

Aurachins, Bacterial Antibiotics Interfering with Electron Transport Processes

Abstract
:1. Introduction
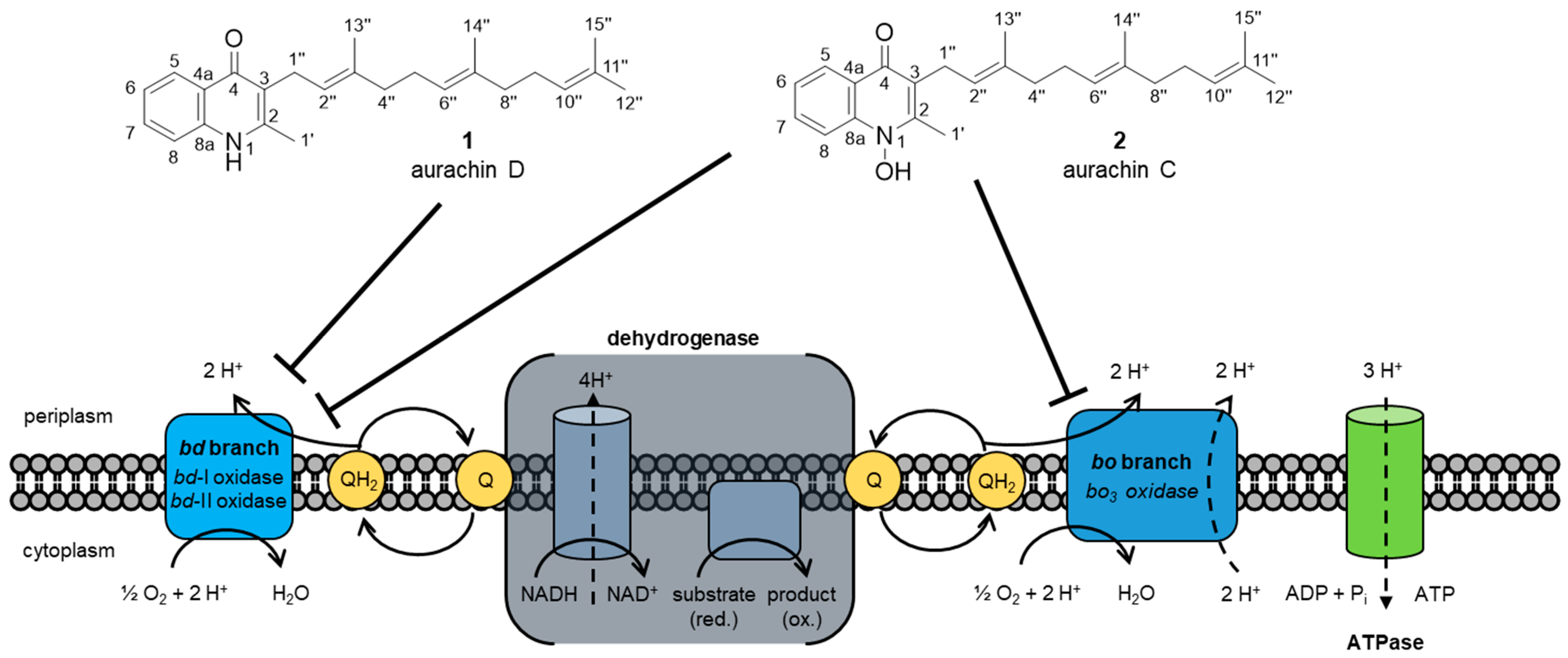
2. Biological Activities
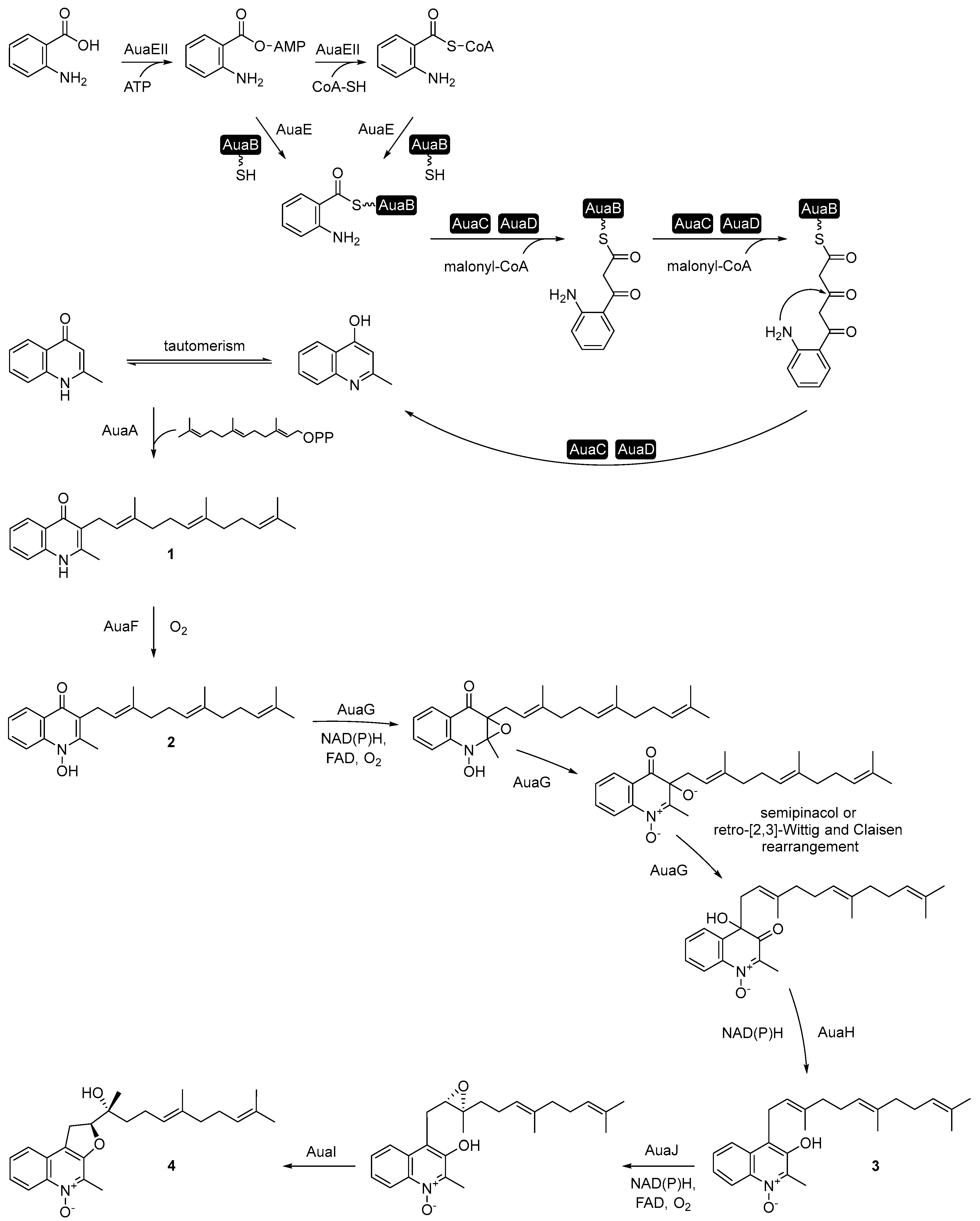
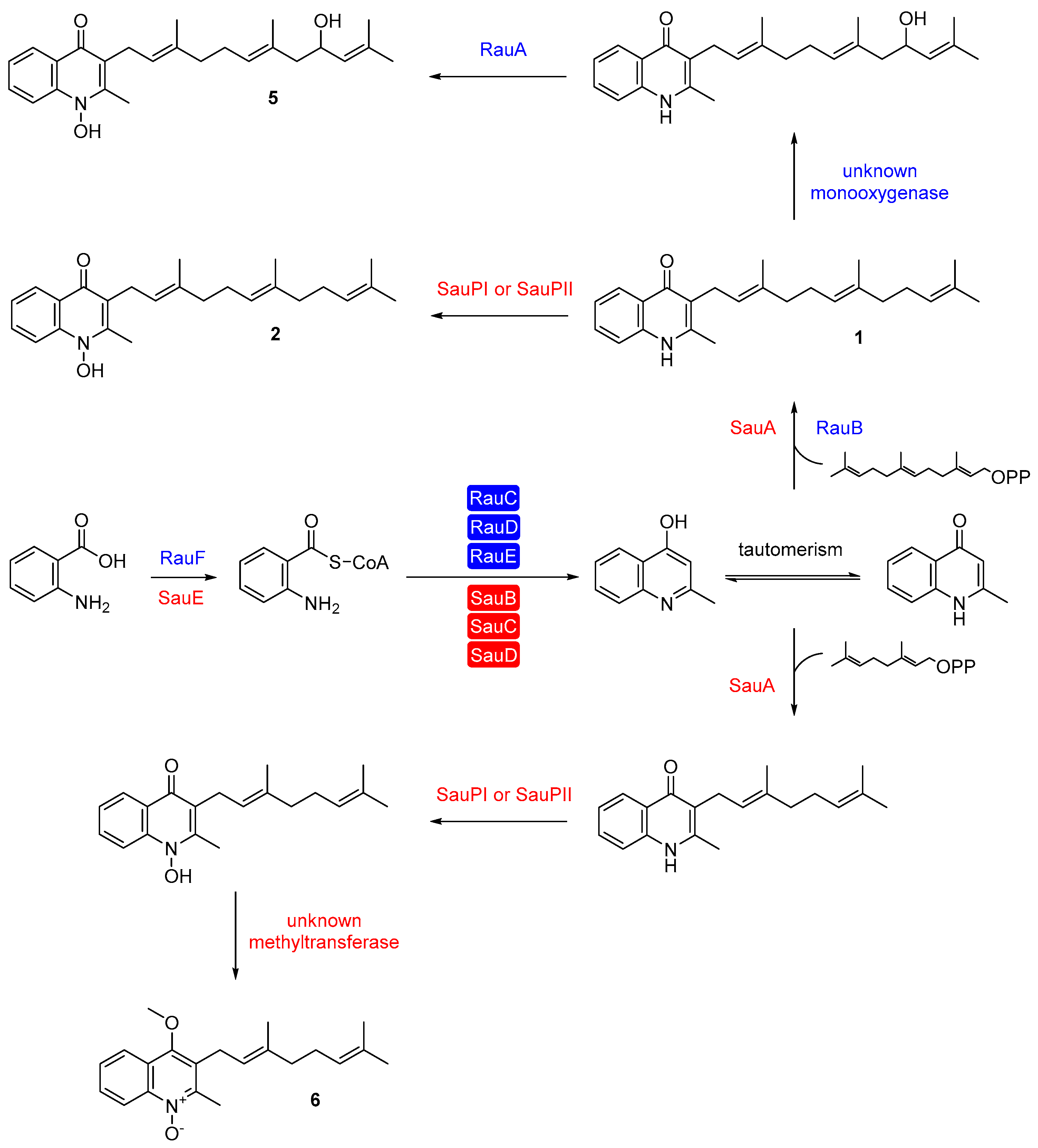
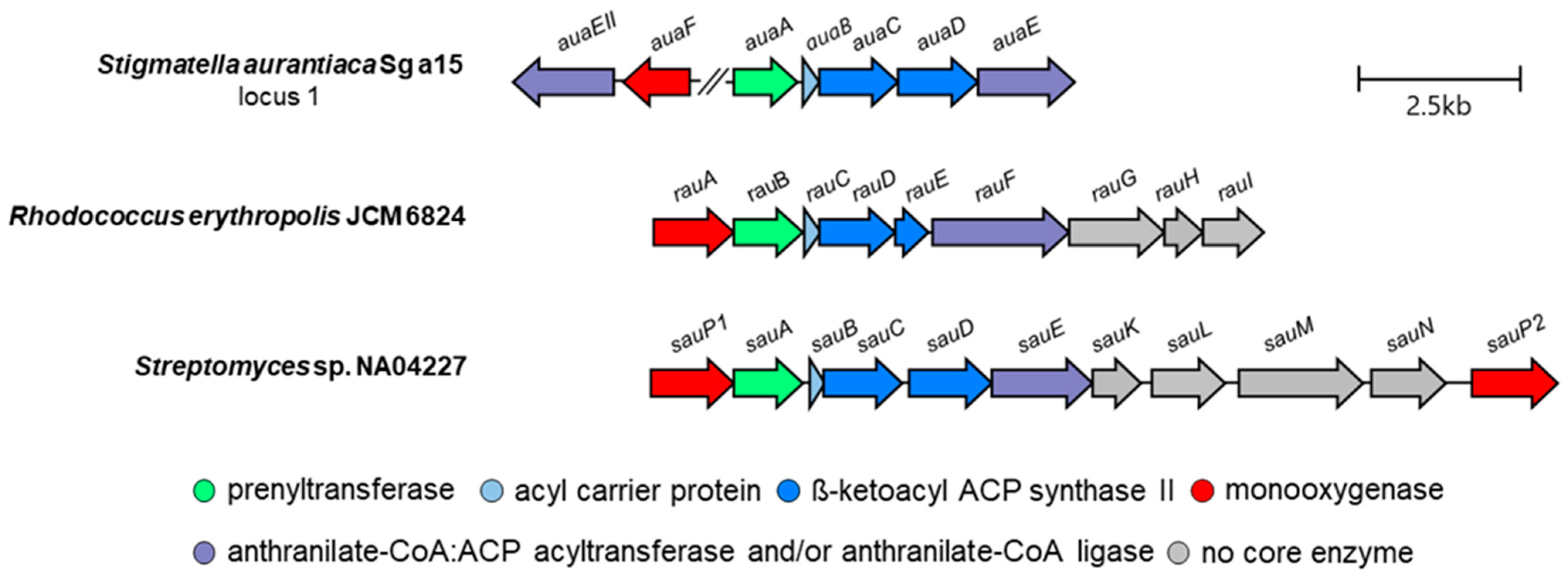
3. Aurachin Biosynthesis
4. Biotechnological Production and Derivatization of Aurachins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. Natural Products as a Source of Drug Leads to Overcome Drug Resistance. Future Microbiol. 2015, 10, 1711–1718. [Google Scholar] [CrossRef]
- Shang, X.F.; Morris-Natschke, S.L.; Liu, Y.Q.; Guo, X.; Xu, X.S.; Goto, M.; Li, J.C.; Yang, G.Z.; Lee, K.H. Biologically Active Quinoline and Quinazoline Alkaloids Part I. Med. Res. Rev. 2018, 38, 775–828. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.F.; Morris-Natschke, S.L.; Yang, G.Z.; Liu, Y.Q.; Guo, X.; Xu, X.S.; Goto, M.; Li, J.C.; Zhang, J.Y.; Lee, K.H. Biologically Active Quinoline and Quinazoline Alkaloids Part II. Med. Res. Rev. 2018, 38, 1614–1660. [Google Scholar] [CrossRef]
- Saalim, M.; Villegas-Moreno, J.; Clark, B.R. Bacterial Alkyl-4-Quinolones: Discovery, Structural Diversity and Biological Properties. Molecules 2020, 25, 5689. [Google Scholar] [CrossRef]
- Schaefer, B. Natural Products in the Chemical Industry; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Kunze, B.; Höfle, G.; Reichenbach, H. The Aurachins, New Quinoline Antibiotics from Myxobacteria: Production, Physico-Chemical and Biological Properties. J. Antibiot. 1987, 40, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Höfle, G.; Irschik, H. Isolation and Biosynthesis of Aurachin P and 5-Nitroresorcinol from Stigmatella erecta. J. Nat. Prod. 2008, 71, 1946–1948. [Google Scholar] [CrossRef]
- Kitagawa, W.; Tamura, T. A Quinoline Antibiotic from Rhodococcus erythropolis JCM 6824. J. Antibiot. 2008, 61, 680–682. [Google Scholar] [CrossRef]
- Nachtigall, J.; Schneider, K.; Nicholson, G.; Goodfellow, M.; Zinecker, H.; Imhoff, J.F.; Süssmuth, R.D.; Fiedler, H.P. Two New Aurachins from Rhodococcus sp. Acta 2259. J. Antibiot. 2010, 63, 567–569. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, C.L.; Xiao, Y.S.; Zhang, B.; Deng, X.Z.; Yang, L.; Shi, J.; Wang, Y.S.; Li, W.; Jiao, R.H.; et al. Aurachin SS, a New Antibiotic from Streptomyces sp. NA04227. J. Antibiot. 2017, 70, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. Semisynthesis and Antiplasmodial Activity of the Quinoline Alkaloid Aurachin E. J. Nat. Prod. 2008, 71, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Herrmann, J.; Zang, Y.; Grellier, P.; Prado, S.; Müller, R.; Nay, B. Synthesis and Biological Activities of the Respirator Chain Inhibitor Aurachin D and New Ring versus Chain Analogues. Beilstein J. Org. Chem. 2013, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruth, S.; Zimmermann, C.J.-M.; Kuhr, K.; Hiller, W.; Lütz, S.; Pietruszka, J.; Kaiser, M.; Nett, M. Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties. Molecules 2023, 28, 1066. [Google Scholar] [CrossRef] [PubMed]
- Oettmeier, W.; Dostatni, R.; Majewski, C.; Höfle, G.; Fecker, T.; Kunze, B.; Reichenbach, H. The Aurachins, Naturally Occurring Inhibitors of Photosynthetic Electron Flow through Photosystem II and the Cytochrome b6/f-Complex. Z. Naturforsch. C J. Biosci. 1990, 45, 322–328. [Google Scholar] [CrossRef]
- Hoefnagel, M.H.N.; Wiskich, J.T.; Madgwick, S.A.; Patterson, Z.; Oettmeier, W.; Rich, P.R. New Inhibitors of the Ubiquinol Oxidase of Higher Plant Mitochondria. Eur. J. Biochem. 1995, 233, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Dejon, L.; Speicher, A. Synthesis of Aurachin D and Isoprenoid Analogues from the Myxobacterium Stigmatella aurantiaca. Tetrahedron Lett. 2013, 54, 6700–6702. [Google Scholar] [CrossRef]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New Inhibitors of the Quinol Oxidation Sites of Bacterial Cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef]
- Miyoshi, H.; Takegami, K.; Sakamoto, K.; Mogi, T.; Iwamura, H. Characterization of the Ubiquinol Oxidation Sites in Cytochromes bo and bd from Escherichia coli Using Aurachin C Analogues. J. Biochem. 1999, 125, 138–142. [Google Scholar] [CrossRef]
- Grauel, A.; Kägi, J.; Rasmussen, T.; Makarchuk, I.; Oppermann, S.; Moumbock, A.F.A.; Wohlwend, D.; Müller, R.; Melin, F.; Günther, S.; et al. Structure of Escherichia coli Cytochrome bd-II Type Oxidase with Bound Aurachin D. Nat. Commun. 2021, 12, 6498. [Google Scholar] [CrossRef]
- Radloff, M.; Elamri, I.; Grund, T.N.; Witte, L.F.; Hohmann, K.F.; Nakagaki, S.; Goojani, H.G.; Nasiri, H.; Miyoshi, H.; Bald, D.; et al. Short-Chain Aurachin D Derivatives Are Selective Inhibitors of E. coli Cytochrome bd-I and bd-II Oxidases. Sci. Rep. 2021, 11, 23852. [Google Scholar] [CrossRef] [PubMed]
- Theßeling, A.; Burschel, S.; Wohlwend, D.; Friedrich, T. The Long Q-Loop of Escherichia coli Cytochrome bd Oxidase Is Required for Assembly and Structural Integrity. FEBS Lett. 2020, 594, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracke, F.; Vassilev, I.; Krömer, J.O. Microbial Electron Transport and Energy Conservation—The Foundation for Optimizing Bioelectrochemical Systems. Front. Microbiol. 2015, 6, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, T.; Wohlwend, D.; Borisov, V.B. Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. Int. J. Mol. Sci. 2022, 23, 3166. [Google Scholar] [CrossRef]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; Van Spanning, R.; Bald, D. The Cytochrome bd-Type Quinol Oxidase Is Important for Survival of Mycobacterium smegmatis under Peroxide and Antibiotic-Induced Stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [Green Version]
- Lawer, A.; Tyler, C.; Hards, K.; Keighley, L.M.; Cheung, C.-Y.Y.; Kierek, F.; Su, S.; Matikonda, S.S.; McInnes, T.; Tyndall, J.D.A.; et al. Synthesis and Biological Evaluation of Aurachin D Analogues as Inhibitors of Mycobacterium tuberculosis Cytochrome bd Oxidase. ACS Med. Chem. Lett. 2022, 13, 1663–1669. [Google Scholar] [CrossRef]
- Dibrov, P.; Dibrov, E.; Pierce, G.N. Na+-NQR (Na+-Translocating NADH:Ubiquinone Oxidoreductase) as a Novel Target for Antibiotics. FEMS Microbiol. Rev. 2017, 41, 653–671. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Murai, M.; Ninokura, S.; Kitazumi, Y.; Mezic, K.G.; Cress, B.F.; Koffas, M.A.G.; Morgan, J.E.; Barquera, B.; Miyoshi, H. Identification of the Binding Sites for Ubiquinone and Inhibitors in the Na+-Pumping NADH-Ubiquinone Oxidoreductase from Vibrio cholerae by Photoaffinity Labeling. J. Biol. Chem. 2017, 292, 7727–7747. [Google Scholar] [CrossRef] [Green Version]
- Masuya, T.; Sano, Y.; Tanaka, H.; Butler, N.L.; Ito, T.; Tosaki, T.; Morgan, J.E.; Murai, M.; Barquera, B.; Miyoshi, H. Inhibitors of a Na+-Pumping NADH-Ubiquinone Oxidoreductase Play Multiple Roles to Block Enzyme Function. J. Biol. Chem. 2020, 295, 12739–12754. [Google Scholar] [CrossRef]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-Virulence, Antiviral and Anti-Parasitic Agents. Adv. Exp. Med. Biol. 2020, 1282, 37–69. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.E.; Marchetti, R.V.; Cowan, A.I.; Howitt, S.M.; Bröer, S.; Kirk, K. Chloroquine Transport via the Malaria Parasite’s Chloroquine Resistance Transporter. Science 2009, 325, 1680–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, K.; Mogi, T.; Tanaka, T.Q.; Hata, M.; Miyoshi, H.; Kita, K. Mitochondrial Dehydrogenases in the Aerobic Respiratory Chain of the Rodent Malaria Parasite Plasmodium yoelii yoelii. J. Biochem. 2009, 145, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Höfle, G.; Kunze, B. Biosynthesis of Aurachins A-L in Stigmatella aurantiaca: A Feeding Study. J. Nat. Prod. 2008, 71, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Korp, J.; Vela Gurovic, M.S.; Nett, M. Antibiotics from Predatory Bacteria. Beilstein J. Org. Chem. 2016, 12, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Phillips, K.E.; Akbar, S.; Stevens, D.C. Concepts and conjectures concerning predatory performance of myxobacteria. Front. Microbiol. 2022, 13, 1031346. [Google Scholar] [CrossRef]
- Sandmann, A.; Dickschat, J.; Jenke-Kodama, H.; Kunze, B.; Dittmann, E.; Müller, R. A Type II Polyketide Synthase from the Gram-Negative Bacterium Stigmatella aurantiaca Is Involved in Aurachin Alkaloid Biosynthesis. Angew. Chem.—Int. Ed. 2007, 46, 2712–2716. [Google Scholar] [CrossRef]
- Pistorius, D.; Li, Y.; Sandmann, A.; Müller, R. Completing the Puzzle of Aurachin Biosynthesis in Stigmatella aurantiaca Sg a15. Mol. Biosyst. 2011, 7, 3308–3315. [Google Scholar] [CrossRef]
- Hertweck, C.; Luzhetskyy, A.; Rebets, Y.; Bechthold, A. Type II Polyketide Synthases: Gaining a Deeper Insight into Enzymatic Teamwork. Nat. Prod. Rep. 2007, 24, 162–190. [Google Scholar] [CrossRef]
- Pistorius, D.; Li, Y.; Mann, S.S.; Müller, R. Unprecedented Anthranilate Priming Involving Two Enzymes of the Acyl Adenylating Superfamily in Aurachin Biosynthesis. J. Am. Chem. Soc. 2011, 133, 12362–12365. [Google Scholar] [CrossRef]
- Stec, E.; Pistorius, D.; Müller, R.; Li, S.M. AuaA, a Membrane-Bound Farnesyltransferase from Stigmatella aurantiaca, Catalyzes the Prenylation of 2-Methyl-4-Hydroxyquinoline in the Biosynthesis of Aurachins. ChemBioChem 2011, 12, 1724–1730. [Google Scholar] [CrossRef]
- Katsuyama, Y.; Harmrolfs, K.; Pistorius, D.; Li, Y.; Müller, R. A Semipinacol Rearrangement Directed by an Enzymatic System Featuring Dual-Function FAD-Dependent Monooxygenase. Angew. Chem.—Int. Ed. 2012, 51, 9437–9440. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, Y.; Li, X.-W.; Müller, R.; Nay, B. Chemically Unprecedented Biocatalytic (AuaG) Retro-[2,3]-Wittig Rearrangement: A New Insight into Aurachin B Biosynthesis. ChemBioChem 2014, 15, 2349–2352. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, W.; Ozaki, T.; Nishioka, T.; Yasutake, Y.; Hata, M.; Nishiyama, M.; Kuzuyama, T.; Tamura, T. Cloning and Heterologous Expression of the Aurachin RE Biosynthesis Gene Cluster Afford a New Cytochrome P450 for Quinoline N-Hydroxylation. ChemBioChem 2013, 14, 1085–1093. [Google Scholar] [CrossRef]
- Yasutake, Y.; Kitagawa, W.; Hata, M.; Nishioka, T.; Ozaki, T.; Nishiyama, M.; Kuzuyama, T.; Tamura, T. Structure of the Quinoline N-Hydroxylating Cytochrome P450 RauA, an Essential Enzyme That Confers Antibiotic Activity on Aurachin Alkaloids. FEBS Lett. 2014, 588, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A New Enzyme Superfamily—The Phosphopantetheinyl Transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Pavlidou, M.; Pross, E.K.; Musiol, E.M.; Kulik, A.; Wohlleben, W.; Weber, T. The Phosphopantetheinyl Transferase KirP Activates the ACP and PCP Domains of the Kirromycin NRPS/PKS of Streptomyces collinus Tü 365. FEMS Microbiol. Lett. 2011, 319, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Gialama, D.; Kostelidou, K.; Michou, M.; Delivoria, D.C.; Kolisis, F.N.; Skretas, G. Development of Escherichia coli Strains That Withstand Membrane Protein-Induced Toxicity and Achieve High-Level Recombinant Membrane Protein Production. ACS Synth. Biol. 2017, 6, 284–300. [Google Scholar] [CrossRef]
- Kruth, S.; Schibajew, L.; Nett, M. Biocatalytic Production of the Antibiotic Aurachin D in Escherichia coli. AMB Express 2022, 12, 138. [Google Scholar] [CrossRef]
- Ghosh, P.; Das, S. Synthesis and Functionalization of 4-Quinolones—A Progressing Story. Eur. J. Org. Chem. 2019, 2019, 4466–4516. [Google Scholar] [CrossRef]
- Sun, M.; Gao, A.X.; Li, A.; Liu, X.; Wang, R.; Yang, Y.; Li, Y.; Liu, C.; Bai, Z. Bicistronic Design as Recombinant Expression Enhancer: Characteristics, Applications, and Structural Optimization. Appl. Microbiol. Biotechnol. 2021, 105, 7709–7720. [Google Scholar] [CrossRef]
- Takyar, S.; Hickerson, R.P.; Noller, H.F. mRNA Helicase Activity of the Ribosome. Cell 2005, 120, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winand, L.; Sester, A.; Nett, M. Bioengineering of Anti-Inflammatory Natural Products. ChemMedChem 2021, 16, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Sester, A.; Stüer-Patowsky, K.; Hiller, W.; Kloss, F.; Lütz, S.; Nett, M. Biosynthetic Plasticity Enables Production of Fluorinated Aurachins. ChemBioChem 2020, 21, 2268–2273. [Google Scholar] [CrossRef]
- Kaila, V.R.I.; Wikström, M. Architecture of Bacterial Respiratory Chains. Nat. Rev. Microbiol. 2021, 19, 319–330. [Google Scholar] [CrossRef]
- Beites, T.; O’Brien, K.; Tiwari, D.; Engelhart, C.A.; Walters, S.; Andrews, J.; Yang, H.-J.; Sutphen, M.L.; Weiner, D.M.; Dayao, E.K.; et al. Plasticity of the Mycobacterium tuberculosis Respiratory Chain and Its Impact on Tuberculosis Drug Development. Nat. Commun. 2019, 10, 4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Huang, T.H.; Yang, S.C.; Chen, C.C.; Fang, J.Y. Nano-Based Drug Delivery or Targeting to Eradicate Bacteria for Infection Mitigation: A Review of Recent Advances. Front. Chem. 2020, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An Overview on Target-Based Drug Design against Kinetoplastid Protozoan Infections: Human African Trypanosomiasis, Chagas Disease and Leishmaniases. Molecules 2021, 26, 4629. [Google Scholar] [CrossRef]
- Ke, H.; Ganesan, S.M.; Dass, S.; Morrisey, J.M.; Pou, S.; Nilsen, A.; Riscoe, M.K.; Mather, M.W.; Vaidya, A.B. Mitochondrial Type II NADH Dehydrogenase of Plasmodium falciparum (PfNDH2) is dispensable in the asexual blood stages. PLoS ONE 2019, 14, e0214023. [Google Scholar] [CrossRef] [Green Version]
- Duarte, M.; Ferreira, C.; Khandpur, G.K.; Flohr, T.; Zimmermann, J.; Castro, H.; Herrmann, J.M.; Tomás, A.M. Leishmania Type II Dehydrogenase is Essential for Parasite Viability Irrespective of the Presence of an Active Complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2103803118. [Google Scholar] [CrossRef]
- Wall, R.J.; Carvalho, S.; Milne, R.; Bueren-Calabuig, J.A.; Moniz, S.; Cantizani-Perez, J.; MacLean, L.; Kessler, A.; Cotillo, I.; Sastry, L.; et al. The Qi Site of Cytochrome b Is a Promiscuous Drug Target in Trypanosoma cruzi and Leishmania donovani. ACS Infect. Dis. 2020, 6, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Ward, V.C.A.; Chatzivasileiou, A.O.; Stephanopoulos, G. Metabolic Engineering of Escherichia coli for the Production of Isoprenoids. FEMS Microbiol. Lett. 2018, 365, fny079. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | Function of Gene Product | B | Function of Gene Product | C | Function of Gene Product |
|---|---|---|---|---|---|
| auaA | prenyltransferase | rauA | cytochrome P450 monooxygenase | sauA | prenyltransferase |
| auaB | acyl carrier protein | rauB | prenyltransferase | sauB | acyl carrier protein |
| auaC | β-ketoacyl ACP synthase II (KSα) | rauC | acyl carrier protein | sauC | β-ketoacyl ACP synthase II |
| auaD | β-ketoacyl ACP synthase II (KSβ) | rauD | β-ketoacyl ACP synthase II | sauD | β-ketoacyl ACP synthase II |
| auaE | anthranilate-CoA:ACP acyltransferase | rauE | β-ketoacyl ACP synthase II | sauE | anthranilate-CoA:ACP acyltransferase and anthranilate-CoA ligase |
| auaEII | anthranilate-CoA ligase | rauF | anthranilate-CoA:ACP acyltransferase and anthranilate-CoA ligase | sauK | thioesterase |
| auaF | Rieske [2Fe–2S] monooxygenase | rauG | efflux protein | sauL | trans-isoprenyl diphosphate synthase |
| auaG | flavin-dependent monooxygenase | rauH | isopentenyl diphosphate isomerase | sauM | DXP synthase (MEP pathway) |
| auaH | reductase | rauI | farnesyl synthase | sauN | HMBDP synthase (MEP pathway) |
| auaJ | flavin-dependent monooxygenase | sauPI | cytochrome P450 monooxygenase | ||
| auaI | steroid δ-isomerase | sauPII | cytochrome P450 monooxygenase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruth, S.; Nett, M. Aurachins, Bacterial Antibiotics Interfering with Electron Transport Processes. Antibiotics 2023, 12, 1067. https://doi.org/10.3390/antibiotics12061067
Kruth S, Nett M. Aurachins, Bacterial Antibiotics Interfering with Electron Transport Processes. Antibiotics. 2023; 12(6):1067. https://doi.org/10.3390/antibiotics12061067
Chicago/Turabian StyleKruth, Sebastian, and Markus Nett. 2023. "Aurachins, Bacterial Antibiotics Interfering with Electron Transport Processes" Antibiotics 12, no. 6: 1067. https://doi.org/10.3390/antibiotics12061067