. [Google Scholar] [PubMed]
- Shapira, Y.; Agmon-Levin, N.; Renaudineau, Y.; Porat-Katz, B.S.; Barzilai, O.; Ram, M.; Youinou, P.; Shoenfeld, Y. Serum markers of infections in patients with primary biliary cirrhosis: Evidence of infection burden. Exp. Mol. Pathol. 2012, 93, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergasa, N.V. Pruritus of Cholestasis. In Itch: Mechanisms and Treatment; Carstens, E., Akiyama, T., Eds.; Frontiers in Neuroscience; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Boca Raton, FL, USA, 2014; ISBN 978-1-4665-0543-8. [Google Scholar]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Heinsen, F.A.; Knecht, H.; Neulinger, S.C.; Schmitz, R.A.; Knecht, C.; Kühbacher, T.; Rosenstiel, P.C.; Schreiber, S.; Friedrichs, A.K.; Ott, S.J. Dynamic changes of the luminal and mucosa-associated gut microbiota during and after antibiotic therapy with paromomycin. Gut Microbes 2015, 6, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.; Guarner, F.; Fernandez, L.B.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; Van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodríguez-Piñeiro, A.M.; Schütte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Bäckhed, F.; Hansson, G.C.; Johansson, M.E.V. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Ianiro, G.; Mullish, B.H.; Kelly, C.R.; Kassam, Z.; Kuijper, E.J.; Ng, S.C.; Iqbal, T.H.; Allegretti, J.R.; Bibbò, S.; Sokol, H.; et al. Reorganisation of faecal microbiota transplant services during the COVID-19 pandemic. Gut 2020, 69, 1555–1563. [Google Scholar] [CrossRef]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.; Kumar, S.; Kapoor, R.K.; Gulati, P. Integrons and antibiotic resistance genes in water-borne pathogens: Threat detection and risk assessment. J. Med. Microbiol. 2019, 68, 679–692. [Google Scholar] [CrossRef]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef] [Green Version]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stripling, J.; Kumar, R.; Baddley, J.W.; Nellore, A.; Dixon, P.; Howard, D.; Ptacek, T.; Lefkowitz, E.J.; Tallaj, J.A.; Benjamin, W.H.; et al. Loss of Vancomycin-Resistant Enterococcus Fecal Dominance in an Organ Transplant Patient With Clostridium difficile Colitis After Fecal Microbiota Transplant. Open Forum Infect. Dis. 2015, 2, ofv078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieërs, G.; Belkhir, L.; Enaud, R.; Leclercq, S.; de Foy, J.-M.P.; Dequenne, I.; de Timary, P.; Cani, P.D. How Probiotics Affect the Microbiota. Front. Cell. Infect. Microbiol. 2020, 9, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Luo, J.; Narbad, A.; Chen, Q. Advances in Lactobacillus Restoration for β-Lactam Antibiotic-Induced Dysbiosis: A System Review in Intestinal Microbiota and Immune Homeostasis. Microorganisms 2023, 11, 179. [Google Scholar] [CrossRef]
- Satokari, R. Modulation of Gut Microbiota for Health by Current and Next-Generation Probiotics. Nutrients 2019, 11, 1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| HEPATIC ENCEPHALOPATHY | SPONTANEOUS BACTERIAL PERITONITIS |
|---|---|
Rifaximin 1200 mg daily
| Prophylaxis
|
| AUTHORS OF THE STUDY | STUDY DESIGN | STUDY POPULATION | AIM OF THE STUDY | PRIMARY ENDPOINTS | SECONDARY ENDPOINTS | RESULTS | |
|---|---|---|---|---|---|---|---|
| Glal et al. (2021) [57] | Randomized double-blind controlled clinical trial | 60 cirrhotic patients | Efficacy and safety of 550 mg twice daily of rifaximin or 500 mg twice daily of Nitazoxanide for 24 weeks | Duration of remission, number of recurrent episodes, evaluation of HE-related clinical symptoms, serum levels of ammonia, TNF-α and octopamine and calculation of Chronic Liver Disease Questionnaire score | NA | Nitazoxanide (faced against rifaximin):
| |
| Bajaj et al. (2013) [48] | Interventional pilot study | 20 cirrhotic patients with MHE | Analysis of the microbiome, metabolome and cognitive improvement after rifaximin | Evaluate the effect of rifaximin on the metabiome (determined by the interaction between phenome, microbiome and metabolome) | NA | Significant improvement in cognition (6/7 tests improved, p < 0.01) and endotoxemia (0.55 to 0.48 Eu/mL, p = 0.02). Increase of serum saturated and unsaturated fatty acids. No significant microbial changes were observed after rifaximin, apart from a modest decrease in Veillonellaceae and increase in Eubacteriaceae. | |
| Bass et al. (2010) [61] | Randomized, double-blind, placebo-controlled trial | 299 cirrhotic patients in remission from recurrent HE | Efficacy and safety of 550 mg twice daily for 6 months in the prevention of HE | Time to the first breakthrough episode of HE | Time to the first hospitalization due to HE | In the rifaximin group:
| |
| Eltawil et al. (2012) [62] | Systematic review of 12 studies | 565 cirrhotic patients | Efficacy of rifaximin in the management of HE | Efficacy and safety of rifaximin for the treatment of patients with at least one episode of HE | Reduction of serum ammonia levels and changes in psychometric parameters (mental status, asterixis, electroencephalographic characteristics and HE sum) after treatment | Rifaximin group:
| |
| Patel et al. (2022) [63] | 2 Meta-analysis of 5 studies | (1) 555 cirrhotic patients (2) 784 cirrhotic patients | Safety and efficacy of rifaximin (1) over systemic antibiotics (oral quinolones) for SBP prevention (2) over placebo for SBP prevention | (1) Comparing rifaximin to systemic antibiotics for the prevention of SBP (2) Comparing rifaximin to placebo for the prevention of SBP | (1) Subgroup analysis comparing rifaximin to systemic antibiotics for primary and secondary SBP prophylaxis (2) Subgroup analysis comparing rifaximin to placebo for primary and secondary SBP prophylaxis | (1) Rifaximin: significantly more protective from SBP that systemic antibiotics (OR 0.38, 95% CI: 0.19–0.76, p = 0.01). OR for primary prophylaxis was 0.59 (95% CI: 0.32–1.09; p = 0.10). OR for secondary prophylaxis was 0.46 (95% CI: 0.09–2.29; p = 0.34). (2) OR for the development of SBP was significantly lower in patients receiving rifaximin compared to no antibiotics at 0.34 (95% CI: 0.11–0.99; p < 0.05). OR for primary prophylaxis was 0.53 (95% CI: 0.28–0.99; p = 0.05) in favour of rifaximin. | |
| Zuo et al. (2017) [64] | Open-label study | 14 cirrhotic patients with MHE | Efficacy of rifaximin in restoring the gut microbiota of patients with MHE | To restore the gut microbiota towards the normal composition and functions | NA | After rifaximin:
| Decrease in the abundance of Firmicutes (more apparent in non-alcoholic patients). Increase in 7 out of 14 patients of Proteobacteria. The remaining half showed unaltered or decreased abundance of Proteobacteria. |
| Kaji et al. (2017) [66] | Open-label study | 20 patients with decompensated cirrhosis | Efficacy and safety of rifaximin 400 mg thrice a day for hepatic encephalopathy with the linkage of gut microbiome | To determine the efficacy of rifaximin for HE, evaluated with serum ammonia level, number connection test (NCT) and endotoxin activity | Effect of rifaximin on the gut microbiome | Rifaximin improves hyperammonia and cognitive impairment, with decreased endotoxin activity | Rifaximin did not alter the diversity and major components of gut microbiome, although the relative abundances of genus Veillonella and Streptococcus were lowered. |
| Kaji et al. (2020) [67] | Observational study | 30 patients with decompensated cirrhosis | Efficacy of rifaximin 1200 mg/daily on intestinal permeability and gut microbiota | MHE symptoms and serum ammonia levels after 4-week rifaximin | Assessment of gut permeability with soluble CD163, soluble mannose receptor (sMR) and zonulin, after 4-week treatment with rifaximin. Assessment of gut microbiota with 16S rRNA gene sequencing, and serum pro-inflammatory cytokines after 4-week rifaximin treatment. | Improvement of MHE and lowering of mean serum ammonia levels (101.9 ± 30.9 µg/dL at baseline vs. 63.3 ± 19.4 µg/dL at RFX; p < 0.01). Serum levels of both sCD163 and sMR were markedly decreased by 4-week rifaximin treatment, while serum zonulin levels were unchanged. | No statistically significant differences in the richness (Chao1 index) (105.0 ± 38.5 at baseline vs. 92.1 ± 26.1 at RFX; p = 0.662) and complexity (Shannon index) (3.857 ± 0.642 at baseline vs. 3.727 ± 0.591; p = 0.776). 90 genera (58 Veillonella decreased significantly after rifaximin (p = 0.031) while the other genera unchanged. Rifaximin did not affect serum levels of TNF-α, IL-6, IFN-γ, and IL-10. |
| AUTHORS OF THE STUDY | STUDY DESIGN | STUDY POPULATION | AIM OF THE STUDY | PRIMARY ENDPOINTS | SECONDARY ENDPOINTS | RESULTS | |
|---|---|---|---|---|---|---|---|
| Kalambokis et al. (2012) [88] | 4-weeks open-label, placebo-controlled, pilot study | 16 cirrhotic patients with ascites and no history of SBP (CPS C) | Efficacy and safety of oral rifaximin 1200 mg daily | WBC, neutrophils and endoxotin levels in ascitic fluis at baseline and 4 weeks | Cytology of ascitic fluid and plasma endotoxin level at baseline and 4 weeks | Rifaximin group: WBC count (−40.00 from baseline, p = 0.004) Neutrophil count (−14,9 from baseline, p = 0.01) Plasma endotoxin (−1.7 from baseline, p = 0.03) | Placebo: WBC count (+11.00 from baseline p = NS) Neutrophil count (+3.3 from baseline p = NS) Plasma endotoxin (+0.1 from baseline p = NS) |
| Dănulescu et al. (2013) [89] | 6 months case—control study | 46 cirrhotic patients with refractory ascites (CPS C) | Safety and efficacy of rifaximin 1200 mg orally daily for SBP prophylaxis | Development of SBP within 6 months | Polymorpho-nucleates (PMN) count in ascitic fluid at 6 months | Rifaximin: One patient developed SBP A significant decreased of PMN was detected in ascitic fluid of 21 of 22 patients | Placebo: SBP was diagnosed in 4 patients An increase of PMN was detected in ascitic fluids of 14 patients |
| Hanouneh et al. (2012) [90] | Retrospective study | 404 cirrhotic patients with ascites | Determine if rifaximin decreases the risk of SBP and improves transplant-free survival in cirrhotic patients with ascites. | Incidence of SBP during follow-up | Transplant-free survival rate | Rifaximin group: incidence rate of SBP was 0.09 per person-year 89% remained SBP free at 4.2 months with 72% SBP reduction in the rifaximin group (hazard ratio = 0.28; 95% confidence interval, 0.11–0.71; p = 0.007) Only 28.6% of patients expired, p = 0.045 | Non-rifaximin group: incidence rate of SBP was 0.4 per person-year 68% remained SBP free at 4.2 months 43.7% of patients expired |
| Mostafa et al. (2015) [91] | 6 months single blinded, randomized, case–control trial | 70 cirrhotic patients with ascites | Safety and efficacy of rifaximin over Norfloxacin for the prevention of SBP | SBP rate after 3 months of therapy | Serum levels of Tumor Necrosis Factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-10 (IL-10) | Rifaximin: No cases of SBP at 3 month Reduced levels of TNF-α and IL-6 (p < 0.05) Increased levels of IL-10 (p < 0.05) | Norfloxacin: Five cases of SBP at 3 months Reduced levels of TNF-α and IL-6 (p < 0.05) Increased levels of IL-10 (p < 0.05) |
| Sidhu et al. (2017) [92] | Systematic review of 5 studies | (1) 70 (2) 86 (3) 334 (4) 262 | Efficacy of rifaximin versus Norfloxacin for the prevention of SBP occurrence/recurrence | Comparing rifaximin with Norfloxacin for SBP prevention of occurrence/recurrence | Mortality benefit with rifaximin as compared to norfloxacin Safety profile of rifaximin as compared to norfloxacin | All studies showed a reduced or equal incidence of SBP in the rifaximin group compared to norfloxacin group, although not always statistically significant | All studies showed a reduced mortality rate in the rifaximin group, although not always statistically significant. No serious adverse events were reported in any of thestudies with either of the drugs. Minor adverse events with similar with the 2 drugs. |
| Goel et al. (2017) [93] | 2 meta-analyses of 5 studies | (1) 555 cirrhotic patients (2) 784 cirrhotic patients | Safety and efficacy of: (1) rifaximin over systemic antibiotics (oral quinolones) for the prevention of SBP (2) rifaximin over placebo for the prevention of SBP | (1) Comparing rifaximin to systemic antibiotics for prevention of SBP (2) Comparing rifaximin to placebo for prevention of SBP | (1) Subgroup analysis comparing rifaximin to systemic antibiotics for primary and secondary SBP prophylaxis (2) Subgroup analysis comparing rifaximin to placebo for primary and secondary SBP prophylaxis | Rifaximin was significantly more protective for SBP that systemic antibiotics (OR 0.38, 95% CI 0.19–0.76, p = 0.01). OR for primary prophylaxis was 0.59 (95% CI: 0.32–1.09; p = 0.10). OR for secondary prophylaxis was 0.46 (95% CI: 0.09–2.29; p = 0.34). | OR for the development of SBP was significantly lower in patients receiving rifaximin compared to no antibiotics at 0.34 (95% CI: 0.11–0.99; p < 0.05). OR for primary prophylaxis was 0.53 (95% CI: 0.28–0.99; p = 0.05) in favour of rifaximin. |
| Faust et al. (2020) [94] | Meta-analysis of 13 studies | 1742 cirrhotic patients | Safety and efficacy of norfloxacin, ciprofloxacin, rifaximin, trimethoprim-sulphamethoxazole over plabebo for the prevention of SBP | Proportion of patients who developed SBP. Diagnosis was baased on a combination of clinical characteristics (fever and abdominal pain), cytologic criteria and ascitic fluid cultures | Risk of death or liver transplantation | All antibiotics were superior to placebo for secondary SBP prophylaxis with this rank: (1) rifaximin, (2) ciprofloxacin, (3) TMP-SMX, (4) norfloxacin and (5) placebo | The rank probability for efficacy of risk reduction of death, in ascending order, is (1) rifaximin, (2) ciprofloxacin, (3) norfloxacin, (4) TMP-SMX and (5) placebo. |
| Menshawy et al. (2019) [95] | Meta-analysis of 6 studies | 973 cirrothic patients | Comparing safety and efficacy of rifaximin and norfloxacin over norfloxacin alone in the prevention of SBP | Prevention of SBP | Mortality rate, hepatorenal syndrome, septic shock, hepatic encephalopaty and GIT bleeding | Rifaximin and norfloxacin group had less incidence of SBP (RR 0.58, 95% CI [0.37, 0.92], p = 0.02) and hepatic encephalopathy (RR 0.38, 95% CI [0.17, 0.84], p = 0.02) compared to the norfloxacin group. No significant difference between rifaximin and norfloxacin in terms of frequency of SBP and success rate of primary prevention of SBP (RR 0.49, 95% CI [0.24, 1.01], p = 0.05; RR1.21, 95% CI [0.95, 1.55], p = 0.13, respectively). | |
| Assem et al. (2016) [96] | 6 months open-label randomized case–control study | 239 chirrotic patients with high SAAG (>1.1) ascites (CPS > 9) | Comparing safety and efficacy of norfloxacin and rifaximin vs. norfloxacin or rifaximin alone as primary prophylaxis for SBP | Development of SBP within 6 months | Overall mortality, incidence of infectious events, hepatorenal syndrome, liver transplantation and adverse event of drugs | Alternating norfloxacin and rifaximin determined lower probability to develop SBP in intention-to-treat (p = 0.016) and per protocol analysis (p = 0.039). No significant differences regarding the incidence or severity of adverse events and the incidence of HRS. | |
 WHEN TO PRESCRIBE WHEN TO PRESCRIBE |
|
 WHEN NOT TO PRESCRIBE WHEN NOT TO PRESCRIBE |
|
 GRAY ZONE GRAY ZONE |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Vincenzo, F.; Nicoletti, A.; Negri, M.; Vitale, F.; Zileri Dal Verme, L.; Gasbarrini, A.; Ponziani, F.R.; Cerrito, L. Gut Microbiota and Antibiotic Treatments for the Main Non-Oncologic Hepato-Biliary-Pancreatic Disorders. Antibiotics 2023, 12, 1068. https://doi.org/10.3390/antibiotics12061068
Di Vincenzo F, Nicoletti A, Negri M, Vitale F, Zileri Dal Verme L, Gasbarrini A, Ponziani FR, Cerrito L. Gut Microbiota and Antibiotic Treatments for the Main Non-Oncologic Hepato-Biliary-Pancreatic Disorders. Antibiotics. 2023; 12(6):1068. https://doi.org/10.3390/antibiotics12061068
Chicago/Turabian StyleDi Vincenzo, Federica, Alberto Nicoletti, Marcantonio Negri, Federica Vitale, Lorenzo Zileri Dal Verme, Antonio Gasbarrini, Francesca Romana Ponziani, and Lucia Cerrito. 2023. "Gut Microbiota and Antibiotic Treatments for the Main Non-Oncologic Hepato-Biliary-Pancreatic Disorders" Antibiotics 12, no. 6: 1068. https://doi.org/10.3390/antibiotics12061068