
Preparation and Characterization of Gluten/SDS/Chitosan Composite Hydrogel Based on Hydrophobic and Electrostatic Interactions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Preparation of the Hydrogel
2.3. Characterization of the Hydrogel
2.3.1. Scanning Electron Microscope (SEM)
2.3.2. Thermal Analysis
2.3.3. Dynamic Rheological Measurements
2.3.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.3.5. X-ray Diffraction (XRD)
2.3.6. Measurement of Intrinsic Fluorescence Spectra
2.3.7. Measurement of UV Spectra
2.3.8. Determination of Surface Hydrophobicity
2.3.9. Particle Size and Zeta-Potential Analysis
2.3.10. Swelling Behavior of the Hydrogels
2.3.11. In Vitro Cytotoxicity
2.3.12. Statistical Analysis
3. Results and Discussion
3.1. Preparation of the Hydrogel
3.2. Characterization of the Hydrogel
3.2.1. Scanning Electron Microscope (SEM)
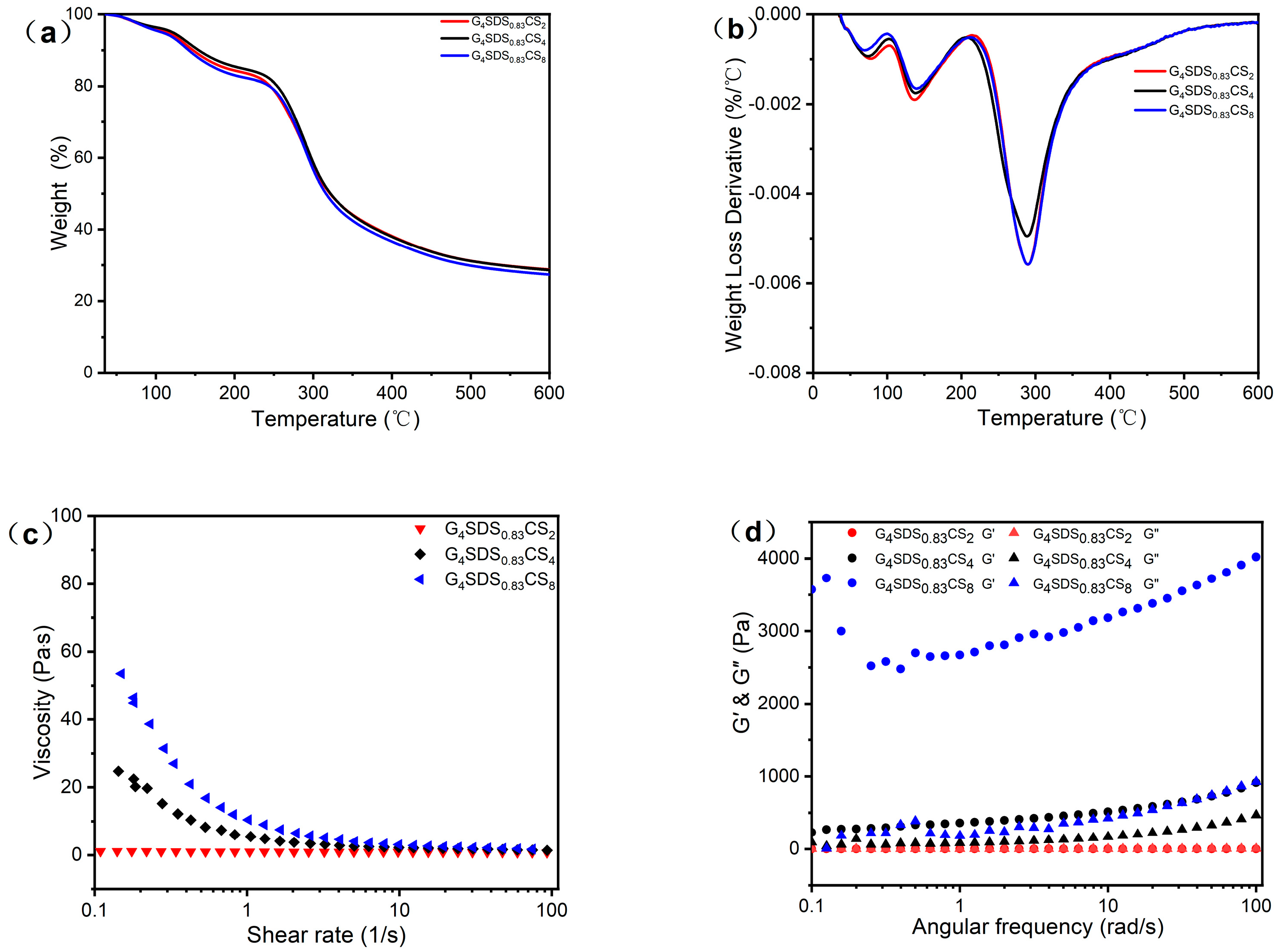
3.2.2. Thermal Analysis
3.2.3. Dynamic Rheological Measurements
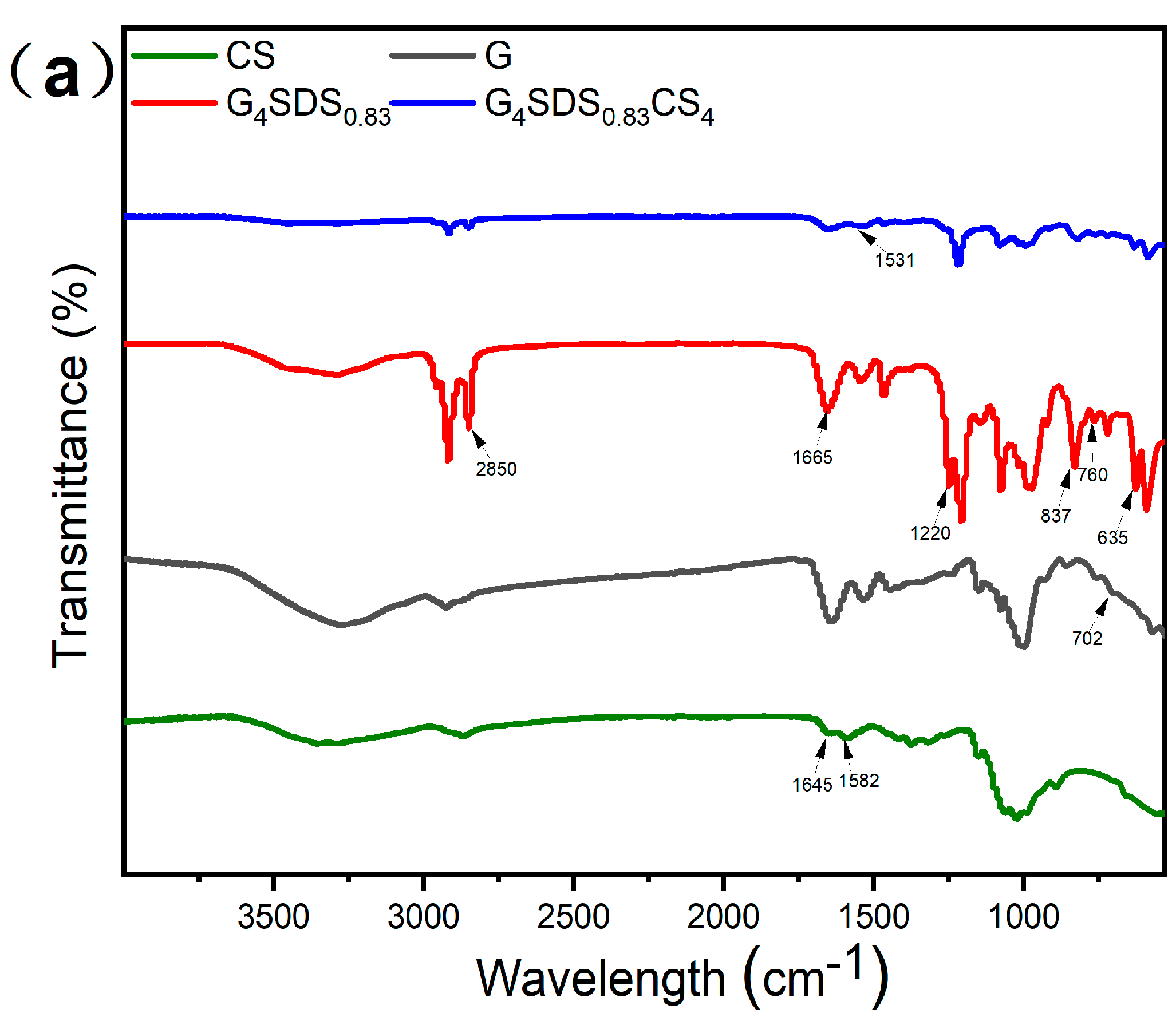
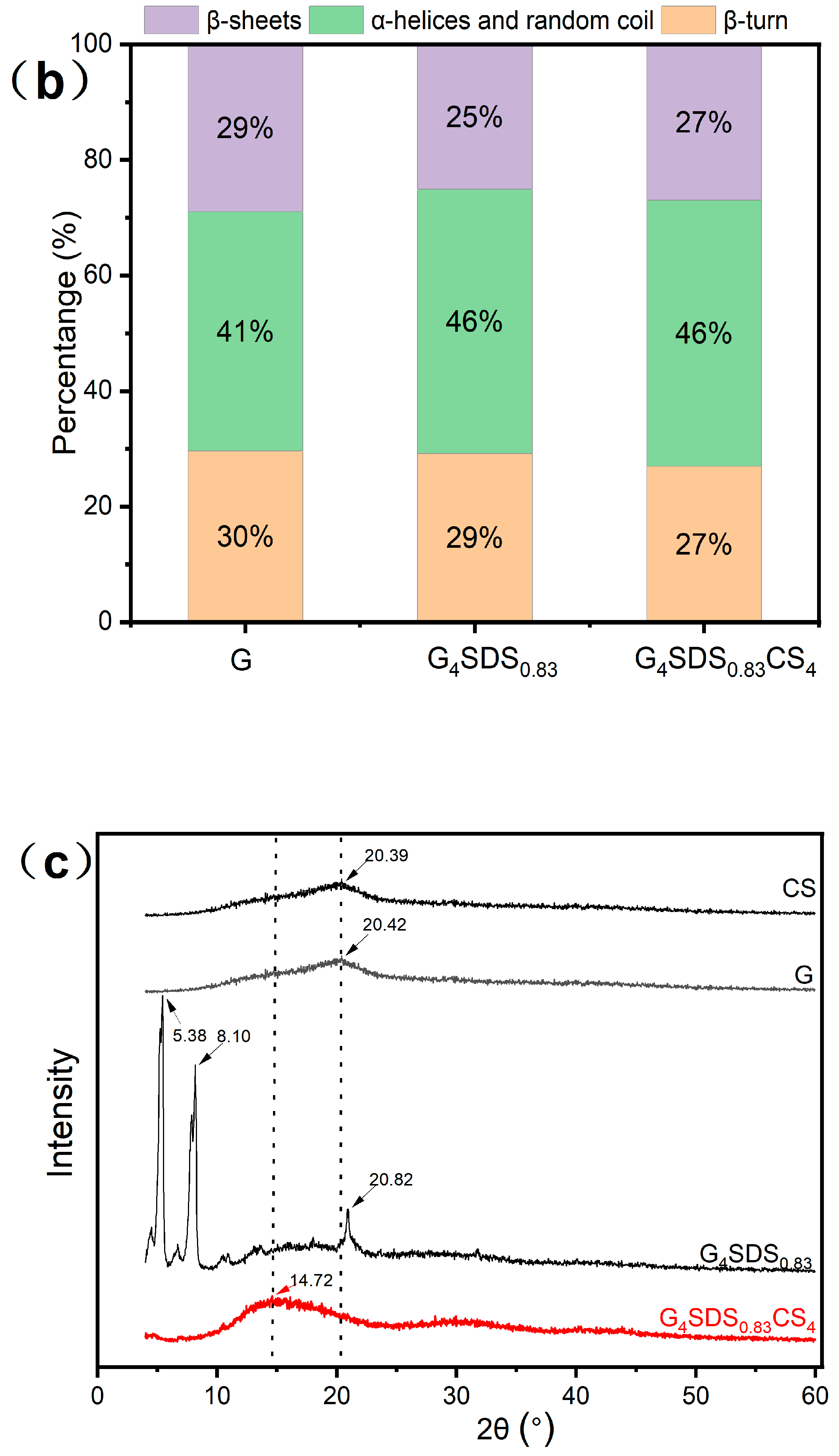
3.2.4. Fourier Transform Infrared Spectroscopy (FTIR)
3.2.5. X-ray Diffraction (XRD)
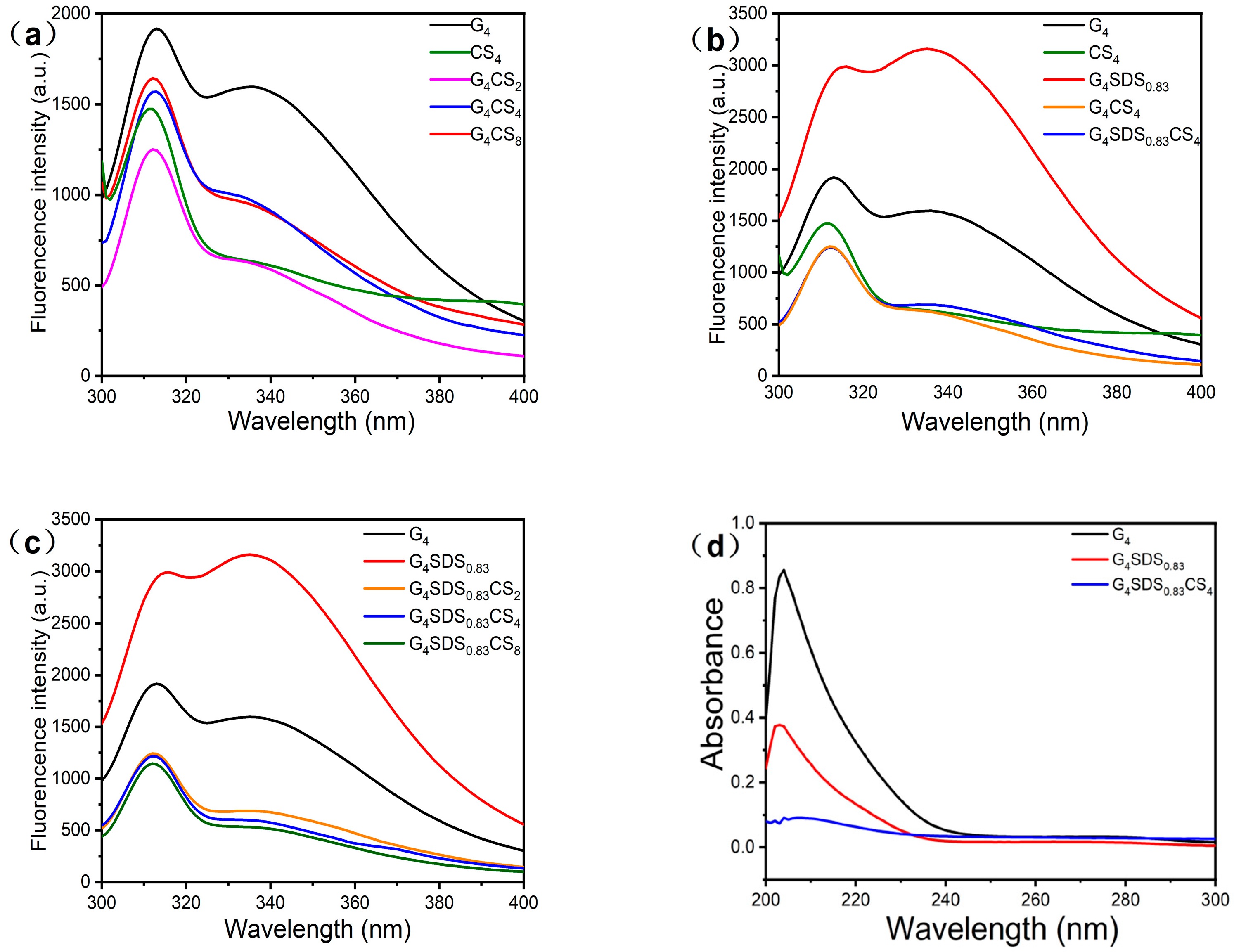
3.2.6. Measurement of Intrinsic Fluorescence Spectra
3.2.7. Measurement of UV Spectra
3.2.8. Determination of Surface Hydrophobicity
3.2.9. Particle Size and Zeta-Potential Analysis
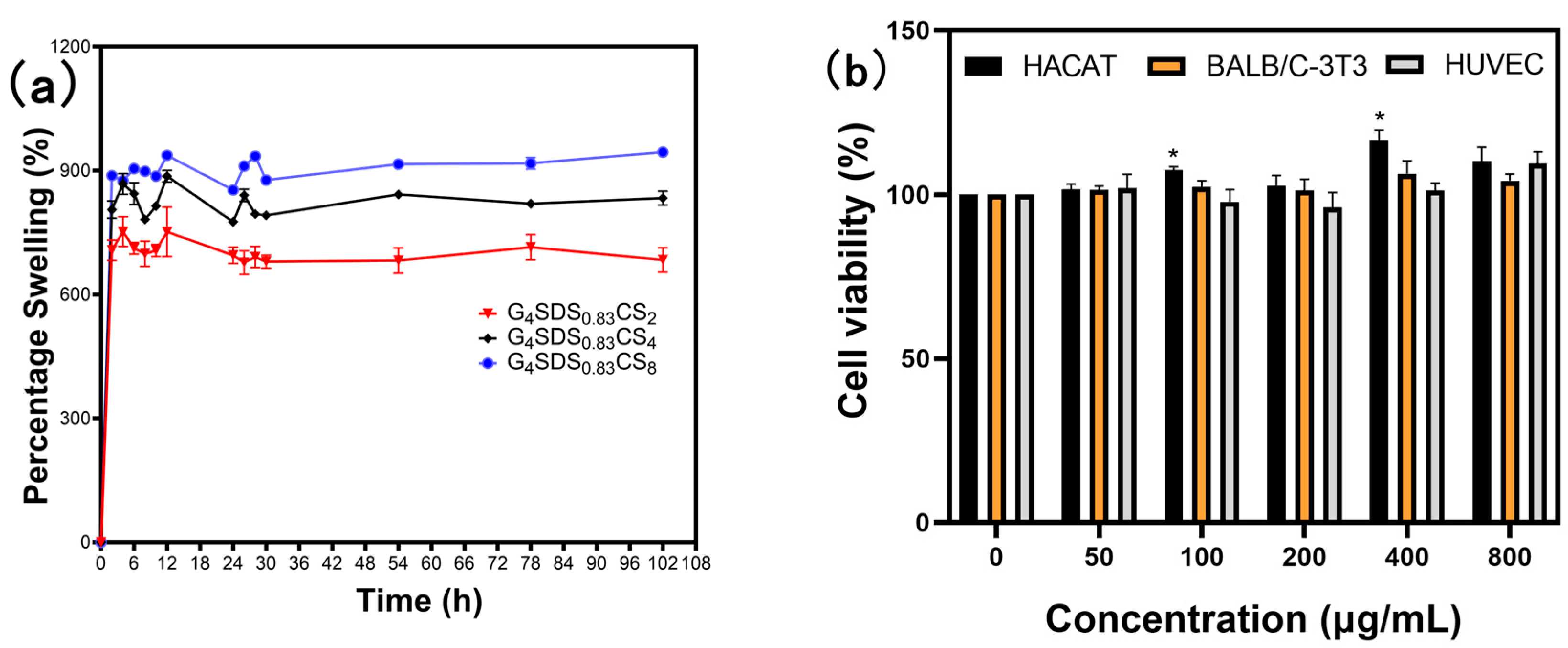
3.2.10. Swelling Behavior of the Hydrogels
3.2.11. In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhao, X.; Wu, H.; Guo, B.; Dong, R.; Qiu, Y.; Ma, P.X. Antibacterial Anti-Oxidant Electroactive Injectable Hydrogel as Self-Healing Wound Dressing with Hemostasis and Adhesiveness for Cutaneous Wound Healing. Biomaterials 2017, 122, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Meng, L.; Cui, C.; Yang, J. An Integrated Self-Healable and Robust Conductive Hydrogel for Dynamically Self-Adhesive and Highly Conformable Electronic Skin. J. Mater. Chem. C 2019, 7, 15208–15218. [Google Scholar] [CrossRef]
- Shay, T.; Dickey, M.D.; Velev, O.D. Hydrogel-Enabled Osmotic Pumping for Microfluidics: Towards Wearable Human-Device Interfaces. Lab Chip 2017, 17, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, C.; Fernandes, S.; Godinho, M.; Borges, J.; Soares, P. Functional Stimuli-Responsive Gels: Hydrogels and Microgels. Gels 2018, 4, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banc, A.; Charbonneau, C.; Dahesh, M.; Appavou, M.-S.; Fu, Z.; Morel, M.-H.; Ramos, L. Small Angle Neutron Scattering Contrast Variation Reveals Heterogeneities of Interactions in Protein Gels. Soft Matter 2016, 12, 5340–5352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, M.; Wehrli, M.C.; Becker, T.; Jekle, M.; Scherf, K.A. Fundamental Characterization of Wheat Gluten. Eur. Food Res. Technol. 2021, 247, 985–997. [Google Scholar] [CrossRef]
- Liu, C.; McClements, D.J.; Li, M.; Xiong, L.; Sun, Q. Development of Self-Healing Double-Network Hydrogels: Enhancement of the Strength of Wheat Gluten Hydrogels by In Situ Metal–Catechol Coordination. J. Agric. Food Chem. 2019, 67, 6508–6516. [Google Scholar] [CrossRef]
- Barak, S.; Mudgil, D.; Khatkar, B.S. Biochemical and Functional Properties of Wheat Gliadins: A Review. Crit. Rev. Food Sci. Nutr. 2015, 55, 357–368. [Google Scholar] [CrossRef]
- Belton, P.S. New Approaches to Study the Molecular Basis of the Mechanical Properties of Gluten. J. Cereal Sci. 2005, 41, 203–211. [Google Scholar] [CrossRef]
- Negm, N.A.; Hefni, H.H.H.; Abd-Elaal, A.A.A.; Badr, E.A.; Abou Kana, M.T.H. Advancement on Modification of Chitosan Biopolymer and Its Potential Applications. Int. J. Biol. Macromol. 2020, 152, 681–702. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Sudheesh Kumar, P.T.; Nair, S.V.; Tamura, H. Biomaterials Based on Chitin and Chitosan in Wound Dressing Applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Mahinroosta, M.; Jomeh Farsangi, Z.; Allahverdi, A.; Shakoori, Z. Hydrogels as Intelligent Materials: A Brief Review of Synthesis, Properties and Applications. Mater. Today Chem. 2018, 8, 42–55. [Google Scholar] [CrossRef]
- Sánchez-Cid, P.; Jiménez-Rosado, M.; Romero, A.; Pérez-Puyana, V. Novel Trends in Hydrogel Development for Biomedical Applications: A Review. Polymers 2022, 14, 3023. [Google Scholar] [CrossRef]
- Cui, C.; Shao, C.; Meng, L.; Yang, J. High-Strength, Self-Adhesive, and Strain-Sensitive Chitosan/Poly (Acrylic Acid) Double-Network Nanocomposite Hydrogels Fabricated by Salt-Soaking Strategy for Flexible Sensors. ACS Appl. Mater. Interfaces 2019, 11, 39228–39237. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Duan, L.; Ren, X.; Gao, G. Hydrophobic Association Hydrogels with Excellent Mechanical and Self-Healing Properties. Eur. Polym. J. 2019, 112, 660–669. [Google Scholar] [CrossRef]
- Yazdani, S.; Khan, M.; Shahzad, A.; Shah, L.A.; Ye, D. Ionic Conductive Hydrogels Formed through Hydrophobic Association for Flexible Strain Sensing. Sens. Actuators A Phys. 2023, 350, 114148. [Google Scholar] [CrossRef]
- Reeve, P.J.; Fallowfield, H.J. Natural and Surfactant Modified Zeolites: A Review of Their Applications for Water Remediation with a Focus on Surfactant Desorption and Toxicity towards Microorganisms. J. Environ. Manag. 2018, 205, 253–261. [Google Scholar] [CrossRef]
- Fatma, I.; Sharma, V.; Thakur, R.C.; Kumar, A. Current Trends in Protein-Surfactant Interactions: A Review. J. Mol. Liq. 2021, 341, 117344. [Google Scholar] [CrossRef]
- Holme, I. Recent Developments in Colorants for Textile Applications. Surf. Coat. Int. Part B Coat. Trans. 2002, 85, 243–264. [Google Scholar] [CrossRef]
- Khan, T.A.; Mahler, H.-C.; Kishore, R.S.K. Key Interactions of Surfactants in Therapeutic Protein Formulations: A Review. Eur. J. Pharm. Biopharm. 2015, 97, 60–67. [Google Scholar] [CrossRef]
- Zhou, Y.; Zeng, G.; Zhang, F.; Luo, J.; Li, X.; Li, J.; Fang, Z. Toward Utilization of Agricultural Wastes: Development of a Novel Keratin Reinforced Soybean Meal-Based Adhesive. ACS Sustain. Chem. Eng. 2021, 9, 7630–7637. [Google Scholar] [CrossRef]
- Li, C.; Chen, G.; Ran, C.; Liu, L.; Wang, S.; Xu, Y.; Tan, Y.; Kan, J. Adlay Starch-Gluten Composite Gel: Effects of Adlay Starch on Rheological and Structural Properties of Gluten Gel to Molecular and Physico-Chemical Characteristics. Food Chem. 2019, 289, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.H.; Nisar, S.; Imtiyaz, K.; Nadeem, M.; Mazumdar, N.; Rizvi, M.M.A.; Ahmad, S. Injectable, Self-Healing, and Biocompatible N, O-Carboxymethyl Chitosan/Multialdehyde Guar Gum Hydrogels for Sustained Anticancer Drug Delivery. Biomacromolecules 2021, 22, 3731–3745. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Sparavigna, A.C.; Pisano, R.; Barresi, A.A. Measuring and Predicting Pore Size Distribution of Freeze-Dried Solutions. Dry. Technol. 2019, 37, 435–447. [Google Scholar] [CrossRef]
- Chen, F.; Gällstedt, M.; Olsson, R.T.; Gedde, U.W.; Hedenqvist, M.S. A Novel Chitosan/Wheat Gluten Biofoam Fabricated by Spontaneous Mixing and Vacuum-Drying. RSC Adv. 2015, 5, 94191–94200. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Nisar, S.; Chauhan, S.; Singh, V.; Rattan, S. Soy Protein-Based Hydrogel under Microwave-Induced Grafting of Acrylic Acid and 4-(4-Hydroxyphenyl) Butanoic Acid: A Potential Vehicle for Controlled Drug Delivery in Oral Cavity Bacterial Infections. ACS Omega 2020, 5, 21610–21622. [Google Scholar] [CrossRef]
- Li, J.; Luo, J.; Li, X.; Yi, Z.; Gao, Q.; Li, J. Soybean Meal-Based Wood Adhesive Enhanced by Ethylene Glycol Diglycidyl Ether and Diethylenetriamine. Ind. Crops Prod. 2015, 74, 613–618. [Google Scholar] [CrossRef]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric Scaffolds in Tissue Engineering Application: A Review. Int. J. Polym. Sci. 2011, 2011, 290602. [Google Scholar] [CrossRef]
- Si, X.; Li, T.; Zhang, Y.; Zhang, W.; Qian, H.; Li, Y.; Zhang, H.; Qi, X.; Wang, L. Interactions between Gluten and Water-Unextractable Arabinoxylan during the Thermal Treatment. Food Chem. 2021, 345, 128785. [Google Scholar] [CrossRef]
- Pan, J.; Li, Y.; Chen, K.; Zhang, Y.; Zhang, H. Enhanced Physical and Antimicrobial Properties of Alginate/Chitosan Composite Aerogels Based on Electrostatic Interactions and Noncovalent Crosslinking. Carbohydr. Polym. 2021, 266, 118102. [Google Scholar] [CrossRef]
- He, H.; Cao, X.; Dong, H.; Ma, T.; Payne, G.F. Reversible Programing of Soft Matter with Reconfigurable Mechanical Properties. Adv. Funct. Mater. 2017, 27, 1605665. [Google Scholar] [CrossRef]
- Lawrie, G.; Keen, I.; Drew, B.; Chandler-Temple, A.; Rintoul, L.; Fredericks, P.; Grøndahl, L. Interactions between Alginate and Chitosan Biopolymers Characterized Using FTIR and XPS. Biomacromolecules 2007, 8, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Valentin, R.; Bonelli, B.; Garrone, E.; Di Renzo, F.; Quignard, F. Accessibility of the Functional Groups of Chitosan Aerogel Probed by FT-IR-Monitored Deuteration. Biomacromolecules 2007, 8, 3646–3650. [Google Scholar] [CrossRef] [PubMed]
- Chakvattanatham, K.; Phattanarudee, S.; Kiatkamjornwong, S. Anionically Surface-modified Pigment/Binder Ink Jet Inks for Silk Fabric Printing. Pigment Resin Technol. 2010, 39, 327–341. [Google Scholar] [CrossRef]
- Skendi, A.; Mouselemidou, P.; Papageorgiou, M.; Papastergiadis, E. Effect of Acorn Meal-Water Combinations on Technological Properties and Fine Structure of Gluten-Free Bread. Food Chem. 2018, 253, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fevzioglu, M.; Ozturk, O.K.; Hamaker, B.R.; Campanella, O.H. Quantitative Approach to Study Secondary Structure of Proteins by FT-IR Spectroscopy, Using a Model Wheat Gluten System. Int. J. Biol. Macromol. 2020, 164, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Zhao, J.; Mu, M.; Jia, F.; Wang, Q.; Liang, Y.; Wang, J. Aggregative and Structural Properties of Wheat Gluten Induced by Pectin. J. Cereal Sci. 2021, 100, 103247. [Google Scholar] [CrossRef]
- Guo, Q.; Su, J.; Shu, X.; Yuan, F.; Mao, L.; Liu, J.; Gao, Y. Fabrication, Structural Characterization and Functional Attributes of Polysaccharide-Surfactant-Protein Ternary Complexes for Delivery of Curcumin. Food Chem. 2021, 337, 128019. [Google Scholar] [CrossRef]
- George, D.; Maheswari, P.U.; Sheriffa Begum, K.M.M.; Arthanareeswaran, G. Biomass-Derived Dialdehyde Cellulose Cross-Linked Chitosan-Based Nanocomposite Hydrogel with Phytosynthesized Zinc Oxide Nanoparticles for Enhanced Curcumin Delivery and Bioactivity. J. Agric. Food Chem. 2019, 67, 10880–10890. [Google Scholar] [CrossRef]
- Tang, C.; Yang, X.-Q.; Chen, Z.; Wu, H.; Peng, Z.-Y. Physicochemical and Structural Characteristics of Sodium Caseinate Biopolymers Induced by Microbial Transglutaminase. J. Food Biochem. 2005, 29, 402–421. [Google Scholar] [CrossRef]
- Wang, K.-Q.; Luo, S.-Z.; Zhong, X.-Y.; Cai, J.; Jiang, S.-T.; Zheng, Z. Changes in Chemical Interactions and Protein Conformation during Heat-Induced Wheat Gluten Gel Formation. Food Chem. 2017, 214, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Stănciuc, N.; Banu, I.; Bolea, C.; Patraşcu, L.; Aprodu, I. Structural and Antigenic Properties of Thermally Treated Gluten Proteins. Food Chem. 2018, 267, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, G.; Zhang, H.; Yu, Q.; Mei, X.; Kan, J. Heat-Induced Inulin-Gluten Gel: Insights into the Influences of Inulin Molecular Weight on the Rheological and Structural Properties of Gluten Gel to Molecular and Physicochemical Characteristics. Food Hydrocoll. 2021, 111, 106397. [Google Scholar] [CrossRef]
- Liu, K.; Huang, R.-L.; Zha, X.-Q.; Li, Q.-M.; Pan, L.-H.; Luo, J.-P. Encapsulation and Sustained Release of Curcumin by a Composite Hydrogel of Lotus Root Amylopectin and Chitosan. Carbohydr. Polym. 2020, 232, 115810. [Google Scholar] [CrossRef]
- Dai, L.; Sun, C.; Wei, Y.; Zhan, X.; Mao, L.; Gao, Y. Formation and Characterization of Zein-Propylene Glycol Alginate-Surfactant Ternary Complexes: Effect of Surfactant Type. Food Chem. 2018, 258, 321–330. [Google Scholar] [CrossRef]
- Xu, Z.; Tang, E.; Zhao, H. An Environmentally Sensitive Silk Fibroin/Chitosan Hydrogel and Its Drug Release Behaviors. Polymers 2019, 11, 1980. [Google Scholar] [CrossRef] [Green Version]
- Gul, K.; Gan, R.-Y.; Sun, C.-X.; Jiao, G.; Wu, D.-T.; Li, H.-B.; Kenaan, A.; Corke, H.; Fang, Y.-P. Recent Advances in the Structure, Synthesis, and Applications of Natural Polymeric Hydrogels. Crit. Rev. Food Sci. Nutr. 2022, 62, 3817–3832. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2012, 65, 157–170. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Hamedi, H.; Moradi, S.; Hudson, S.M.; Tonelli, A.E. Chitosan Based Hydrogels and Their Applications for Drug Delivery in Wound Dressings: A Review. Carbohydr. Polym. 2018, 199, 445–460. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | G (w/v) a | SDS (mM) | CS (v/v) a |
|---|---|---|---|
| G2SDS0.83CS2 | 2% | 8.3 | 2% |
| G2SDS0.83CS4 | 2% | 8.3 | 4% |
| G2SDS0.83CS8 | 2% | 8.3 | 8% |
| G4SDS0.83CS2 | 4% | 8.3 | 2% |
| G4SDS0.83CS4 | 4% | 8.3 | 4% |
| G4SDS0.83CS8 | 4% | 8.3 | 8% |
| G4 | 4% | 0% | 0% |
| G4SDS0.83 | 4% | 8.3 | 0% |
| CS4 | 0% | 0% | 4% |
| G2CS2 | 2% | 0% | 2% |
| G2CS4 | 2% | 0% | 4% |
| G2CS8 | 2% | 0% | 8% |
| G4CS2 | 4% | 0% | 2% |
| G4CS4 | 4% | 0% | 4% |
| G4CS8 | 4% | 0% | 8% |
| Samples | T10% a (°C) | T30% a (°C) | T50%a (°C) | Td b (°C) | Residual Mass (wt % ) |
|---|---|---|---|---|---|
| G4 | 276 | 306 | 329 | 308 | 25.10 |
| G4SDS0.83 | 255 | 290 | 343 | 281 | 29.77 |
| G4CS4 | 176 | 284 | 353 | 292 | 41.41 |
| G4SDS0.83CS2 | 147 | 274 | 321 | 290 | 30.15 |
| G4SDS0.83CS4 | 154 | 279 | 322 | 289 | 30.29 |
| G4SDS0.83CS8 | 142 | 278 | 316 | 288 | 28.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Lan, N.; Huang, Y.; Mo, C.; Wang, Q.; Wu, C.; Wang, Y. Preparation and Characterization of Gluten/SDS/Chitosan Composite Hydrogel Based on Hydrophobic and Electrostatic Interactions. J. Funct. Biomater. 2023, 14, 222. https://doi.org/10.3390/jfb14040222
Li G, Lan N, Huang Y, Mo C, Wang Q, Wu C, Wang Y. Preparation and Characterization of Gluten/SDS/Chitosan Composite Hydrogel Based on Hydrophobic and Electrostatic Interactions. Journal of Functional Biomaterials. 2023; 14(4):222. https://doi.org/10.3390/jfb14040222
Chicago/Turabian StyleLi, Guangfeng, Ni Lan, Yanling Huang, Chou Mo, Qiaoli Wang, Chaoxi Wu, and Yifei Wang. 2023. "Preparation and Characterization of Gluten/SDS/Chitosan Composite Hydrogel Based on Hydrophobic and Electrostatic Interactions" Journal of Functional Biomaterials 14, no. 4: 222. https://doi.org/10.3390/jfb14040222