Ruthenium-Anchored Carbon Sphere-Customized Sensor for the Selective Amperometric Detection of Melatonin

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of CSs and Ru–CSs
2.3. Fabrication of Ru–CS/SPCE
2.4. Instrumentation and Measurements
2.5. Preparation of Pharmaceutical Samples
3. Results and Discussion
3.1. Morphology/Microstructure and Elemental Analysis
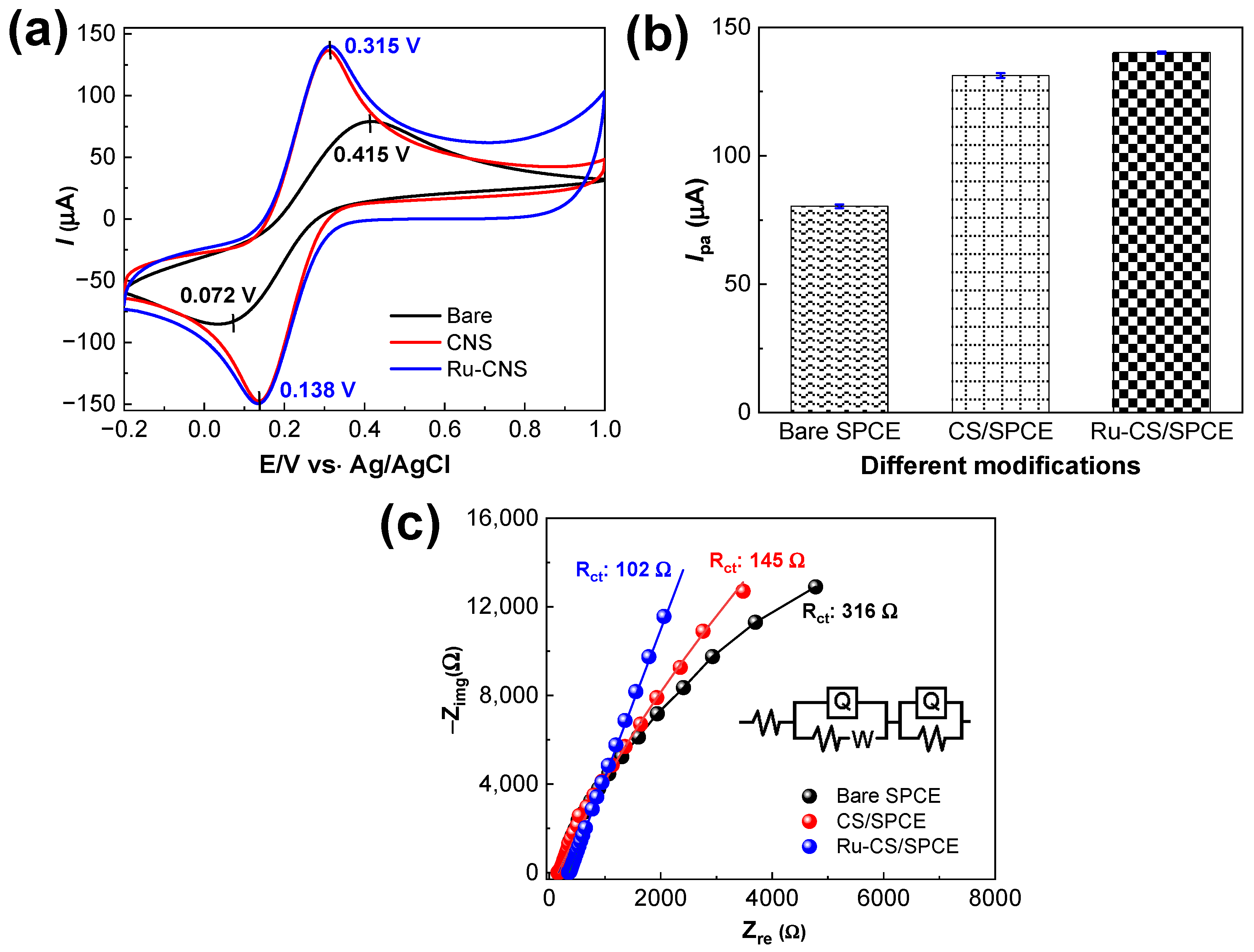
3.2. Electrochemical Performance of the Sensors
3.3. Effect of Scan Rate on the Ru–CS/SPCE
3.4. Optimization of Solution pH
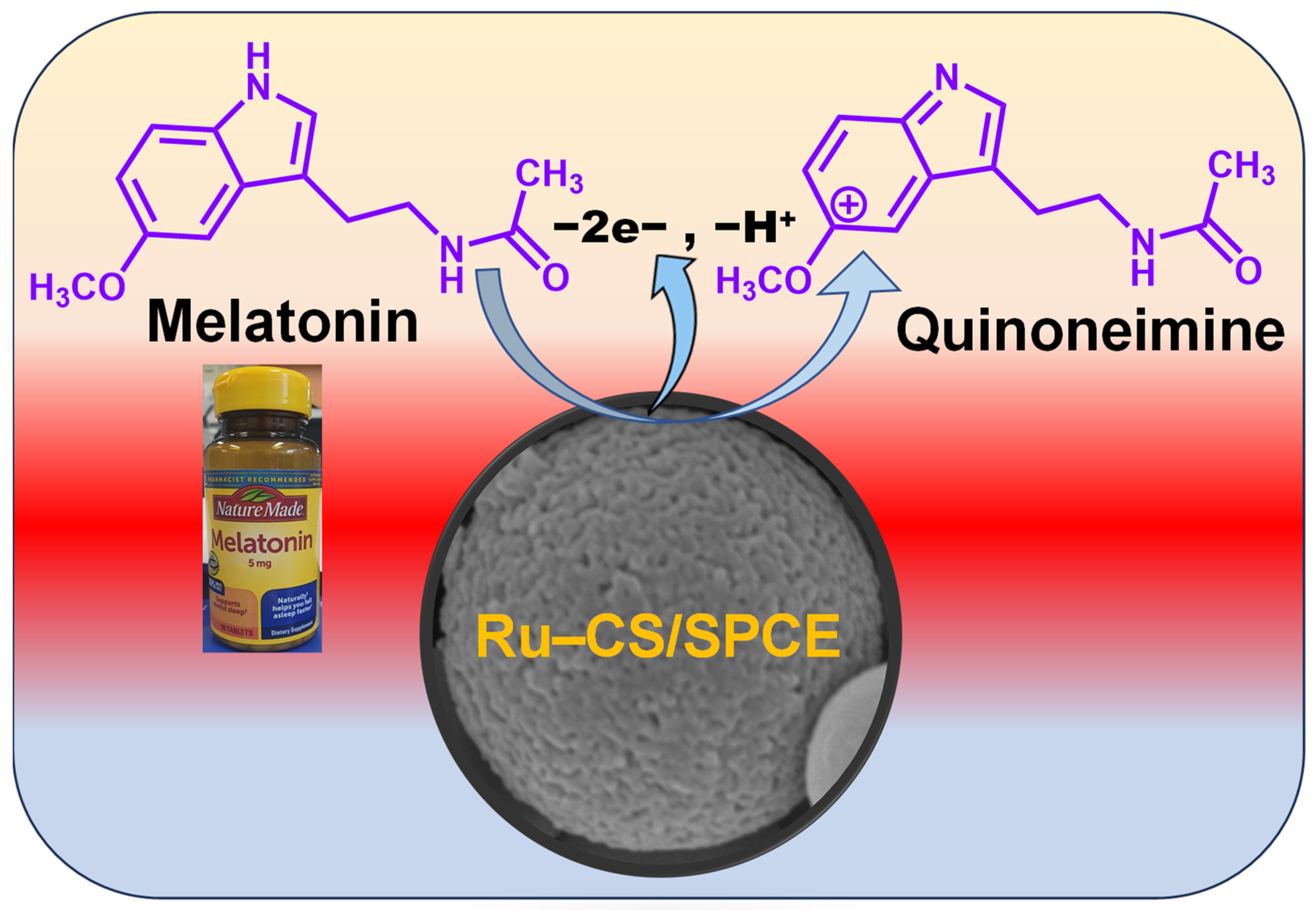
3.5. Optimization of Working Potential; Selectivity and Sensing Mechanism at the Ru–CS/SPCE
3.6. Amperometric Estimation of MT on the Ru–CS/SPCE
3.7. Reproducibility and Stability
3.8. Analysis of Pharmaceutical Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hossain, M.F.; Uddin, M.S.; Uddin, G.M.S.; Sumsuzzman, D.M.; Islam, M.S.; Barreto, G.E.; Mathew, B.; Ashraf, G.M. Melatonin in Alzheimer’s Disease: A Latent Endogenous Regulator of Neurogenesis to Mitigate Alzheimer’s Neuropathology. Mol. Neurobiol. 2019, 56, 8255–8276. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Sun, Q.; Qiu, H.; Yang, K.; Boya, X.; Xia, T.; Wang, A.; Gao, H.; Zhang, S. Melatonin protects against developmental PBDE-47 neurotoxicity by targeting the AMPK/mitophagy axis. J. Pineal Res. 2023, 75, e12871. [Google Scholar] [CrossRef] [PubMed]
- Cecon, E.; Liu, L.; Jockers, R. Melatonin receptor structures shed new light on melatonin research. J. Pineal Res. 2019, 67, e12606. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Rajarathinam, T.; Chang, S.-C. Disposable Sensor with Copper-Loaded Carbon Nanospheres for the Simultaneous Determination of Dopamine and Melatonin. Chemosensors 2023, 11, 254. [Google Scholar] [CrossRef]
- Daneshvar Kakhaki, R.; Ostadmohammadi, V.; Kouchaki, E.; Aghadavod, E.; Bahmani, F.; Tamtaji, O.R.; Reiter, J.R.; Mansournia, M.A.; Asemi, Z. Melatonin supplementation and the effects on clinical and metabolic status in Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Neurol. Neurosurg. 2020, 195, 105878. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Zheng, Z.-H.; Liu, T.-T.; Lin, L. Contralateral retinal dopamine decrease, and melatonin increase in progression of hemiparkinsonium rat. Neurochem. Res. 2012, 37, 1050–1056. [Google Scholar] [CrossRef]
- Carvalho, L.A.; Gorenstein, C.; Moreno, R.A.; Markus, R.P. Melatonin levels in drug-free patients with major depression from the southern hemisphere. Psychoneuroendocrinology 2006, 31, 761–768. [Google Scholar] [CrossRef]
- Maarman, G.J. Melatonin Against Pulmonary Arterial Hypertension: Is it ready for testing in patients? Cardiovasc. J. Afr. 2021, 32, 111–112. [Google Scholar] [CrossRef]
- Zuccari, D.A.P.D.C.; Reiter, R.J.; Chuffa, L.G.D.A.; Novais, A.A.; Silva, R.F.D.; Fernandes, F.L.C.; De Castro, T.B.D.; Mosqueta-Pinheiro, M.G.; Neto, R.J.B.; Sartorelli, L.S. Blood melatonin level can serve as a potential biomarker for prostate and hepatocellular carcinomas. Melatonin Res. 2021, 4, 253–269. [Google Scholar]
- Apetrei, I.M.; Apetrei, C. Voltammetric determination of melatonin using a graphene-based sensor in pharmaceutical products. Int. J. Nanomed. 2016, 11, 1859–1866. [Google Scholar] [CrossRef]
- Tapp, E.; Skinner, R.G.; Phillips, V. Radioimmunoassay for melatonin. J. Neural Transm. 1980, 48, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.S.; Zaky, M.; El-Beshbeshy, A.M. Colorimetric Estimation of Melatonin in Pharmaceutical Formulations. Mikrochim. Acta 2000, 135, 81–85. [Google Scholar] [CrossRef]
- Li, Y.; Cassone, V.M. A simple, specific high-throughput enzyme-linked immunosorbent assay (ELISA) for quantitative determination of melatonin in cell culture medium. Int. Immunopharmacol. 2015, 28, 230–234. [Google Scholar] [CrossRef]
- Bongiorno, D.; Ceraulo, L.; Mele, A.; Panzeri, W.; Selva, A.; Turco Liveri, V. Structural and physicochemical characterization of the inclusion complexes of cyclomaltooligosaccharides (cyclodextrins) with melatonin. Carbohydr. Res. 2002, 337, 743–754. [Google Scholar] [CrossRef]
- Yin, B.; Li, T.; Li, Z.; Dang, T.; He, P. Determination of Melatonin and Its Metabolites in Biological Fluids and Eggs Using High-Performance Liquid Chromatography with Fluorescence and Quadrupole-Orbitrap High-Resolution Mass Spectrometry. Food Anal. Methods 2015, 9, 1142–1149. [Google Scholar] [CrossRef]
- Alessa, H.; Saber, A.L.; Althakafy, J.T. Up-to-date studies regarding the determination of melatonin by chromatographic methods. Arch. Pharm. 2022, 355, e2100378. [Google Scholar] [CrossRef] [PubMed]
- Barreiro Astray, S.; Barbosa-Pereira, L.; Lage-Yusty, M.A.; López-Hernández, J. Comparison of Analytical Methods for the Rapid Determination of Melatonin in Food Supplements. Food Anal. Methods 2021, 14, 734–741. [Google Scholar] [CrossRef]
- Aboul-Enein, H.Y.; Doneanu, C.; Covaci, A. Capillary gc-ms determination of melatonin in several pharmaceutical tablet formulations. Biomed. Chromatogr. 1999, 13, 24–26. [Google Scholar] [CrossRef]
- Levent, A. Electrochemical determination of melatonin hormone using a boron-doped diamond electrode. Diam. Relat. Mater. 2012, 21, 114–119. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal Res. 2012, 52, 365–375. [Google Scholar] [CrossRef]
- Zhang, Y.; Waterhouse, G.I.N.; Xiang, Z.-P.; Che, J.; Chen, C.; Sun, W. A highly sensitive electrochemical sensor containing nitrogen-doped ordered mesoporous carbon (NOMC) for voltammetric determination of L-tryptophan. Food Chem. 2020, 326, 126976. [Google Scholar] [CrossRef] [PubMed]
- Walcarius, A. Mesoporous materials and electrochemistry. Chem. Soc. Rev. 2013, 42, 4098–4140. [Google Scholar] [CrossRef] [PubMed]
- Eivazzadeh-Keihan, R.; Bahojb Noruzi, E.; Chidar, E.; Jafari, M.; Davoodi, F.; Kashtiaray, A.; Ghafori Gorab, M.; Masoud Hashemi, S.; Javanshir, S.; Ahangari Cohan, R.; et al. Applications of carbon-based conductive nanomaterials in biosensors. J. Chem. Eng. 2022, 442, 136183. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Liu, Q.; Zhang, H.; Li, Z.; Jing, X.; Yuan, Y.; Zhang, H.; Liu, P.; Wang, J. Ionic liquids combined with Pt-modified ordered mesoporous carbons as electrolytes for the oxygen sensing. Sens. Actuators B Chem. 2018, 254, 490–501. [Google Scholar] [CrossRef]
- Yarar Kaplan, B.; Haghmoradi, N.; Jamil, E.; Merino, C.; Alkan Gürsel, S. Platinum nanoparticles decorated carbon nanofiber hybrids as highly active electrocatalysts for polymer electrolyte membrane fuel cells. Int. J. Energy Res. 2020, 44, 10251–10261. [Google Scholar] [CrossRef]
- Li, Y.; Shi, L.; Cai, X.; Zhao, H.; Niu, X.; Lan, M. Construction of non-enzymatic sensor based on porous carbon matrix loaded with Pt and Co nanoparticles for real-time monitoring of cellular superoxide anions. Electrochim. Acta 2019, 294, 304–311. [Google Scholar] [CrossRef]
- Yu, H.; Wu, L.; Ni, B.; Chen, T. Research Progress on Porous Carbon-Based Non-Precious Metal Electrocatalysts. Materials 2023, 16, 3283. [Google Scholar] [CrossRef]
- Veerakumar, P.; Salamalai, K.; Thanasekaran, P.; Lin, K.-C. Simple Preparation of Porous Carbon-Supported Ruthenium: Propitious Catalytic Activity in the Reduction of Ferrocyanate(III) and a Cationic Dye. ACS Omega 2018, 3, 12609–12621. [Google Scholar] [CrossRef]
- Kim, S.G.; Lee, J.S. Ruthenium Nanoparticle-Immobilized Porous Carbon Nanofibers for Nonenzymatic Dopamine Sensing. ACS Appl. Nano Mater. 2021, 4, 13683–13691. [Google Scholar] [CrossRef]
- Ye, H.; Mohar, J.; Wang, Q.; Catalano, M.; Kim, M.J.; Xia, X. Peroxidase-like properties of Ruthenium nanoframes. Sci. Bull. 2016, 61, 1739–1745. [Google Scholar] [CrossRef]
- Blume, R.; Havecker, M.; Zafeiratos, S.; Teschner, D.; Kleimenov, E.; Knop-Gericke, A.; Schlögl, R.; Barinov, A.; Dudin, P.; Kiskinova, M. Catalytically active states of Ru(0001) catalyst in CO oxidation reaction. J. Catal. 2006, 239, 354–361. [Google Scholar] [CrossRef]
- Veerakumar, P.; Hung, S.-T.; Hung, P.-Q.; Lin, K.-C. Review of the Design of Ruthenium-Based Nanomaterials and Their Sensing Applications in Electrochemistry. J. Agric. Food Chem. 2022, 70, 8523–8550. [Google Scholar] [CrossRef]
- Axet, M.R.; Philippot, K. Catalysis with Colloidal Ruthenium Nanoparticles. Chem. Rev. 2020, 120, 1085–1145. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, C.; Chen, Q.; Wang, Y.; Zhang, Y.; Fan, G. Electrochemical performance of ruthenium nanoparticles decorated on nitride carbon for non-enzymatic detection of hydrogen peroxide. Analyst 2019, 144, 6706–6711. [Google Scholar] [CrossRef] [PubMed]
- Rio, S.; Peru, G.; Léger, B.; Kerdi, F.; Besson, M.; Pinel, C.; Monflier, E.; Ponchel, A. Supported ruthenium nanoparticles on ordered mesoporous carbons using a cyclodextrin-assisted hard-template approach and their applications as hydrogenation catalysts. J. Catal. 2020, 383, 343–356. [Google Scholar] [CrossRef]
- Chang, S.C.; Rawson, K.; McNeil, C.J. Disposable tyrosinase-peroxidase bi-enzyme sensor for amperometric detection of phenols. Biosens. Bioelectron. 2002, 17, 1015–1023. [Google Scholar] [CrossRef]
- Han, A.; Chen, W.; Zhang, S.; Zhang, M.; Han, Y.; Zhang, J.; Ji, S.; Zheng, L.; Wang, Y.; Gu, L.; et al. A Polymer Encapsulation Strategy to Synthesize Porous Nitrogen-Doped Carbon-Nanosphere-Supported Metal Isolated-Single-Atomic-Site Catalysts. Adv. Mater. 2018, 30, e1706508. [Google Scholar] [CrossRef]
- Yang, Y.-Q.; Xiao, L.-P.; Xiao, W.-Z.; Li, X.-Y.; Wang, Q.; Sun, R.-C. Nitrogen-doped carbon anchored ruthenium nanoparticles for biofuel upgrade. Fuel 2022, 314, 123100. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, Z.; Chen, Y.; He, H.; Li, X.; Qin, T.; Wang, Y. High-performance supercapacitors of ruthenium-based nanohybrid compounds. J. Alloys Compd. 2020, 842, 155798. [Google Scholar] [CrossRef]
- Sun, L.; Jiang, X.; Jin, Z. Interfacial engineering of porous SiOx@C composite anodes toward high-performance lithium-ion batteries. Chem. Eng. J. 2023, 474, 145960. [Google Scholar] [CrossRef]
- Tavakkoli, N.; Soltani, N.; Shahdost-Fard, F.; Ramezani, M.; Salavati, H.; Jalali, M.R. Simultaneous voltammetric sensing of acetaminophen, epinephrine and melatonin using a carbon paste electrode modified with zinc ferrite nanoparticles. Mikrochim. Acta 2018, 185, 479. [Google Scholar] [CrossRef] [PubMed]
- Devadas, B.; Madhu, R.; Chen, S.-M.; Veeramani, V.; Rajkumar, M. Electrochemical Preparation of a Reduced Graphene Oxide/Ruthenium Oxide Modified Electrode and Its Application to the Simultaneous Determination of Serotonin and Melatonin. Sci. Adv. Mater. 2015, 7, 654–662. [Google Scholar] [CrossRef]
- Tadayon, F.; Sepehri, Z. A new electrochemical sensor based on a nitrogen-doped graphene/CuCo2O4 nanocomposite for simultaneous determination of dopamine, melatonin and tryptophan. RSC Adv. 2015, 5, 65560–65568. [Google Scholar] [CrossRef]
- Castagnola, E.; Woeppel, K.; Golabchi, A.; McGuier, M.; Chodapaneedi, N.; Metro, J.; Taylor, I.M.; Cui, X.T. Electrochemical detection of exogenously administered melatonin in the brain. Analyst 2020, 145, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Rajarathinam, T.; Kang, M.; Hong, S.; Chang, S.-C. Nanocomposite-based electrochemical sensors for neurotransmitters detection in neurodegenerative diseases. Chemosensors 2023, 11, 103. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Huang, Y.-C.; Chen, S.-M. Manganese hexacyanoferrate with poly (3,4-ethylenedioxythiophene) hybrid film modified electrode for the determination of catechin and melatonin. Int. J. Electrochem. Sci. 2011, 6, 3238–3253. [Google Scholar] [CrossRef]
- Radi, A.; Bekhiet, G.E. Voltammetry of melatonin at carbon electrodes and determination in capsules. Bioelectrochemistry Bioenerg. 1998, 45, 275–279. [Google Scholar] [CrossRef]
- Saber, A.L. Novel Potentiometric Sensors for Determination of Melatonin and Oxomemazine in Biological Samples and in Pharmaceutical Formulations. Electroanalysis 2010, 22, 2997–3002. [Google Scholar] [CrossRef]
- Gamal, O.; Alaa, S. Voltammetric determination of melatonin in tablet dosage forms and human serum. Lat. Am. J. Pharm. 2010, 29, 1235–1241. [Google Scholar]
- Sebastian, N.; Yu, W.-C.; Balram, D.; Chen, Q.; Shiue, A.; Noman, M.T.; Amor, N. Porous hematite embedded C and Fe codoped graphitic carbon nitride for electrochemical detection of pineal gland hormone melatonin. Mater. Today Chem. 2023, 29, 101406. [Google Scholar] [CrossRef]
- Freitas, R.C.; Orzari, L.O.; Ferreira, L.M.C.; Paixão, T.R.L.C.; Coltro, W.K.T.; Vicentini, F.C.; Janegitz, B.C. Electrochemical determination of melatonin using disposable self-adhesive inked paper electrode. J. Electroanal. Chem. 2021, 897, 115550. [Google Scholar] [CrossRef]
- Gomez, F.J.V.; Martín, A.; Silva, M.F.; Escarpa, A. Screen-printed electrodes modified with carbon nanotubes or graphene for simultaneous determination of melatonin and serotonin. Microkhim. Acta 2015, 182, 1925–1931. [Google Scholar] [CrossRef]
- Chen, G.; Ding, X.; Cao, Z.; Ye, J. Determination of melatonin and pyridoxine in pharmaceutical preparations for health-caring purposes by capillary electrophoresis with electrochemical detection. Anal. Chim. Acta 2000, 408, 249–256. [Google Scholar] [CrossRef]
- Kumar, N.; Goyal, R.N. Nanopalladium grained polymer nanocomposite based sensor for the sensitive determination of Melatonin. Electrochim. Acta 2016, 211, 18–26. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrochemical Sensor | Linear Range (µM) | LOD (µM) | Electrochemical Technique | Tested Real Samples | Reference |
|---|---|---|---|---|---|
| CPE | 3.0–550 | 2.3 | CV, LSV | Pharmaceutical samples | [47] |
| BDD | 0.5–4.0 | 0.11 | CV, SWV | Pharmaceutical and human urine samples | [19] |
| BT-drug ion pair PVC sensor | 1.0–1000 | 7.0 | Potentiometry | Pharmaceutical samples | [48] |
| GCE | 10.0–500 | 1.48 | CV, DPV, SWV | Pharmaceutical and human serum samples | [49] |
| ZnFe2O4/CPE | 50.0–500 | 3.0 | SWV | Fruits, human urine, and pharmaceutical samples | [50] |
| Graphene/SPCE | 1.00–300 | 0.87 | CA | Pharmaceutical samples | [10] |
| Gr-Av | 10.0–100.0 | 0.47 | SWV | Synthetic urine, human blood serum, and pharmaceutical samples | [51] |
| GO nanoribbons/SPCE | 5.0–3000 | 1.1 | CV, DPV | Pharmaceutical samples and herb extracts | [52] |
| Carbon disc electrode | 2.5–1000 | 1.3 | CE-ED | Pharmaceutical samples | [53] |
| MnHCF-PEDOT/GCE | 100–4600 | 100 | CV, CA | – | [46] |
| Polymer-PdNPs-rGO/GCE | 5.0–100 | 0.09 | CV, SWV | Pharmaceutical and human urine samples | [54] |
| rGO/RuO2/GCE | 2.0–20.0 | 0.18 | DPV | – | [42] |
| Ru–CS/SPCE | 1.25–20.0 | 0.27 | CV, CA | Pharmaceutical samples | This study |
| Pharmaceutical Samples | MT Taken (mg) | MT Found (mg) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Nature Made MT | 5.0 | 4.99 | 97.4 | 1.1 |
| 10.0 | 9.63 | 95.2 | 2.9 | |
| 15.0 | 14.9 | 97.6 | 2.1 | |
| 20.0 | 19.03 | 94.6 | 0.7 | |
| Melatonina (Cosmopharm) | 3.0 | 3.11 | 102.2 | 2.9 |
| 6.0 | 6.13 | 101.4 | 2.3 | |
| 9.0 | 9.09 | 100.6 | 1.6 | |
| 12.0 | 12.10 | 102.0 | 2.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayaraman, S.; Rajarathinam, T.; Jang, H.-G.; Thirumalai, D.; Lee, J.; Paik, H.-J.; Chang, S.-C. Ruthenium-Anchored Carbon Sphere-Customized Sensor for the Selective Amperometric Detection of Melatonin. Biosensors 2023, 13, 936. https://doi.org/10.3390/bios13100936
Jayaraman S, Rajarathinam T, Jang H-G, Thirumalai D, Lee J, Paik H-J, Chang S-C. Ruthenium-Anchored Carbon Sphere-Customized Sensor for the Selective Amperometric Detection of Melatonin. Biosensors. 2023; 13(10):936. https://doi.org/10.3390/bios13100936
Chicago/Turabian StyleJayaraman, Sivaguru, Thenmozhi Rajarathinam, Hyeon-Geun Jang, Dinakaran Thirumalai, Jaewon Lee, Hyun-Jong Paik, and Seung-Cheol Chang. 2023. "Ruthenium-Anchored Carbon Sphere-Customized Sensor for the Selective Amperometric Detection of Melatonin" Biosensors 13, no. 10: 936. https://doi.org/10.3390/bios13100936