
Evaluating the Performance of Multiple Sequence Alignment Programs with Application to Genotyping SARS-CoV-2 in the Saudi Population

Abstract
:1. Introduction
1.1. General Background
1.2. Problem Statement
1.3. Research Questions and Significance of the Study
- How do individual MSAs, specifically Clustal, MAFFT, and MUSCLE, compare in terms of accuracy and efficiency?
- What is the comparative performance of MSAs assembled using MergeAlign against individual MSA solutions?
- How does a reference sequence such as the SARS-CoV-2 Wuhan-Hu-1 isolate compare with consensus sequences in terms of their effectiveness for genotyping?
- How many SNPs and mutations occur specifically within the coding regions of the Saudi SARS-CoV-2 dataset, and how does this compare with results published globally?
- How do the genetic relationships and transmission dynamics of the Saudi SARS-CoV-2 dataset differ from the global SARS-CoV-2 dataset?
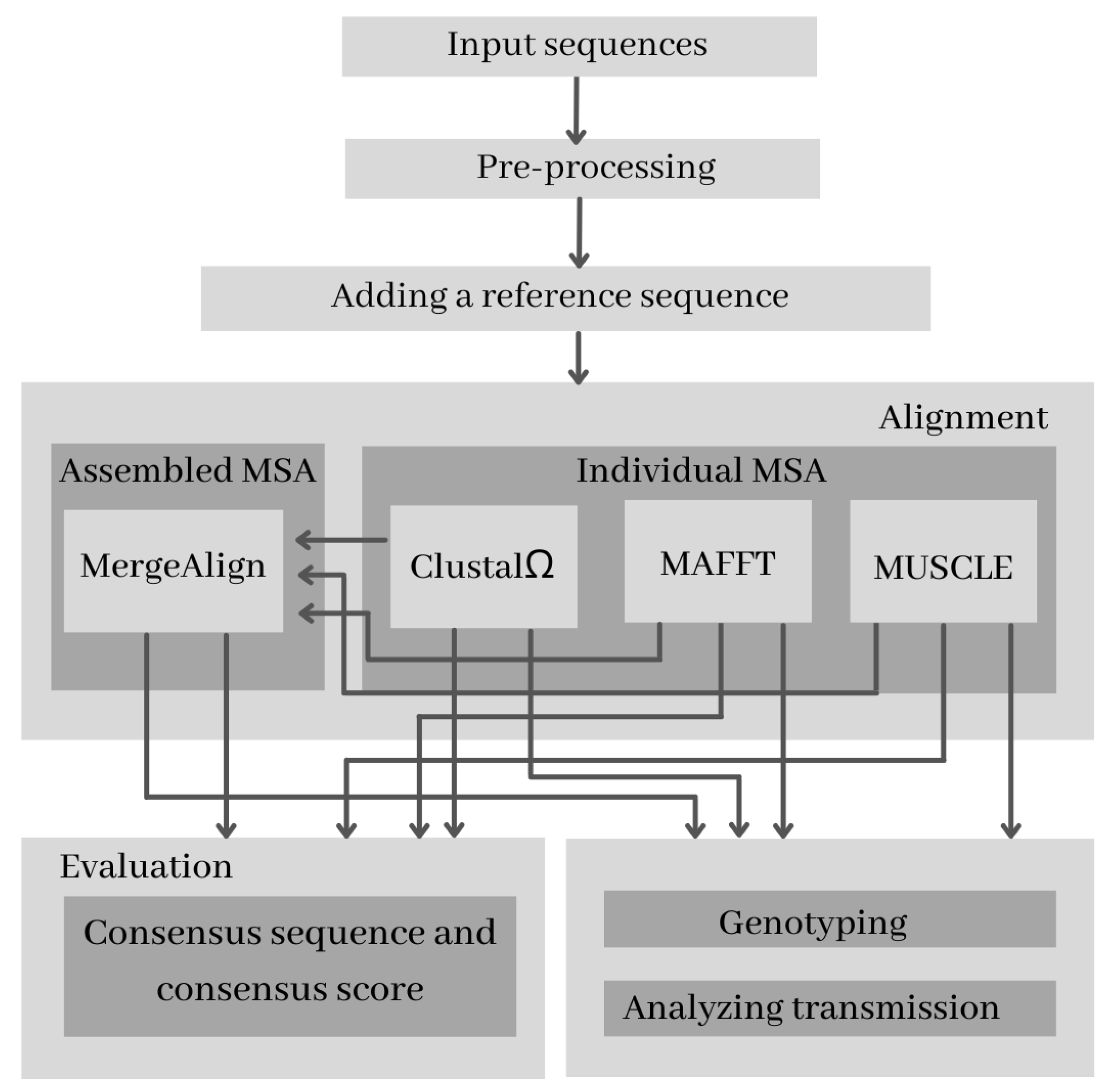
2. Methodology
2.1. Dataset and Preprocessing
2.2. Reference Sequence Selection Approaches
2.2.1. Reference-Based Approach
2.2.2. Consensus-Based Approach
2.3. Alignment Selection Approaches
2.3.1. Individual MSA Alignment
2.3.2. Assembled MSA Alignment
2.4. Genotyping
2.5. Analyzing Transmissions
2.6. Performance Evaluation Metrics
2.7. k-Tuple Size Selection
2.8. Experimental Environment
3. Results and Discussion
3.1. Evaluating the Performance of MSA Programs
3.1.1. Computational Resources Usage
3.1.2. Accuracy of MSA Programs
3.1.3. The Impact of the k Value
3.2. Genotyping SARS-CoV-2 for the Saudi Population
3.2.1. Discovered SNPs and Mutations
Identified SNPs and Mutations
Identified SNPs and Mutations Inside Coding Regions
Identified High-Frequency SNPs
Identified High-Frequency SNPs Inside Coding Regions
3.2.2. Comparing High-Frequency SNPs between the Saudi and Global SARS-CoV-2 Datasets
3.2.3. Analyzing Transmissions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pearson, W. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Bucka-Lassen, K.; Caprani, O.; Hein, J. Combining many multiple alignments in one improved alignment. Bioinformatics 1999, 15, 122–130. [Google Scholar] [CrossRef]
- Collingridge, P.; Kelly, S. MergeAlign: Improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. BMC Bioinform. 2012, 13, 117. [Google Scholar] [CrossRef]
- Chatzou, M.; Magis, C.; Chang, J.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Briefings Bioinform. 2015, 17, 1009–1023. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Saha, I.; Ghosh, N.; Pradhan, A.; Sharma, N.; Maity, D.; Mitra, K. Whole genome analysis of more than 10000 SARS-CoV-2 virus unveils global genetic diversity and target region of NSP6. Briefings Bioinform. 2021, 22, 1106–1121. [Google Scholar] [CrossRef]
- Mok, P.; Koh, A.; Farhana, A.; Alsrhani, A.; Alam, M.; Suresh Kumar, S. Computational drug screening against the SARS-CoV-2 Saudi Arabia isolates through a multiple-sequence alignment approach. Saudi J. Biol. Sci. 2021, 28, 2502–2509. [Google Scholar] [CrossRef]
- Nour, I.; Alenazi, I.; Hanif, A.; Eifan, S. Molecular adaptive evolution of SARS-COV-2 spike protein in Saudi Arabia. Saudi J. Biol. Sci. 2021, 28, 3325–3332. [Google Scholar] [CrossRef] [PubMed]
- Sallam, M.; Ababneh, N.; Dababseh, D.; Bakri, F.; Mahafzah, A. Temporal increase in D614G mutation of SARS-CoV-2 in the Middle East and North Africa. Heliyon 2021, 7, e06035. [Google Scholar] [CrossRef]
- Wang, L. Algorithms for Multiple Sequences Alignment, Comparison of Trees, and Steiner Trees. Ph.D. Thesis, McMaster University, Hamilton, ON, Canada, 1995. [Google Scholar]
- Wang, Y.; Wu, H.; Cai, Y. A benchmark study of sequence alignment methods for protein clustering. BMC Bioinform. 2018, 19, 529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q.; Zhou, J.; Zou, Q. A survey on the algorithm and development of multiple sequence alignment. Briefings Bioinform. 2022, 23, bbac069. [Google Scholar] [CrossRef] [PubMed]
- Pais, F.; Ruy, P.; Oliveira, G.; Coimbra, R. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol. Biol. 2014, 9, 2014. [Google Scholar] [CrossRef]
- Ballouz, S.; Dobin, A.; Gillis, J. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.; Heger, A.; Ponting, C. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.; Wang, W.; Song, Z.; Hu, Y.; Tao, Z.; Tian, J.; Pei, Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Saha, I.; Ghosh, N.; Maity, D.; Sharma, N.; Mitra, K. Inferring the genetic variability in Indian SARS-CoV-2 genomes using consensus of multiple sequence alignment techniques. Infect. Genet. Evol. 2020, 85, 104522. [Google Scholar] [CrossRef]
- Gusfield, D. Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology; Cambridge University Press: Cambridge, MA, USA, 1997. [Google Scholar]
- Karki, R.; Pandya, D.; Elston, R.; Ferlini, C. Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med Genom. 2015, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.; Antao, T.; Chang, J.; Chapman, B.; Cox, C.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Shu, Y.; Cai, Y. Genetic differences among ethnic groups. BMC Genom. 2015, 16, 1093. [Google Scholar] [CrossRef]
- Choudhury, A.; Hazelhurst, S.; Meintjes, A.; Achinike-Oduaran, O.; Aron, S.; Gamieldien, J.; Jalali Sefid Dashti, M.; Mulder, N.; Tiffin, N.; Ramsay, M. Population-specific common SNPs reflect demographic histories and highlight regions of genomic plasticity with functional relevance. BMC Genom. 2014, 15, 437. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Abbreviation | Definition |
|---|---|
| MSA | Multiple sequence alignment. |
| k-tuple | A sequence of k adjacent items or elements in a set, often used in sequence analysis algorithms. |
| k-tuple Algorithm | It is a heuristic method for finding the similarity of sequences by first finding shared k-tuple(s). |
| Individual MSA alignment | An MSA alignment produced by a single MSA program. |
| Assembled MSA alignment | An MSA alignment produced by combining the results of multiple MSA programs. |
| Well-known Reference sequence | A widely recognized and accepted sequence used as a standard or reference. |
| Consensus sequence | A sequence derived from the consensus of multiple sequences. |
| Consensus score | A measure of how similar an MSA alignment is to its consensus sequence. |
| Reference-Based Approach | An approach to MSA that uses a well-known reference sequence to align other sequences. |
| Consensus-Based approach | An approach to MSA that uses a consensus sequence to align other sequences. |
| SNPs | Single nucleotide polymorphisms. |
| High-frequency SNPs | SNPs that occur frequently in a set of sequences (e.g., more than 5% or 10%). |
| SNP profile | A collection of information about the occurrence and characteristics of all SNPs pairs found between a specific sequence. and the reference sequence. |
| Clustal | Clustal Omega a popular MSA program [2]. |
| MAFFT | Multiple Alignment using Fast Fourier Transform a popular MSA program [3]. |
| MUSCLE | Multiple Sequence Comparison by Log Expectation a popular MSA program [4]. |
| MergeAlign | A program for merging the results of multiple MSA programs [6]. |
| Directed Jaccard Distance | A measure of the mutual relationship between two SNP profiles. |
| Characteristics | Raw Dataset | Processed Dataset |
|---|---|---|
| Number of Sequences | 912 | 641 |
| Maximum length | 30,643 | 30,643 |
| Minimum length | 29,300 | 29,300 |
| Average Length | 29,882 | 29,882 |
| Completeness | No | Yes |
| High coverage | No | Yes |
| Contains stretch of ‘NNNNN’ | Yes | No |
| Program | ET | MAX RAM |
|---|---|---|
| Clustal | s h | |
| MAFFT | 283 s min | |
| MUSCLE | s h | |
| MergeAlign | 97 s min |
| Program | ||
|---|---|---|
| Clustal | ||
| MUSCLE |
| Program | Ref.Seq | Cons.Seq | Enhancement |
|---|---|---|---|
| Clustal | 13,934.5 | 13,920.5 | |
| MAFFT | 13,201.5 | 13,178.5 | |
| MUSCLE | 13,238.5 | 13,232.5 | |
| MergeAlign | 13,188.5 | 13,176.5 |
| Program | ||||
|---|---|---|---|---|
| Clustal | 2764.5 | |||
| MAFFT | ||||
| MUSCLE | ||||
| MergeAlign |
| Program | ||||
|---|---|---|---|---|
| Clustal | 29,042 s ≈ 8 h | 18,228 s ≈ 5 h | 13,571 s ≈ 3.77 h | 10,582 s ≈ 2.93 h |
| MAFFT | 60 s ≈ 1 min | 47 s | 31 s | 23 s |
| MUSCLE | 11,875 s ≈ 3.29 h | 9245 s ≈ 2.56 h | 7683 s ≈ 2.13 hrs | 5946 s ≈ 1.65 h |
| MergeAlign | 41 s | 27 s | 22 s | 18 s |
| Program | ||||
|---|---|---|---|---|
| Clustal | ||||
| MAFFT | ||||
| MUSCLE | ||||
| MergeAlign |
| Reference-Based | Consensus-Based | |||
|---|---|---|---|---|
| Program | SNPs | Mutations | SNPs | Mutations |
| Clustal | 35 | 631 | 35 | 631 |
| MAFFT | 32 | 612 | 32 | 613 |
| MUSCLE | 43 | 591 | 38 | 572 |
| MergeAlign | 32 | 612 | 32 | 612 |
| Reference-Based | Consensus-Based | |||
|---|---|---|---|---|
| Program | SNPs | Mutations | SNPs | Mutations |
| Clustal | 29 | 545 | 29 | 545 |
| MAFFT | 29 | 545 | 29 | 545 |
| MUSCLE | 36 | 541 | 34 | 548 |
| MergeAlign | 29 | 545 | 29 | 545 |
| Reference-Based | Consensus-Based | |||
|---|---|---|---|---|
| Program | SNPs | SNPs | SNPs | SNPs |
| Clustal | 15 | 12 | 17 | 10 |
| MAFFT | 16 | 12 | 16 | 10 |
| MUSCLE | 31 | 28 | 25 | 18 |
| MergeAlign | 16 | 12 | 16 | 10 |
| Reference-Based | Consensus-Based | |||
|---|---|---|---|---|
| Program | SNPs | SNPs | SNPs | SNPs |
| Clustal | 12 | 11 | 14 | 10 |
| MAFFT | 14 | 11 | 14 | 10 |
| MUSCLE | 26 | 23 | 22 | 16 |
| MergeAlign | 14 | 11 | 14 | 10 |
| Global [8] (#Seq = 558) | Global [9] (#Seq = 10,098) | Indian [9] (#Seq = 566) | Saudis [Our result](#Seq = 641) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. Pos | SNPs | Freq | RF | Ref. Pos | SNPs | Freq | RF | Ref. Pos | SNPs | Freq | RF | Ref. Pos | SNPs | Freq | RF |
| 241 | 178 | ||||||||||||||
| 1059 | 2048 | ||||||||||||||
| 3037 | 182 | 3037 | 6768 | 3037 | 339 | 3037 | 545 | ||||||||
| 6312 | 177 | ||||||||||||||
| 8782 | 138 | 8782 | 1212 | ||||||||||||
| 11,083 | 115 | 11,083 | 1107 | 11,083 | 189 | ||||||||||
| 184 | |||||||||||||||
| 182 | 14,408 | 6753 | 14,408 | 332 | 14,408 | 603 | |||||||||
| 18,060 | 62 | ||||||||||||||
| 18,877 | 117 | 18,877 | 346 | ||||||||||||
| 19,557 | 2246 | 19,557 | 218 | 0.39 | |||||||||||
| 19,558 | 2260 | 19,558 | 218 | ||||||||||||
| 22,444 | 69 | 22,444 | 130 | ||||||||||||
| 22,506 | 99 | ||||||||||||||
| 22,507 | 99 | ||||||||||||||
| 23,403 | 183 | 23,403 | 6780 | 334 | 23,403 | 483 | |||||||||
| 23,929 | 165 | ||||||||||||||
| 25,563 | 2489 | 25,563 | 122 | 25,563 | 459 | ||||||||||
| 26,735 | 112 | 26,735 | 465 | ||||||||||||
| 28,144 | 140 | 28,144 | 1262 | ||||||||||||
| 174 | |||||||||||||||
| 71 | 28,854 | 125 | |||||||||||||
| 28,881 | 74 | 28,881 | 2098 | 28,881 | 115 | ||||||||||
| 28,882 | 74 | 28,882 | 2087 | 28,882 | 115 | ||||||||||
| 28,883 | 74 | 28,883 | 2086 | 28,883 | 115 | ||||||||||
| Saudis SNP Co-Mutations | Descendants |
|---|---|
| (241, C, T), (3037, C, T), (14408, C, T), (25563, G, T), (26735, C, T) | 187 |
| (241, C, T), (14408, C, T), (23403, A, G), (25563, G, T), (26735, C, T) | 145 |
| (241, C, T), (3037, C, T), (14408, C, T), (23403, A, G), (25563, G, T), (26735, C, T) | 132 |
| (241, C, T), (3037, C, T), (14408, C, T), (18877, C, T), (25563, G, T), (26735, C, T) | 118 |
| (241, C, T), (14408, C, T), (18877, C, T), (23403, A, G) | 111 |
| (241, C, T), (14408, C, T), (18877, C, T), (23403, A, G), (25563, G, T), (26735, C, T) | 104 |
| (241, C, T), (3037, C, T), (14408, C, T), (18877, C, T), (23403, A, G), (26735, C, T) | 100 |
| (241, C, T), (3037, C, T), (14408, C, T), (18877, C, T), (23403, A, G), (25563, G, T), (26735, C, T) | 98 |
| (241, C, T), (14408, C, T), (28881, G, A), (28882, G, A), (28883, G, C) | 74 |
| (241, C, T), (3037, C, T), (14408, C, T), (28881, G, A), (28882, G, A), (28883, G, C) | 65 |
| Globe SNP Co-Mutations [8] | Descendants |
| (8782, C, T), (28144,T, C), (18060,T, C) | 54 |
| (241, C, T), (3037, C, T), (23403, A, G), (28144, T, C) | 145 |
| (241, C, T), (3037, C, T), (14408, C, T), (23403, A, G) | 132 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, A.; Almutairy, M. Evaluating the Performance of Multiple Sequence Alignment Programs with Application to Genotyping SARS-CoV-2 in the Saudi Population. Computation 2023, 11, 212. https://doi.org/10.3390/computation11110212
Alqahtani A, Almutairy M. Evaluating the Performance of Multiple Sequence Alignment Programs with Application to Genotyping SARS-CoV-2 in the Saudi Population. Computation. 2023; 11(11):212. https://doi.org/10.3390/computation11110212
Chicago/Turabian StyleAlqahtani, Aminah, and Meznah Almutairy. 2023. "Evaluating the Performance of Multiple Sequence Alignment Programs with Application to Genotyping SARS-CoV-2 in the Saudi Population" Computation 11, no. 11: 212. https://doi.org/10.3390/computation11110212