
Novel PEPPSI-Type NHC Pd(II) Metallosurfactants on the Base of 1H-Imidazole-4,5-dicarboxylic Acid: Synthesis and Catalysis in Water–Organic Media
 , , ,
, , ,
Abstract
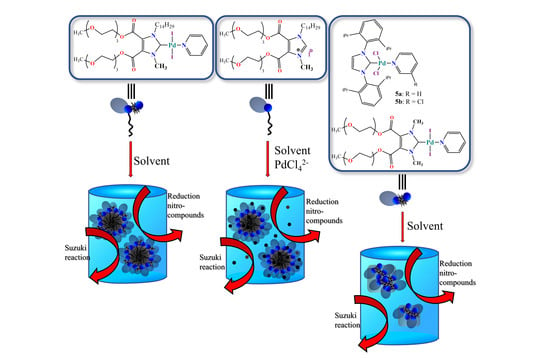
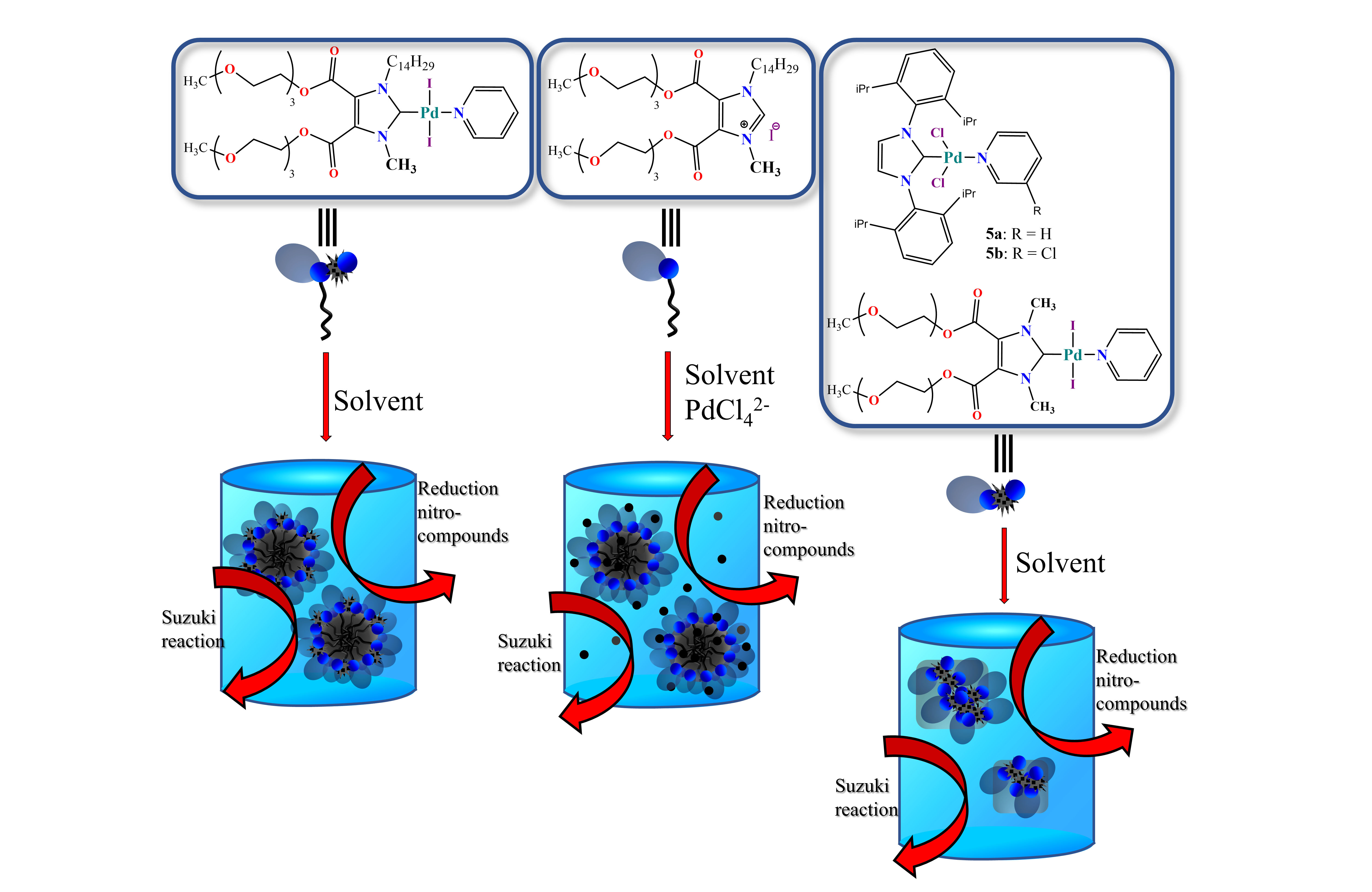
:
1. Introduction
2. Materials and Methods
2.1. Characterisation Methods
2.2. Reagents
2.3. Microscopy
2.4. Dynamic Light Scattering
2.5. Model Reduction Reaction
2.6. Model Suzuki-Miyaura Reaction
2.7. Gas Chromatography-Mass Spectrometry
3. Results
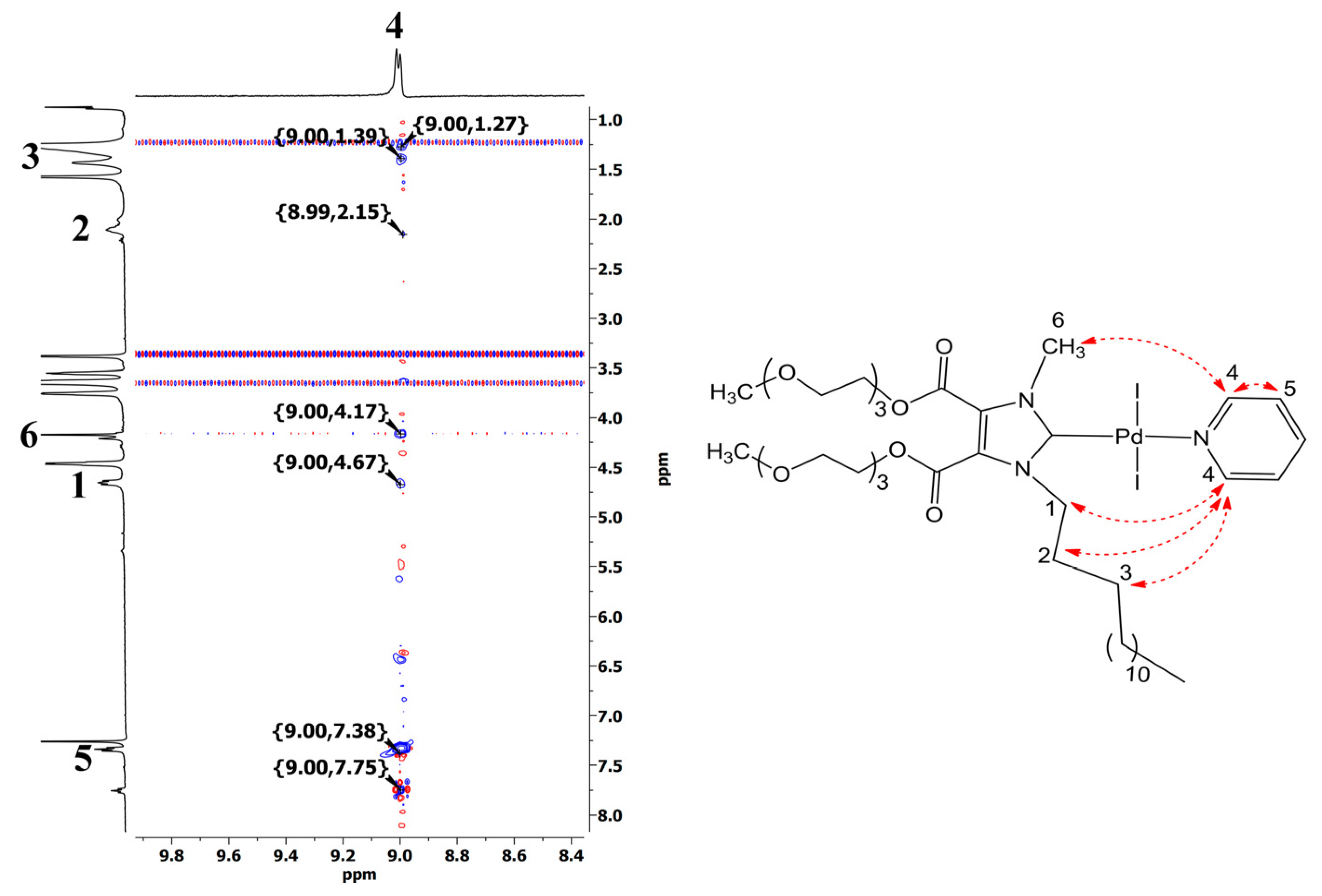
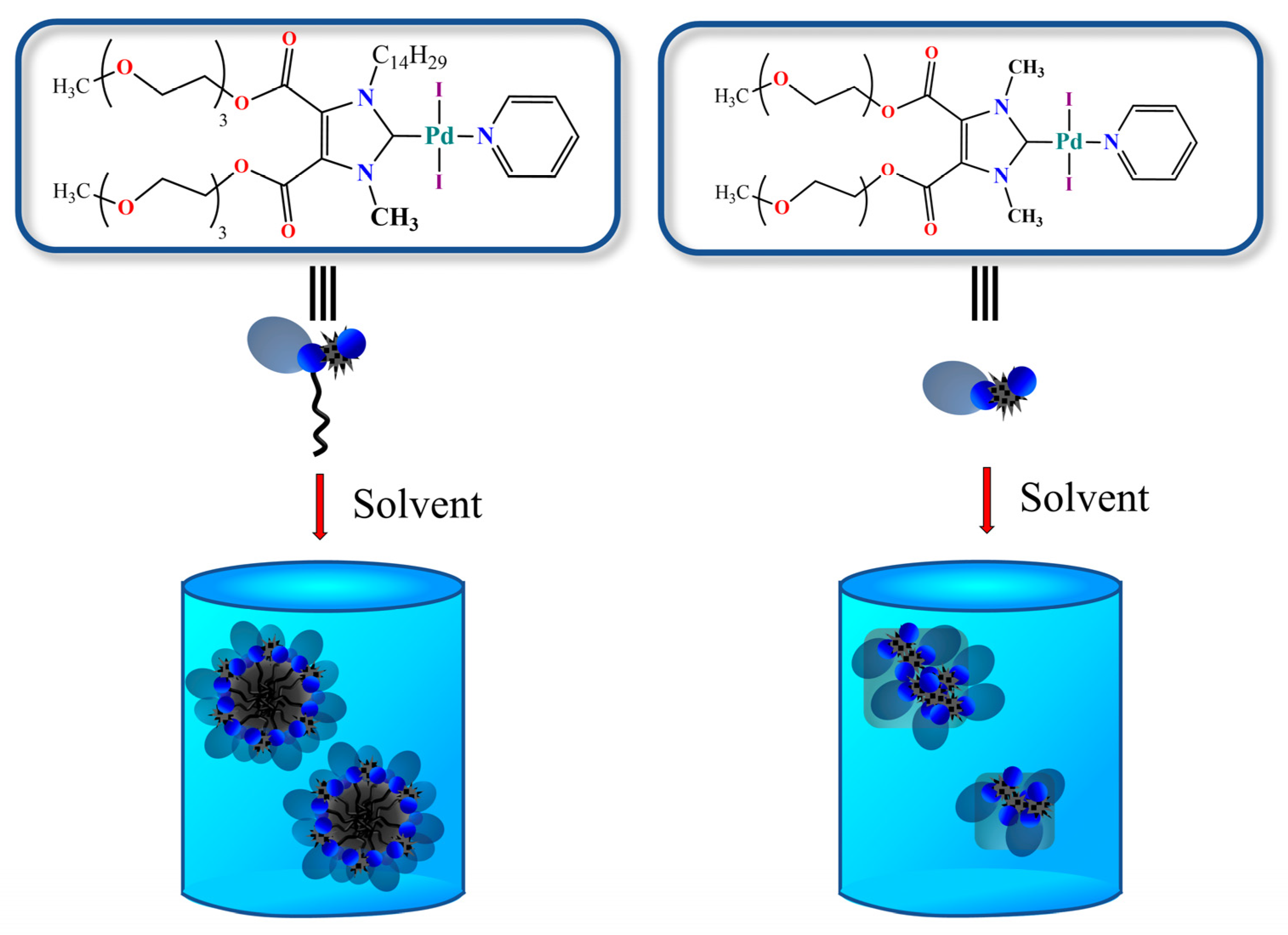
3.1. Synthesis
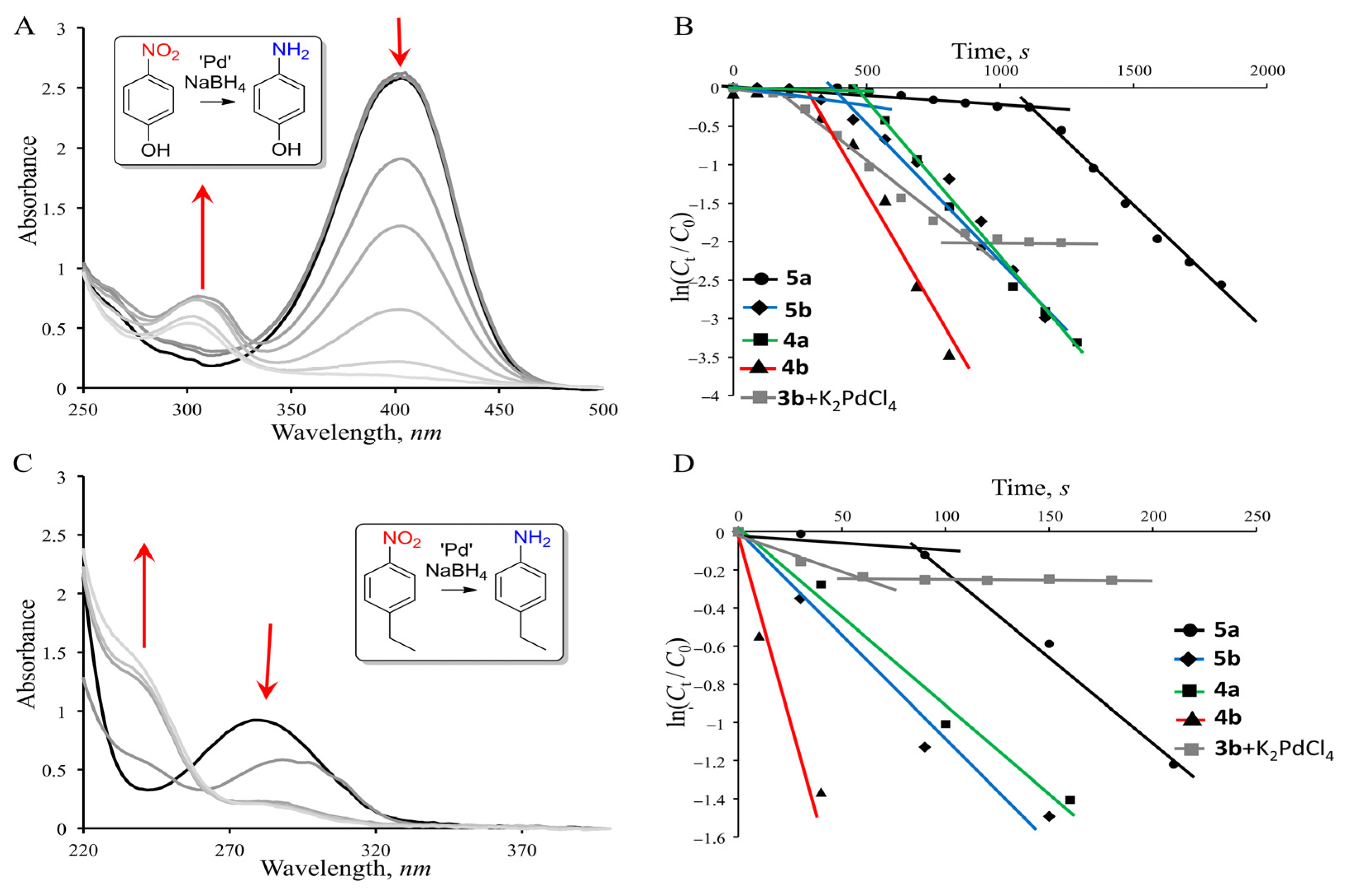
3.2. Catalytic Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, D.; Gulati, V.; Guleria, M.; Singh, S.P.; Agarwal, J. Aqueous Synthesis of 2-aryl-3-nitro-2H-chromenes via l-prolinamide Mediated Tandem Oxa-Michael Henry reactions. J. Mol. Struct. 2022, 1265, 133341. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent Advances in Catalysis in Micellar Media. Green Chem. 2015, 17, 644–683. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Cortes-Clerget, M. The Hydrophobic Effect Applied to Organic Synthesis: Recent Synthetic Chemistry “in Water”. Chem. Eur. J. 2018, 24, 6672–6695. [Google Scholar] [CrossRef]
- Polarz, S.; Landsmann, S.; Klaiber, A. Hybrid Surfactant Systems with Inorganic Constituents. Angew. Chem. Int. Ed. 2014, 53, 946–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattschneider, C.; Doniz Kettenmann, S.; Hinojosa, S.; Heinrich, J.; Kulak, N. Biological Activity of Amphiphilic Metal Complexes. Coord. Chem. Rev. 2019, 385, 191–207. [Google Scholar] [CrossRef]
- Taira, T. Metallosurfactants Consisting of Amphiphilic Ligands and Transition Metals: Structure, Bonding, Reactivity, and Self-assembling Property. J. Oleo Sci. 2022, 71, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Meng, X.-G.; Zeng, X.-C.; Yu, X.-Q. Metallomicellar Supramolecular Systems and their Applications in Catalytic Reactions. Coord. Chem. Rev. 2009, 253, 2166–2177. [Google Scholar] [CrossRef]
- Kaur, R.; Mehta, S.K. Self Aggregating Metal Surfactant Complexes: Precursors for Nanostructures. Coord. Chem. Rev. 2014, 262, 37–54. [Google Scholar] [CrossRef]
- Hondow, N.; Harowfield, J.; Koutsantonis, G.; Nealon, G.; Saunders, M. Metallosurfactants in the Preparation of Mesoporous Silicas. Microporous Mesoporous Mater. 2012, 151, 264–270. [Google Scholar] [CrossRef]
- Griffiths, P.C.; Fallis, I.A.; Chenpratoom, T.; Watanesk, R. Metallosurfactants: Interfaces and Micelles. Adv. Colloid Interface Sci. 2006, 122, 107–117. [Google Scholar] [CrossRef]
- Valls, E.; Solsona, A.; Suades, J.; Mathieu, R.; Comelles, F.; López-Iglesias, C. Synthesis and Characterization of New Amphiphilic Phosphines and Palladium Metallosurfactants. Organometallics 2002, 21, 2473–2480. [Google Scholar] [CrossRef]
- Parera, E.; Comelles, F.; Barnadas, R.; Suades, J. New Surfactant Phosphine Ligands and Platinum (II) Metallosurfactants. Influence of Metal Coordination on the Critical Micelle Concentration and Aggregation Properties. Langmuir 2010, 26, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Hamasaka, G.; Muto, T.; Uozumi, Y. A Novel Amphiphilic Pincer Palladium Complex: Design, Preparation and Self-assembling behavior. Dalton Trans. 2011, 40, 8859–8868. [Google Scholar] [CrossRef]
- Hamasaka, G.; Muto, T.; Uozumi, Y. Molecular-Architecture-Based Administration of Catalysis in Water: Self-Assembly of an Amphiphilic Palladium Pincer Complex. Angew. Chem. Int. Ed. 2011, 50, 4876–4878. [Google Scholar] [CrossRef]
- Smith, C.A.; Narouz, M.R.; Lummis, P.A.; Singh, I.; Nazemi, A.; Li, C.-H.; Crudden, C.M. N-Heterocyclic Carbenes in Materials Chemistry. Chem. Rev. 2019, 119, 4986–5056. [Google Scholar] [CrossRef]
- Koy, M.; Bellotti, P.; Das, M.; Glorius, F. N-Heterocyclic carbenes as tunable ligands for catalytic metal surfaces. Nat. Catal. 2021, 4, 352–363. [Google Scholar] [CrossRef]
- Taira, T.; Yanagimoto, T.; Sakai, K.; Sakai, H.; Imura, T. Au (I)-, Ag (I)-, and Pd (II)-coordination-driven Diverse Self-assembly of an N-heterocyclic Carbene-based Amphiphile. RSC Adv. 2021, 11, 17865–17870. [Google Scholar] [CrossRef]
- Taira, T.; Yanagimoto, T.; Fouquet, T.; Sakai, K.; Sakai, H.; Imura, T. Synthesis of an N-Heterocyclic Carbene-based Au (I) Coordinate Surfactant: Application for Alkyne Hydration Based on Au Nanoparticle Formation. J. Oleo Sci. 2020, 69, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Yanagimoto, T.; Sakai, K.; Sakai, H.; Endo, A.; Imura, T. Self-assembling Properties of an N-Heterocyclic Carbene-based Metallosurfactant: Pd-Coordination Induced Formation of Reactive Interfaces in Water. J. Oleo Sci. 2018, 67, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, T.; Yanagimoto, T.; Sakai, K.; Sakai, H.; Endo, A.; Imura, T. Synthesis of Surface-active N-heterocyclic Carbene Ligand and its Pd-catalyzed Aqueous Mizoroki–Heck Reaction. Tetrahedron 2016, 72, 4117–4122. [Google Scholar] [CrossRef]
- Xie, Q.; Li, J.; Wen, X.; Huang, Y.; Hu, Y.; Huang, Q.; Xu, G.; Xie, Y.; Zhou, Z. Carbohydrate-substituted N-heterocyclic Carbenes Palladium Complexes: High Efficiency Catalysts for Aqueous Suzuki–Miyaura Reaction. Carbohydr. Res. 2022, 512, 108516. [Google Scholar] [CrossRef] [PubMed]
- Donner, A.; Trepka, B.; Theiss, S.; Immler, F.; Traber, J.; Polarz, S. NHC-Metallosurfactants as Active Polymerization Catalysts. Langmuir 2019, 35, 16514–16520. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.-G.; Gu, J.-Z.; Jiang, L.; Tan, M.; Lu, T.-B. Achiral and Chiral Coordination Polymers Containing Helical Chains: The Chirality Transfer Between Helical Chains. Cryst. Growth Des. 2008, 8, 192–199. [Google Scholar] [CrossRef]
- Serrao, E.; Xu, Z.-L.; Debnath, B.; Christ, F.; Debyser, Z.; Long, Y.-Q.; Neamati, N. Discovery of a Novel 5-carbonyl-1H-imidazole-4-carboxamide Class of Inhibitors of the HIV-1 Integrase-LEDGF/p75 Interaction. Bioorg. Med. Chem. 2013, 21, 5963–5972. [Google Scholar] [CrossRef] [Green Version]
- Furó, I. NMR Spectroscopy of Micelles and Related Systems. J. Mol. Liq. 2005, 117, 117–137. [Google Scholar] [CrossRef]
- Teng, Q.; Huynh, H.V. A Unified Ligand Electronic Parameter Based on 13C NMR Spectroscopy of N-Heterocyclic Carbene Complexes. Dalton Trans. 2017, 46, 614–627. [Google Scholar] [CrossRef]
- Huynh, H.V.; Han, Y.; Jothibasu, R.; Yang, J.A. 13C NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium (II). Organometallics 2009, 28, 5395–5404. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G.R.J. Metal Complexes of N-Heterocyclic Carbenes—A New Structural Principle for Catalysts in Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2371–2374. [Google Scholar] [CrossRef]
- Kostyukovich, A.Y.; Tsedilin, A.M.; Sushchenko, E.D.; Eremin, D.B.; Kashin, A.S.; Topchiy, M.A.; Asachenko, A.F.; Nechaev, M.S.; Ananikov, V.P. In Situ Transformations of Pd/NHC Complexes with N-Heterocyclic Carbene Ligands of Different Nature into Colloidal Pd Nanoparticles. Inorg. Chem. Front. 2019, 6, 482–492. [Google Scholar] [CrossRef]
- Kaufhold, S.; Petermann, L.; Staehle, R.; Rau, S. Transition Metal Complexes with N-Heterocyclic Carbene Ligands: From Organometallic Hydrogenation Reactions Towards Water Splitting. Coord. Chem. Rev. 2014, 304–305, 73–87. [Google Scholar] [CrossRef]
- Denisova, E.A.; Kostyukovich, A.Y.; Fakhrutdinov, A.N.; Korabelnikova, V.A.; Galushko, A.S.; Ananikov, V.P. “Hidden” Nanoscale Catalysis in Alkyne Hydrogenation with Well-Defined Molecular Pd/NHC Complexes. ACS Catal. 2022, 12, 6980–6996. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Hadei, C.V.N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC = N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Hervés, P.; Pérez-Lorenzo, M.; Liz-Marzán, L.M.; Dzubiella, J.; Lu, Y.; Ballauff, M. Catalysis by Metallic Nanoparticles in Aqueous Solution: Model Reactions. Chem. Soc. Rev. 2012, 41, 5577–5587. [Google Scholar] [CrossRef]
- Chen, A.; Ostrom, C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999–12044. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Xu, S.; Wang, L.; Liu, Z.; Dong, X.; Wang, L.; Zheng, A.; Menga, X.; Xiao, F.-S. Fish-in-hole: Rationally positioning palladium into traps of zeolite crystals for sinter-resistant catalysts. Chem. Commun. 2018, 54, 3274–3277. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ward, T.R. Recent Advances in the Palladium Catalyzed Suzuki–Miyaura Cross-Coupling Reaction in Water. Catal. Lett. 2016, 146, 820–840. [Google Scholar] [CrossRef] [Green Version]
- Kaloğlu, N.; Özdemir, İ. PEPPSI-Pd-NHC catalyzed Suzuki-Miyaura cross-coupling reactions in aqueous media. Tetrahedron 2019, 75, 2306–2313. [Google Scholar] [CrossRef]
- Nasielski, J.; Hadei, N.; Achonduh, G.; Kantchev, E.A.B.; O’Brien, C.J.; Lough, A.; Organ, M.G. Structure–Activity Relationship Analysis of Pd–PEPPSI Complexes in Cross-Couplings: A Close Inspection of the Catalytic Cycle and the Precatalyst Activation Model. Chem. Eur. J. 2010, 16, 10844–10853. [Google Scholar] [CrossRef] [PubMed]
- Sayah, M.; Lough, A.J.; Organ, M.G. Sulfination by Using Pd-PEPPSI Complexes: Studies into Precatalyst Activation, Cationic and Solvent Effects and the Role of Butoxide Base. Chem. Eur. J. 2013, 19, 2749–2756. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
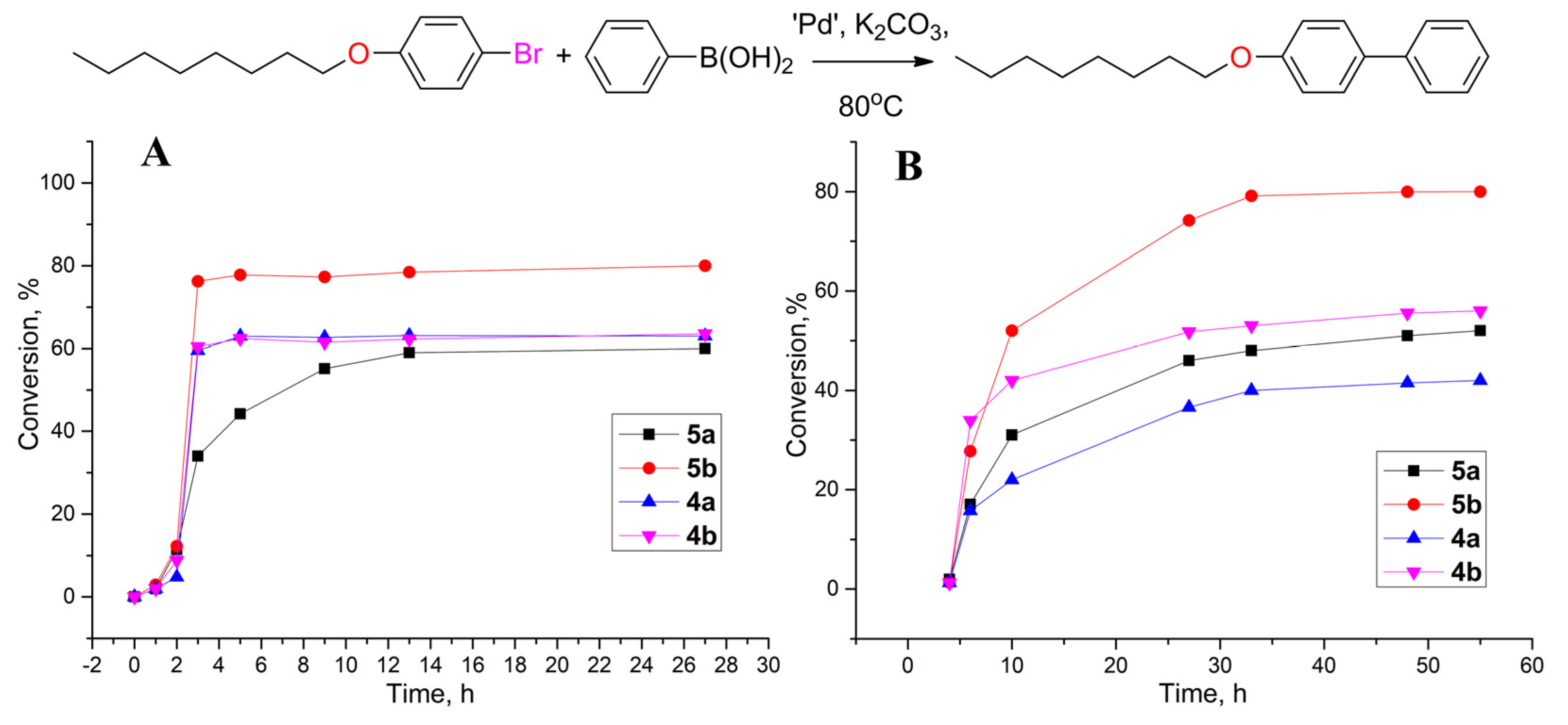{kind=link}
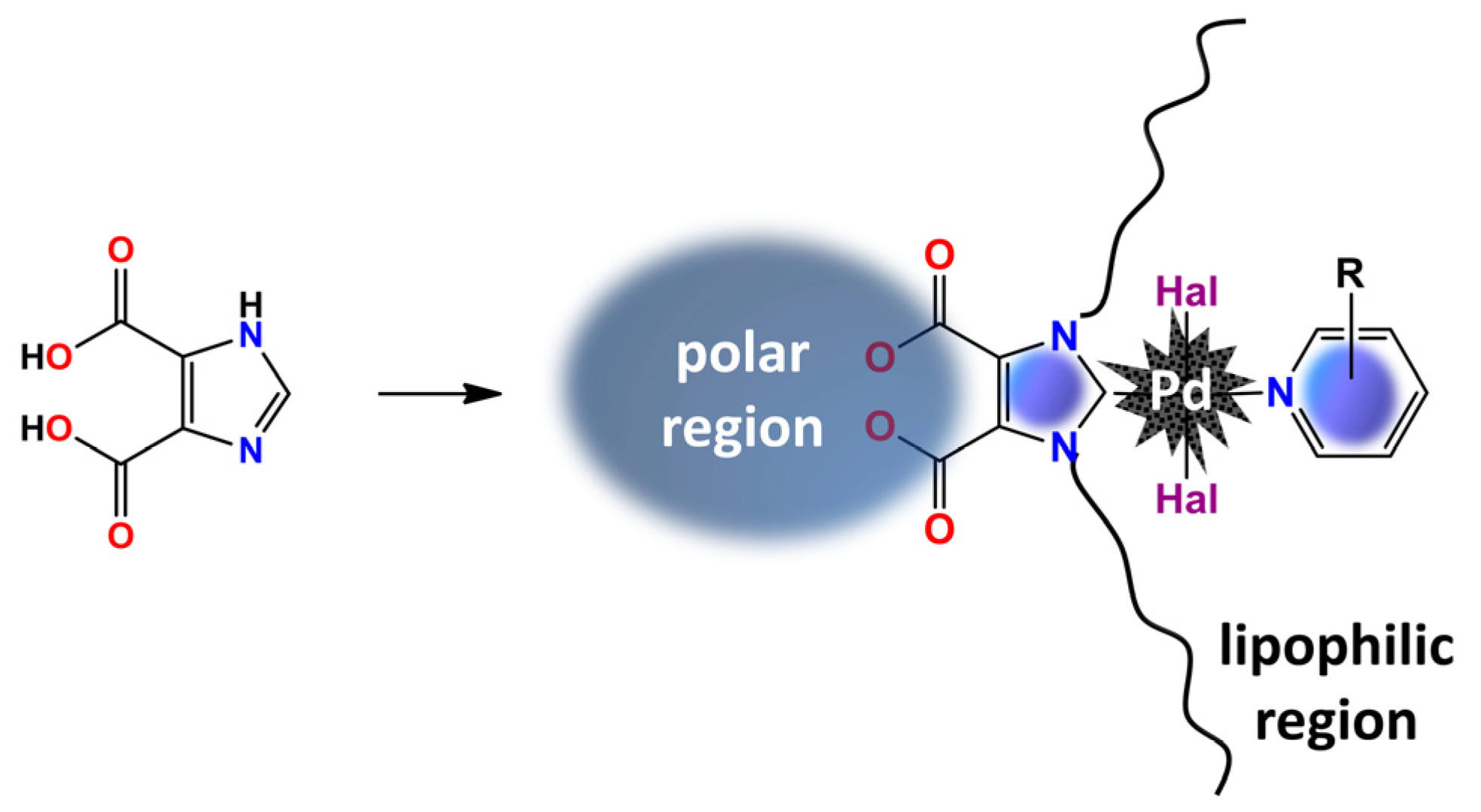{kind=link}
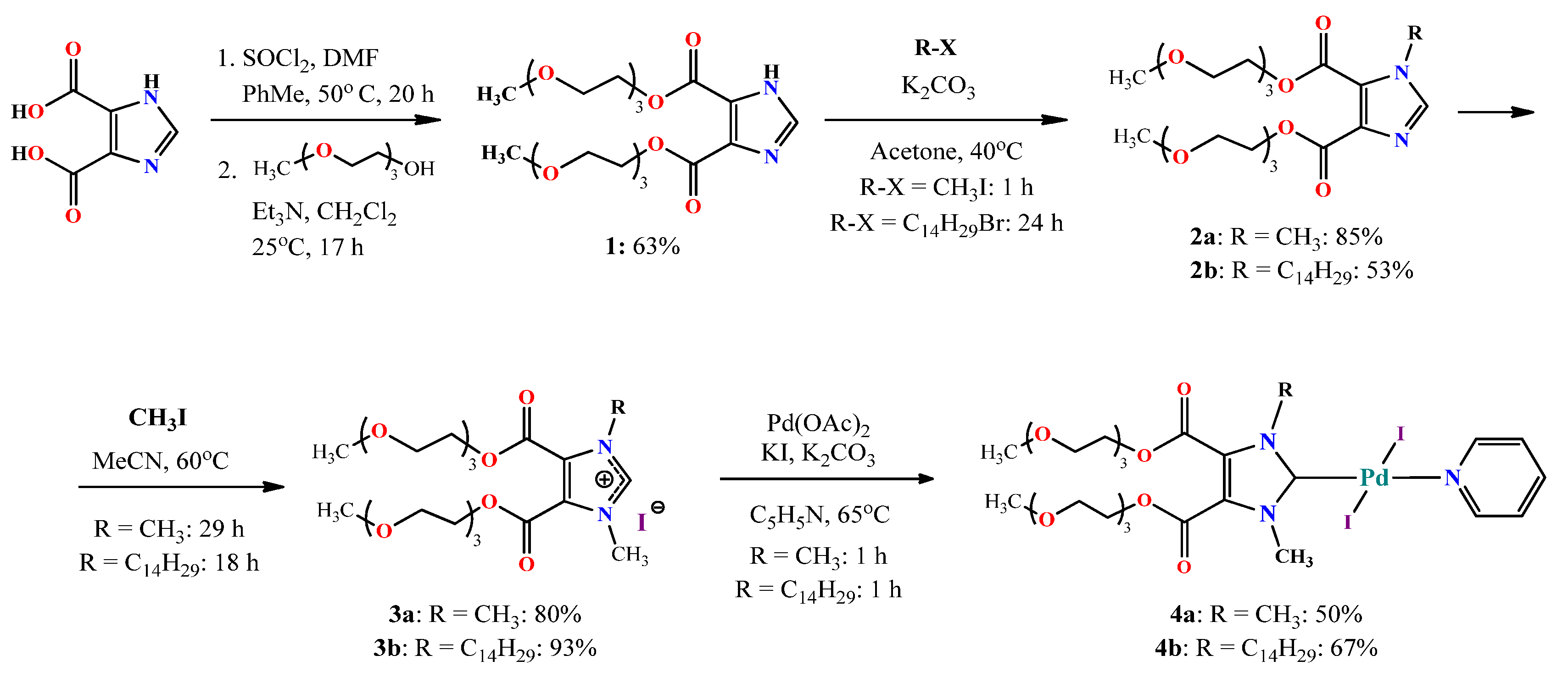{kind=link}
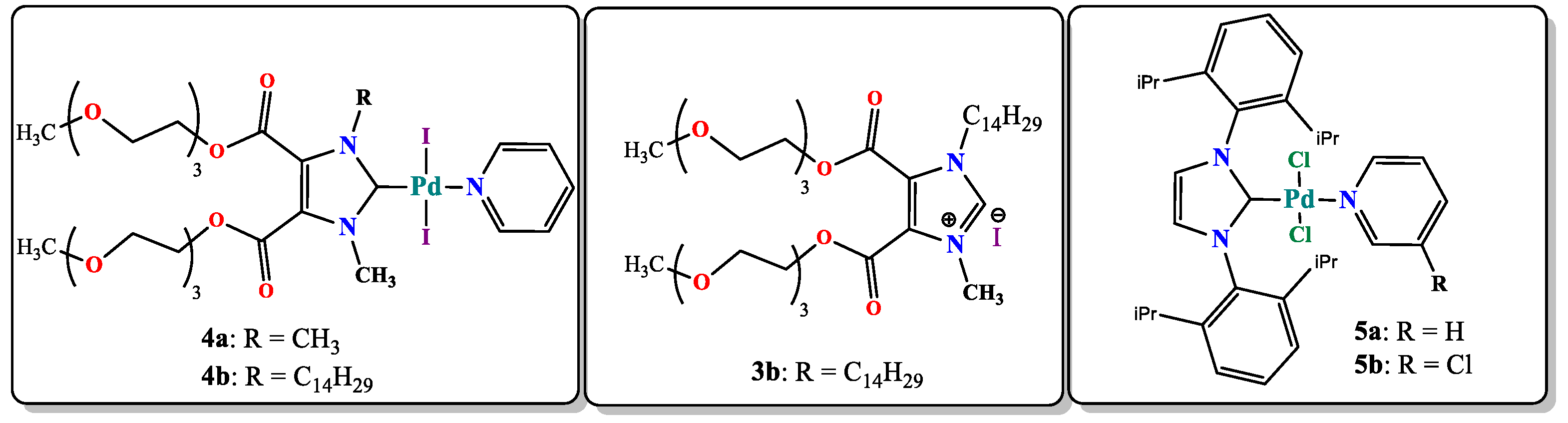{kind=link}
{kind=link}
| System | p-Nitrophenol | p-Ethylnitrobenzene | ||
|---|---|---|---|---|
| Apparent Rate Constant, k, s−1 | Specific Catalytic Activity, Ka, ×105 mol1s−1 | Apparent Rate Constant, k, s−1 | Specific Catalytic Activity, Ka, ×105 mol1s−1 | |
| 5a | 3.4 × 10−3 | 1.7 | 12 × 10−3 | 6.0 |
| 5b | 3.5 × 10−3 | 1.75 | 10 × 10−3 | 5.1 |
| 4a | 4.1 × 10−3 | 2.05 | 9.2 × 10−3 | 4.6 |
| 4b | 6.7 × 10−3 | 3.35 | 32 × 10−3 | 16.3 |
| 3b + K2PdCl4 | 2.7 × 10−3 | 1.35 | 2.8 × 10−3 | 1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burilov, V.; Radaev, D.; Sultanova, E.; Mironova, D.; Duglav, D.; Evtugyn, V.; Solovieva, S.; Antipin, I. Novel PEPPSI-Type NHC Pd(II) Metallosurfactants on the Base of 1H-Imidazole-4,5-dicarboxylic Acid: Synthesis and Catalysis in Water–Organic Media. Nanomaterials 2022, 12, 4100. https://doi.org/10.3390/nano12224100
Burilov V, Radaev D, Sultanova E, Mironova D, Duglav D, Evtugyn V, Solovieva S, Antipin I. Novel PEPPSI-Type NHC Pd(II) Metallosurfactants on the Base of 1H-Imidazole-4,5-dicarboxylic Acid: Synthesis and Catalysis in Water–Organic Media. Nanomaterials. 2022; 12(22):4100. https://doi.org/10.3390/nano12224100
Chicago/Turabian StyleBurilov, Vladimir, Dmitriy Radaev, Elza Sultanova, Diana Mironova, Daria Duglav, Vladimir Evtugyn, Svetlana Solovieva, and Igor Antipin. 2022. "Novel PEPPSI-Type NHC Pd(II) Metallosurfactants on the Base of 1H-Imidazole-4,5-dicarboxylic Acid: Synthesis and Catalysis in Water–Organic Media" Nanomaterials 12, no. 22: 4100. https://doi.org/10.3390/nano12224100