1. Introduction
Drug delivery and its encapsulation efficiency are major concerns for medical research. Many conventional medical treatments have a low proficiency and may have adverse side effects; targeted drug delivery offers an alternative to effectively defeat such diseases. There are many approaches to the technology of drug delivery, including the encapsulation of drugs using nanotubes [
1,
2,
3] and the functionalization and trapping of drugs in various nanocarriers [
4,
5], especially on graphene and graphene oxide [
6,
7,
8,
9].
A frequently adopted anti-cancer drug is Doxorubicin (DOX), which tends to show a significant improvement in cancer treatment. One derivative of DOX is a doxorubicin nitrate, (DOXH)NO
, and it is believed to be approximately 20 times more active in resistant cells than DOX [
10]. However, DOX and its derivatives have a limited therapeutic index and cause the development of multiple drug resistance [
11,
12]. The distribution of free DOX in the cell nucleus may produce considerable cytotoxicity [
13,
14,
15].
In terms of the delivery process, the binding between DOX and graphene has been widely studied, and we refer the reader to a comprehensive review by Sanchez et al. [
16] for the interaction between biomolecules and graphene. In particular, Vovusha et al. [
17] theoretically investigate the binding of DOX to graphene and to graphene oxide and conclude that graphene is a better binder of DOX compared to graphene oxide. Further, Tone et al. [
18] study the interaction of DOX with pristine graphene by utilizing the density functional theory framework and obtain five stable configurations. The interaction mechanism between DOX and chitosan-decorated graphene has been investigated by Shen et al. [
19] using molecular dynamics simulations. They discover that the pH of the fluid affects how DOX is loaded and released. Recently, Song et al. [
20] have also employed the density function theory method to investigate the affinity of hydroxyl and epoxy groups and found that the loading of DOX on graphene mainly depends on the hydroxyl groups. This could be viewed as a design for biological or chemical molecular machines [
21].
Artificial intelligence (AI) has also been utilized in the area of nanotechnology to obtain an optimal structure or a stable system. Specifically, metaheuristic algorithms can be applied directly if the objective function and the constraints are defined. For example, using genetic algorithm, Cuckoo search, Symbiotic organism search and Firefly algorithm tunes hyperparameters for the multi-layer perceptron artificial neural network and models nanovector in the system of drug delivery [
22,
23]. Non-dominated sorting genetic algorithm II (NSGA-II), a multi-objective heuristic algorithm, is an effective optimization algorithm for multi-objective problems [
24]. NSGA-II works well not only on multi-objective problems but also on single-objective problems and shows better performance than using common single-objective optimization algorithms [
25]. Recently, unified non-dominated sorting genetic algorithm III (U-NSGA-III), a modified NSGA-III, has been developed to handle all types of problems: single, multiple and many objectives, which has fewer tuning parameters than the existing algorithms. To maintain diversity among solutions, NSGA-II uses crowding distance, NSGA-III uses reference directions and U-NSGA-III increases the performance of NSGA-III by adding more tournament pressure. The applications of NSGA-II and U-NSGA-III are discussed as good algorithms for solving real-world problems [
26,
27]. In this study, NSGA-II and U-NSGA-III are employed to calculate the energy of the nano-scaled system.
Here, we focus on facilitating the stability of the system by investigating the interaction and activity between DOXHs and a flat graphene sheet using a mathematical model and heuristic algorithm techniques. The Lennard-Jones potential function is utilized to determine the non-bonded interaction energy between two materials. This energy function has been successfully used to determine the interaction between DOXs and bio-molecules, including liposome and peptide nanotubes [
28,
29]. The analytical expression derived from the Lennard-Jones potential function and the continuous approximation between a point and an infinite flat plane will be utilized to reduce the computational calculation time. The discrete-discrete atomic positions are defined for the interaction between two DOXH molecules, and the discrete-continuous description is represented for the interaction between a DOXH and the graphene.
The methodology, including molecular description, mathematical derivation for the interaction energy between two non-bonded molecules and the use of NSGA-II and U-NSGA-III, is given in
Section 2. In
Section 3, all numerical results are presented. The discussion of our finding is given in
Section 4 and the summary is made in
Section 5. The
Supplementary Material is also provided for the calculation details.
2. Methodology
The stabilities of the three systems, which are (i) the interaction energy between two DOXH molecules, (ii) the interaction energy between a DOXH and a flat graphene sheet and (iii) the interaction energy between two DOXHs and a flat graphene sheet, are investigated. The Lennard-Jones potential function is exploited to measure the energy of the system. In each system, NSGA-II and U-NSGA-III algorithms are utilized to determine the stability, and 30 experiments fixing seeds from 1 to 15 in each system are reported. NSGA-II and U-NSGA-III algorithms are employed from the open-source multi-objective optimization framework in Python [
30], and all numeric calculations, as well as graph visualizations, are operated via the Python program in Jupyter Notebook. The 3D molecular figures are created by Avogadro 2 [
31]. Then the optimized results are tested for the well-being of the local minimum by varying an offset distance and an offset rotational angle with respect to the fixed DOXH molecule. In order to refer to seed
of NSGA-II and seed
of U-NSGA-III, we define notations II-
n and III-
m, respectively, where
and
.
2.1. Molecular Description
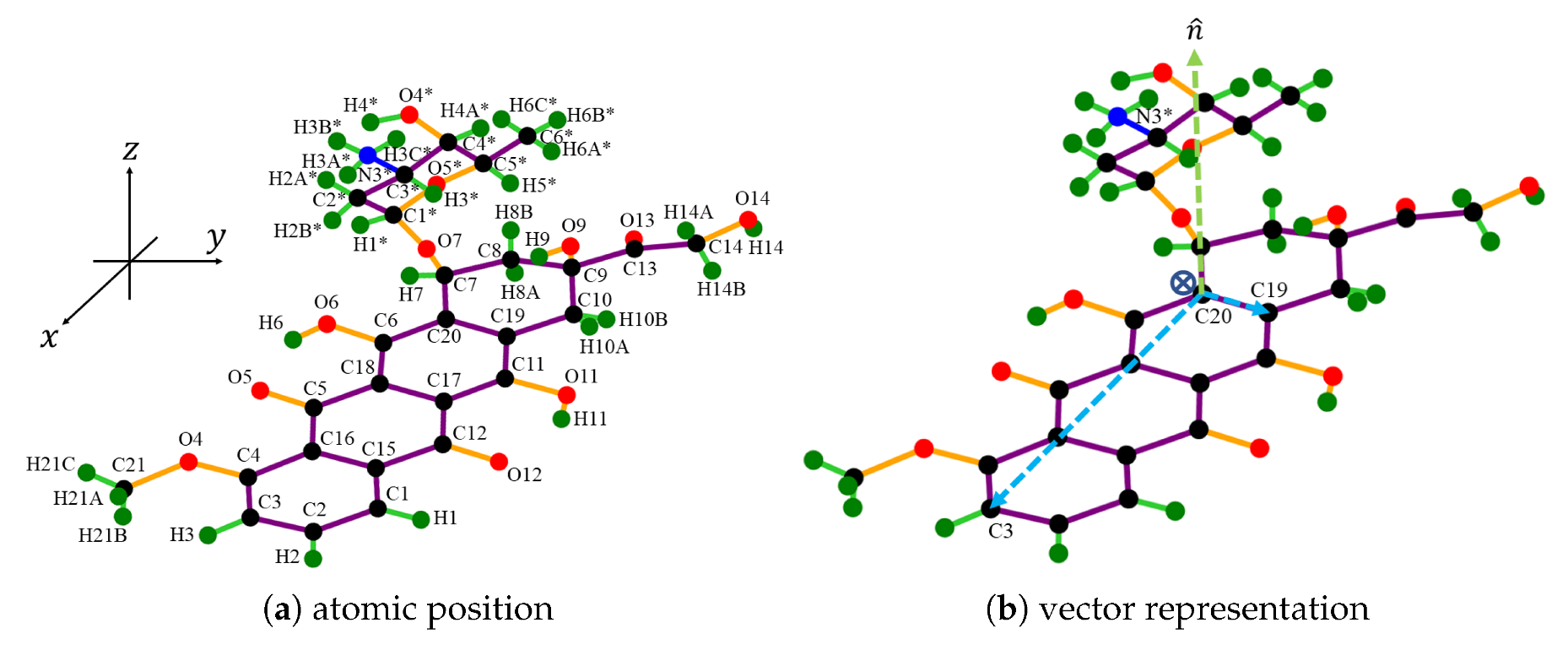
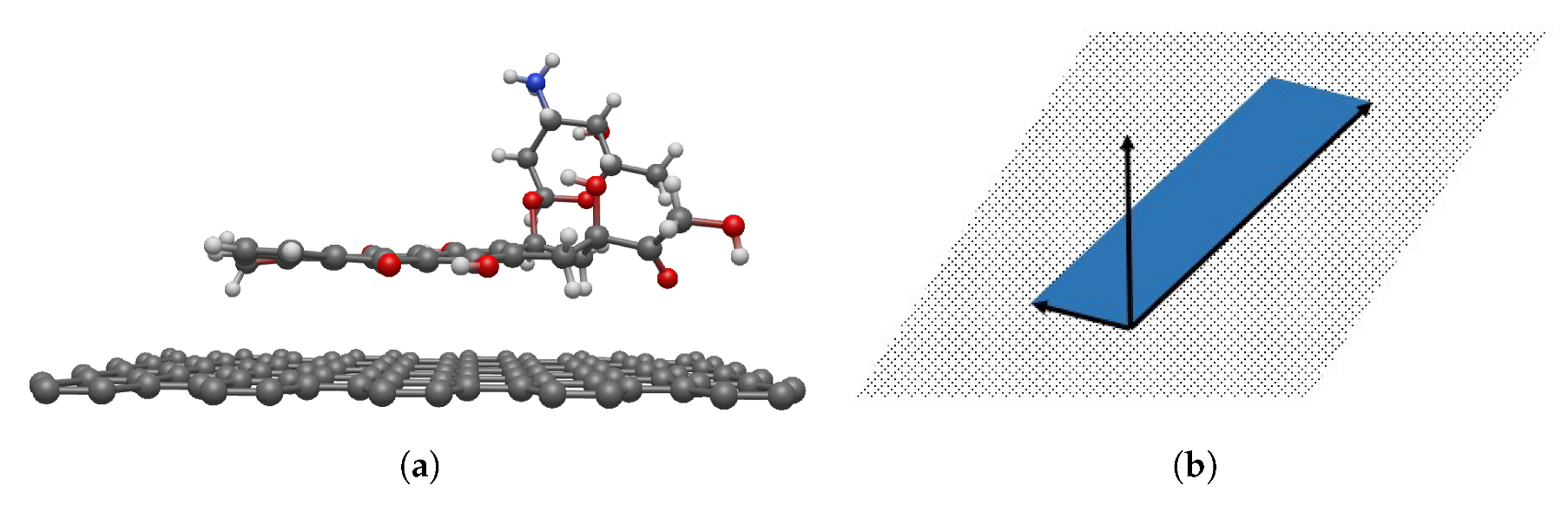
The atomic structure of DOXH is depicted in
Figure 1a, where carbon atoms are colored black, oxygen atoms are red, hydrogen atoms are green, and the nitrogen atom is blue (see color online). There is a total of 69 atoms, and the atomic positions are obtained from the work of Mathivathanan et al. [
32] by transforming the fractional coordinate to the Cartesian coordinate. Assuming a fixed crystal structure, the DOXH is treated as a solid molecule.
Furthermore, in each DOXH molecule, we create a point and three vectors, as shown in
Figure 1b. Two vectors are made by setting the coordinate of C
atom as an initial point and C
and C
are the terminal points. The third vector is the normal vector
. By this representation, the plane in real vector space spanned from vectors
and
pretends to pass the same normal vector as the carbon ring plane. Further, the cross circle in the middle (near the atom C
) indicates the center of mass of the DOXH.
In terms of a graphene sheet, we assume a perfect flat hexagonal lattice located on the -plane. It is further modeled as an infinite flat plane with the distribution of 0.3812 carbon atoms per square angstrom on its surface.
2.2. Interaction Energy and Parameter Values
We can use either the Lennard-Jones potential or Buckingham potential to describe the non-bonded interaction between graphene and drugs. The polynomial function of the Lennard-Jones potential is an appropriate option because our goal is to identify the analytical expression for the interaction. Additionally, because the electronic structure of a molecule is outside the scope, the density functional theory method can be disregarded.
The 6-12 Lennard-Jones function for two non-bonded atoms can be written as
where
denotes the distance between two typical points, and
A and
B are attractive and repulsive Lennard-Jones constants, respectively. Further,
denotes a well depth and
is the van der Waals diameter, from which we may deduce
and
, where
and
are taken from the work of Rappe et al. [
33].
Moreover, the mixing rule is utilized in the system of two atomic species, which are
and
. We aim to determine the interaction energy between each atom in the DOXH molecule with the graphene sheet; therefore, the Lennard-Jones parameters and the corresponding Lennard-Jones constants are given in
Table 1.
For two separated (non-bonded) molecular structures, the interaction energy can be evaluated using either a discrete atom–atom formulation or by a continuous approach. Thus, the non-bonded interaction energy may be obtained as a summation of the interaction energy between each atom pair, namely
where
is the potential function for atoms
i and
j located a distance
apart on two distinct molecular structures.
In the interest of modeling irregularly shaped molecules, such as drugs, an alternative hybrid discrete-continuous approximation can also be used, which is given by
where
is the surface density of atoms on the molecule that is considered continuous,
is the distance between a typical surface element
on the continuously modeled molecule and atom
i in the molecule that is modeled as discrete. The energy is obtained by summing all atoms in the drug that are represented discretely.
For conveniencem in the case of hybrid discrete-continuous approximation, we define
First, we consider the interaction energy between a point
P located at
and an infinite plane
. The distance between the point
P to the plane is
, and the integral
becomes
On using the substitution and changing the limit of integration (see [
34] for the integration details), we may deduce
Hence, the interaction energy between a point and the infinite plane is given by
where
is the mean atomic surface density of the graphene, and it is given by 0.3812 atom/Å
, and
is the distance between each atom on DOXH and the flat graphene sheet.
Hence, the total interaction energy between a DOXH and an infinite graphene plane is
where the Lennard-Jones constants for each interaction are given by
Table 1, and there is a total of 69 terms in the summations corresponding to 69 atoms in the DOXH.
For the case of the interaction between two drug molecules, the discrete atom–atom formulation is employed, and we may deduce
where
is the distance between atom
i on the first molecule and atom
j on the second molecule. Further,
and
are the Lennard-Jones constants depending on the atomic types for each pair of the interaction.
2.3. Optimization Setting
First, we let the DOXH structure transforme from [
32] to be a default structure and represent it by the reference coordinate
. Next, we assume the reference coordinate together with rotational angles on the
x- and
y-axis,
and
, to be decision variables; there are a total of
decision variables
where
n is the number of DOXH molecules. During the optimizing process, all coordinates of the atoms are computed from the
variables. For bounds or constraints in variables, all components of reference coordinates are restricted within the box of the side range
Å, two rotational angles are varied in
. Then, we set the total energy to be the objective function. Moreover, in the system with a graphene sheet, we fix the graphene sheet on the
-plane where
.
Both NSGA-II and U-NSGA-III consist of sampling, tournament selection, crossover and mutation as common processes. In terms of the difference between these two algorithms, NSGA-II uses the rank and crowding distance to obtain the next generation, which gives a more stable configuration as the final process. U-NSGA-III uses, instead, reference directions as the final process and improves the parent selection for mono-objective. The population sizes of the three systems and of both NSGA-II and U-NSGA-III are set as presented in
Table 2. Other parameters in the processes, such as sampling, crossover, mutation and eliminate_duplicates of NSGA-II and reference directions of U-NSGA-III, are set to default.
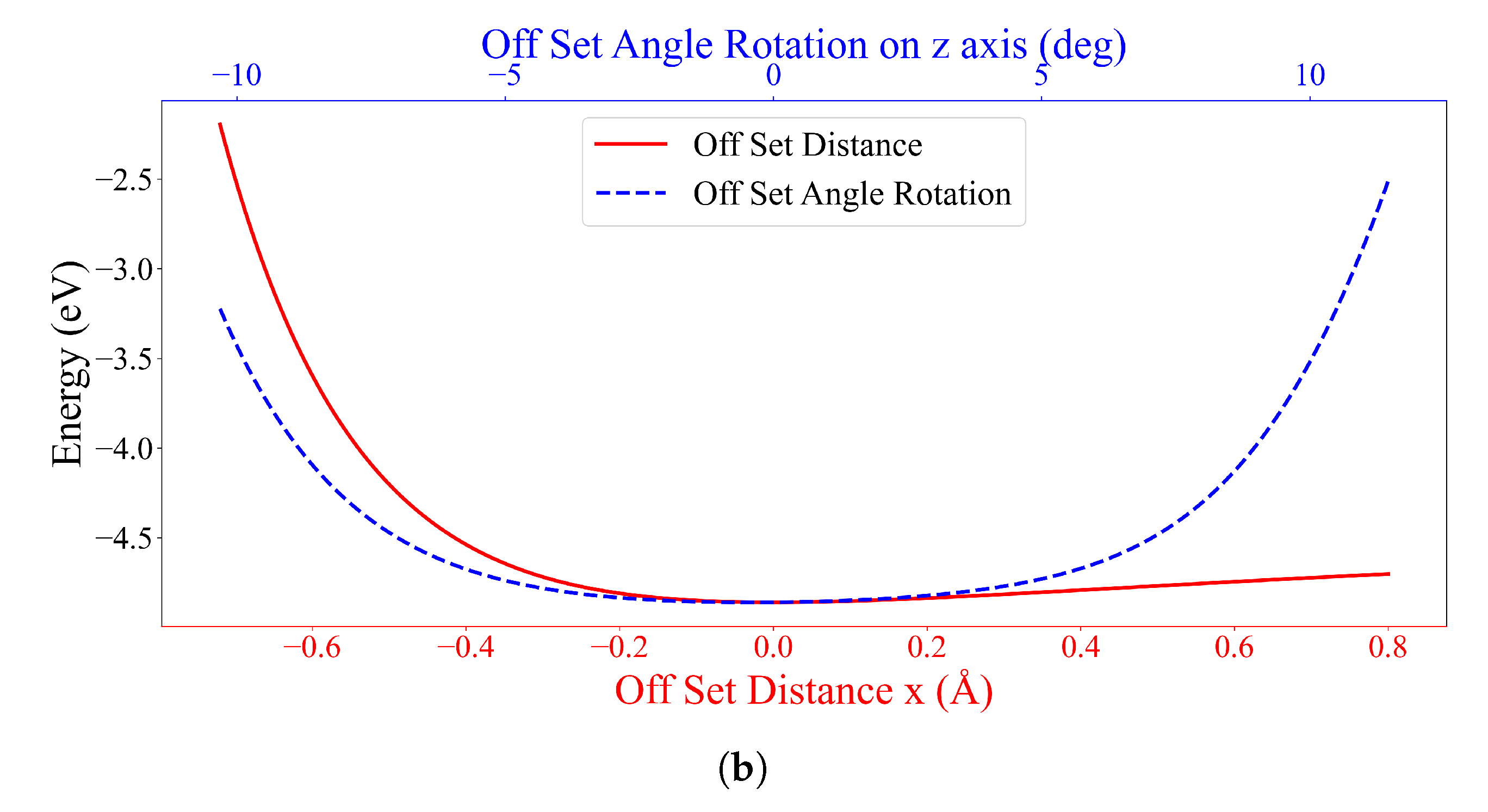
After finishing the optimization process, each solution from the experiment consisting of the objective value, which is the total energy (eV), and the coordinate points of 69 atoms are stored so as to investigate the obtained numerical results. Moreover, the solutions from each generation of experiments are accumulated by saving the history module in order to plot the learning curve and to see how the solution converges to a stable point.
4. Discussion
A number of theoretical studies have been carried out to investigate the attachment of DOX or DOXH to graphene or to graphene oxide. Most of the works have undertaken molecular dynamic simulation. Here, we propose the use of a heuristic algorithm to determine the stable configuration for the interaction between the DOXH molecule and the flat graphene sheet.
Tone et al. [
18] studied the interaction of DOX with pristine graphene by utilizing the density functional theory. They obtained five stable configurations of DOX; our numerical results from
Section 3.2 are compatible with their fifth configuration, which is a parallel configuration between DOX and graphene. In their work, H
is reported to be the atom that is closest to the graphene with a distance of
Å. However, in our study, H
is the closest atom to the graphene, with a distance of
Å, and H
has a distance of
Å from the graphene. Additionally, they concluded that the most stable configuration is the parallel configuration, which is in excellent agreement with our results.
Mirhosseini et al. [
35] studied the loading of the DOX drug and functionalizing it to graphene as a nanocarrier. They used molecular dynamics simulations to observe the chemical functionality and interaction energy. They also studied the neat graphene with DOX and reported the energy value at the stability of
eV (converted from
kcal/mol). Comparing to our study, it is well nigh to our finding of the energy value
eV presented in
Section 3.2.
According to the work by Song et al. [
20], they studied similar systems to those of Mirhosseini et al. [
35] and found that the most stable configuration calculated from the density functional theory is the parallel pattern with the binding energy of
eV and the separation distance of DOX–graphene is
Å.
Since there is no definite definition of the separation distance between DOX and graphene, we define
as an average distance between 20 carbon atoms on the DOXH carbon ring plane, C
to C
, to the flat graphene sheet. In our study, we obtain
Å. In terms of the energy value, our finding differs from the work of Song et al. [
20] by around
eV.
The results given in
Section 3.3 can also be indirectly used in the discussion of interactions between DOXH and graphene, as given in
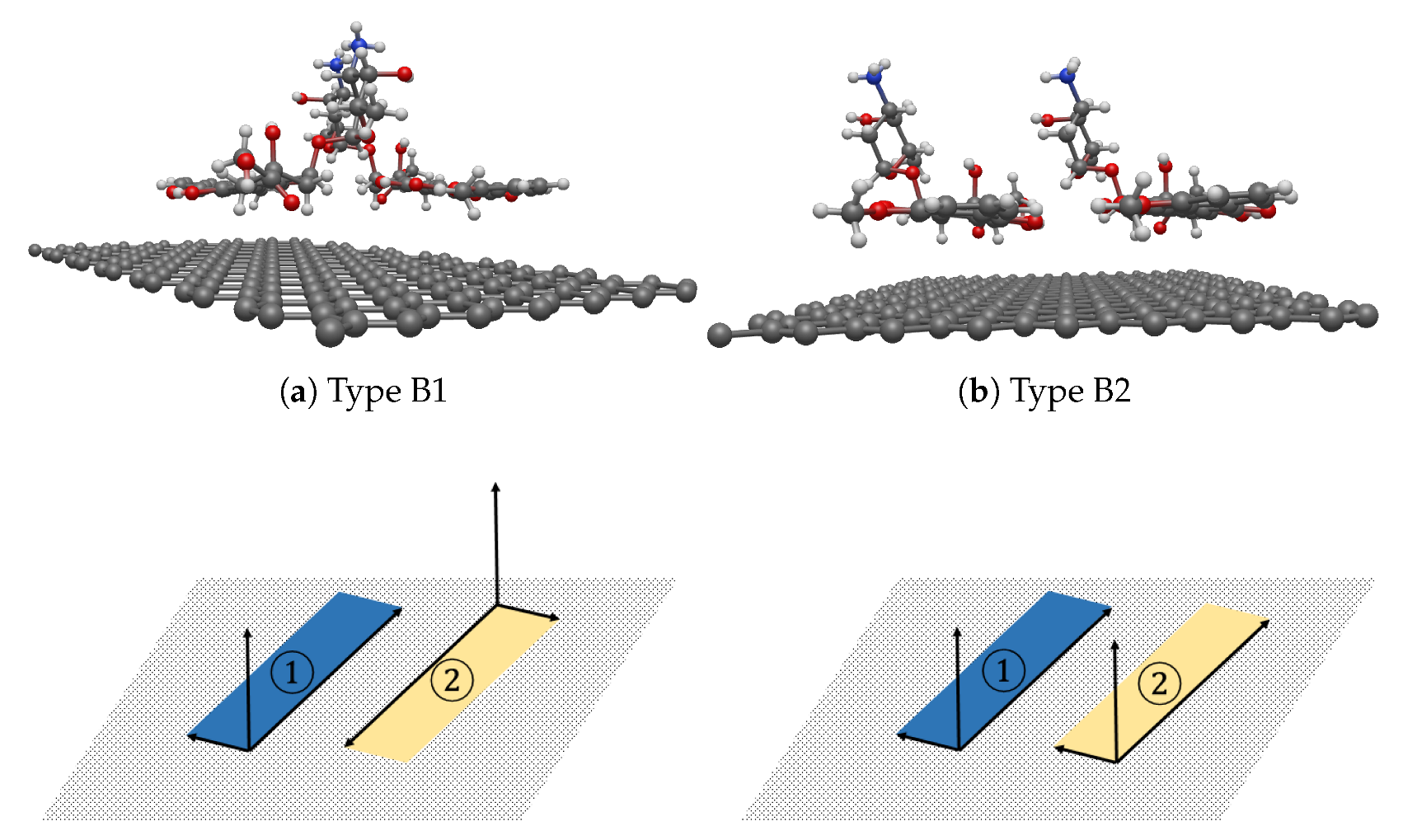
Section 3.2. Both stable configurations, Type B1 and B2, show parallel configurations but in a different direction from the carbon ring planes. For seed III-9, representing Type B1, the closest atoms from each of the two DOXHs to the graphene sheet are H
s with the same distance of
Å (
Å obtained in
Section 3.2). Further, the H
atoms from both DOXHs have a distance of
Å from the graphene sheet (
Å obtained in
Section 3.2). At the steady state, the average distance
from the graphene to each DOXH molecule was
Å, which is exactly the same as obtained in
Section 3.2.
For seed III-8, representing Type B2, we achieve the distance between the H
s and the sheet as
Å and
Å, and those from the H
s to the graphene as
Å and
Å for the first and second DOXH, respectively. The average distances
between each of the DOXH carbon ring planes to the graphene are
Å and
Å. The values obtained here are all comparable with the previous studies but use different techniques [
18,
20,
35].
In terms of the energy, the total energy of the two systems of a DOXH interacting with a graphene sheet is
eV, which is greater than the energy obtained in
Section 3.3,
to
eV. Therefore, the results are precise, and we achieve a more stable system. Additionally, the energy of the most stable configuration of two DOXHs obtained in
Section 3.1 is around
eV, the interaction energy between a DOXH and graphene reported in
Section 3.2 is
eV, and the addition of the energies from these two systems indicates a possible configuration of a two-DOXH and graphene system, as obtained in
Section 3.3. It gives a possible energy value of
eV, which is greater than
eV. Hence, the energy values of these two possible, stable configurations, Type B1 and Type B2, do not conflict with previous results.
The main discrepancy in our work from others is the use of DOXH instead of using DOX. This causes only a minor difference in the drug structure and the atomic positions; however, our findings are in good agreement and have proximate values in both distance and energy. Therefore, using a metaheuristic algorithm, we used NSGA-II and U-NSGA-III, is another approach to obtaining a stable configuration of nanoparticles. The concept of these algorithms is quite different from well-known methods, such as density function theory and molecular dynamic simulation. The density function theory investigates the electronic structure of a group of molecules to form a stable configuration, and molecular dynamic simulation measures the energy of molecules by simulating the movements of atoms and molecules using the valet algorithm. Here NSGA-II and U-NSGA-III aim to optimize and get the best solution by adapting over generations.
5. Summary
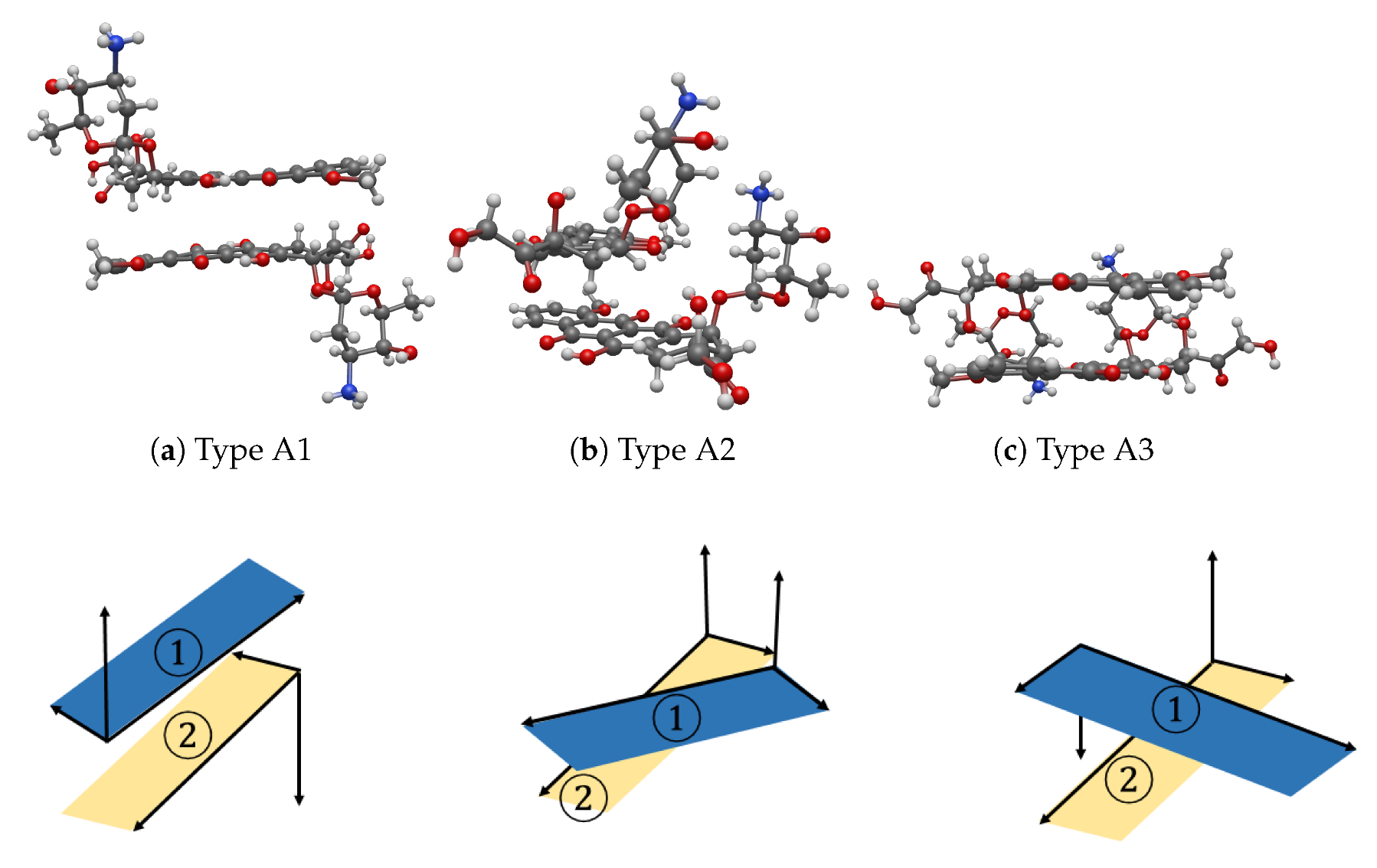
We have studied three systems: (i) the interaction energy between two DOXH molecules, (ii) the interaction energy between a DOXH and a flat graphene sheet and (iii) the interaction energy between two DOXHs and a flat graphene. Each system consists of 30 experiments using NSGA-II and U-NSGA-III algorithms to find the most stable structure. All systems show that the stable configurations have remarkable relationship with the inclined angle and the rotated angle between two carbon ring planes of DOXHs and the graphene sheet.
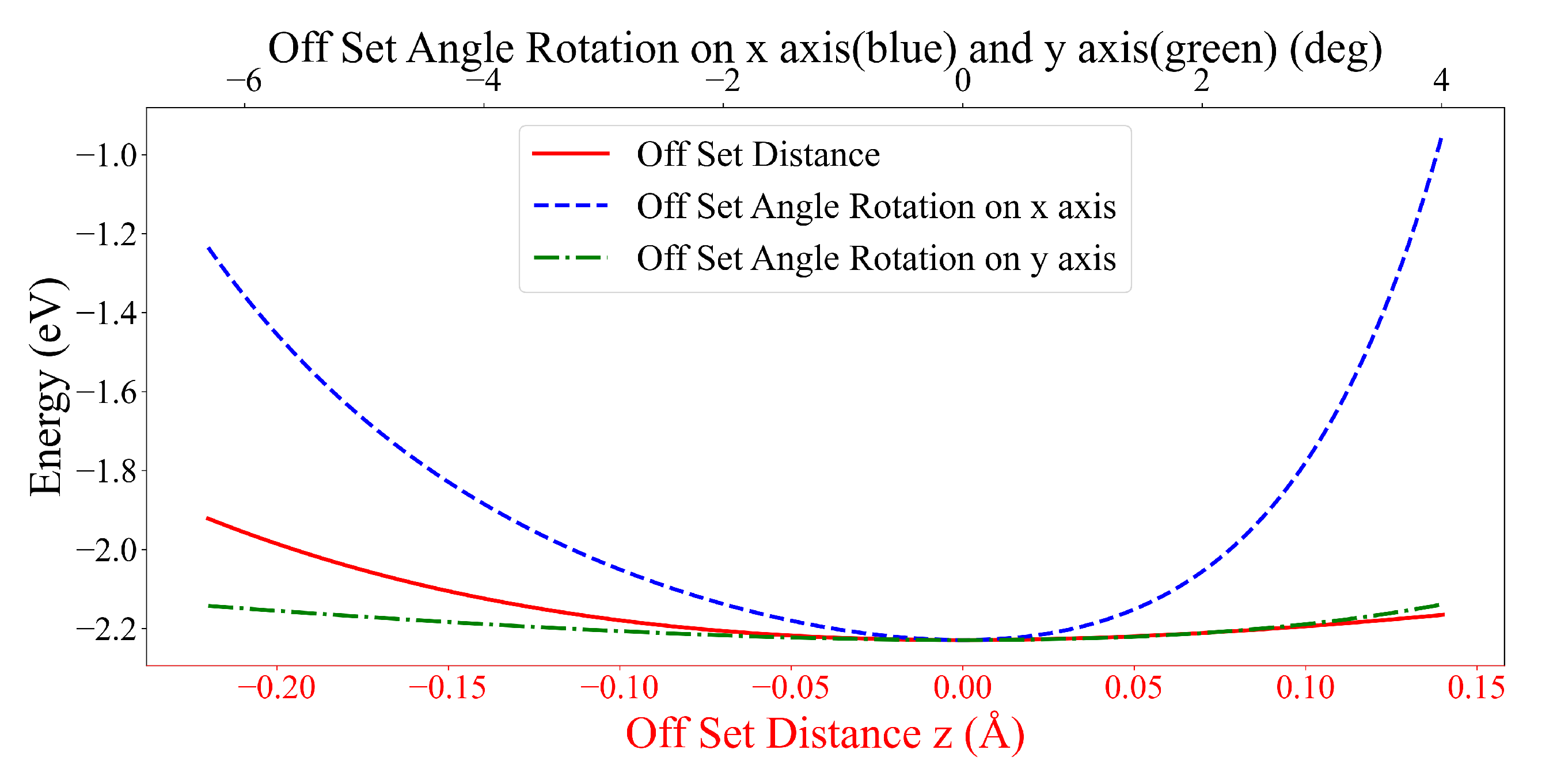
System (i) shows three possible, stable configurations where their carbon ring planes of two DOXHs are oppositely parallel, overlapping or perpendicular. The perpendicular configuration gives the lowest energy, of, on average, around eV, which has and in the inclined and the rotated angles, respectively. The DOXH molecules are about Å away from their center of masses. The other two configurations show small differences in the energy values, which are eV for parallel and eV for overlapping configurations.
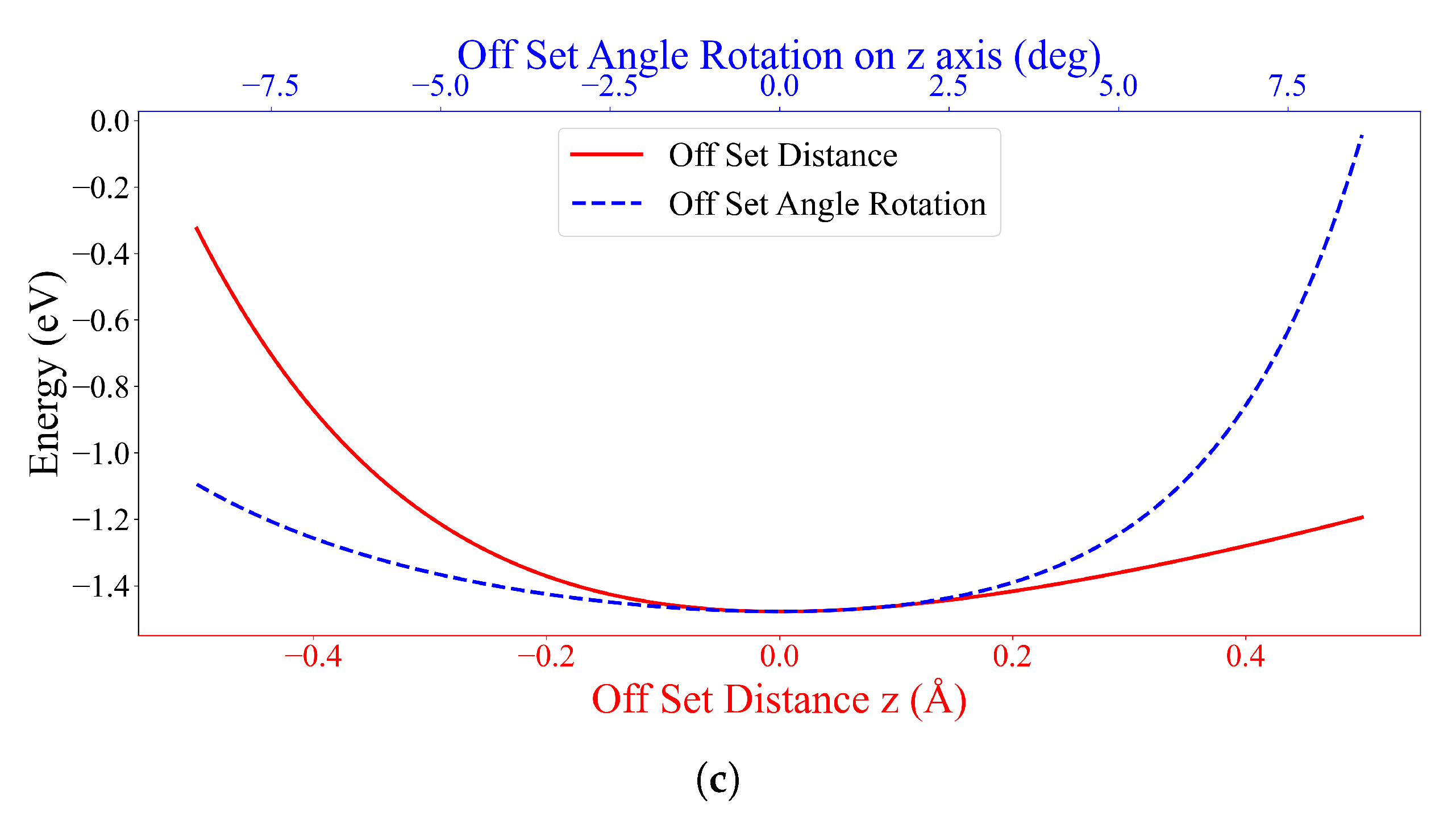
All experiments on system (ii) result in the same outcome, where the most stable configuration is the parallel form of the DOXH carbon ring plane and the graphene with in the inclined angle and an energy of eV. The DOXH is 4.59 Å away from the graphene, and its closest atom to the graphene is H, with a distance of 2.57 Å. Lastly, system (iii) shows two equivalently stable configurations, the opposite direction and the same direction of parallel DOXH molecules. In both cases, two DOXHs are parallel to the graphene with an energy of to eV.
Our findings are based on an elementary mathematical derivation, and the use of heuristic algorithms are comparable with previous studies where they employ expensive computational calculations. Therefore, this theoretical study can be thought of as a first step in designing a DOXH interaction with graphene in the drug delivery system, which can reduce the time of calculation.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}