

Identification and Analysis of Phosphatidylethanolamine-Binding Protein Family Genes in the Hangzhou White Chrysanthemum (Chrysanthemum morifolium Ramat)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Abiotic Treatments
2.2. RNA Isolation and cDNA Synthesis
2.3. Identification of PEBP Family Genes
2.4. Analysis of Sequences
2.5. Gene Expression Analysis Using qRT-PCR
3. Results
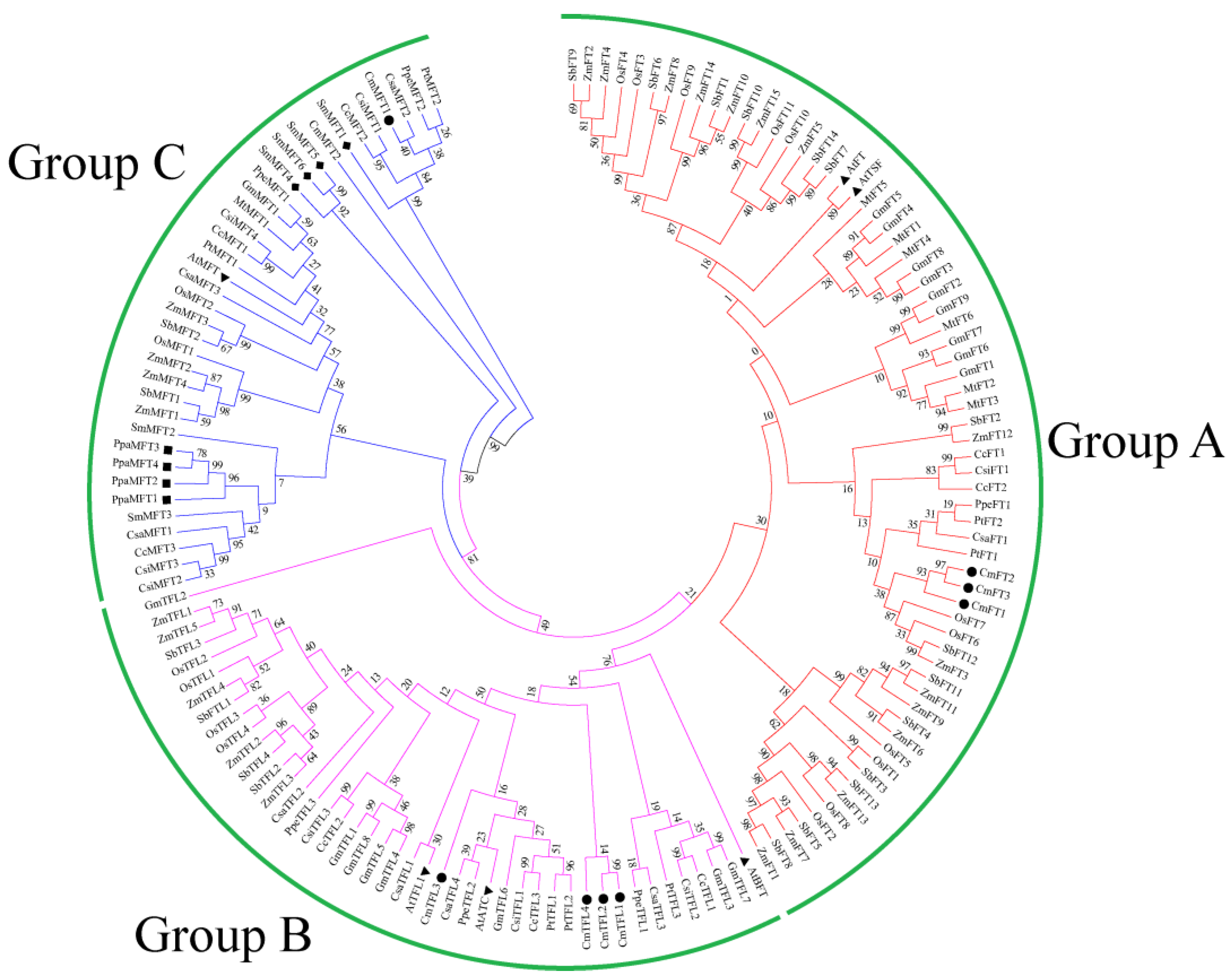
3.1. Isolation and Identification of the PEBP Family Genes in the Chrysanthemum
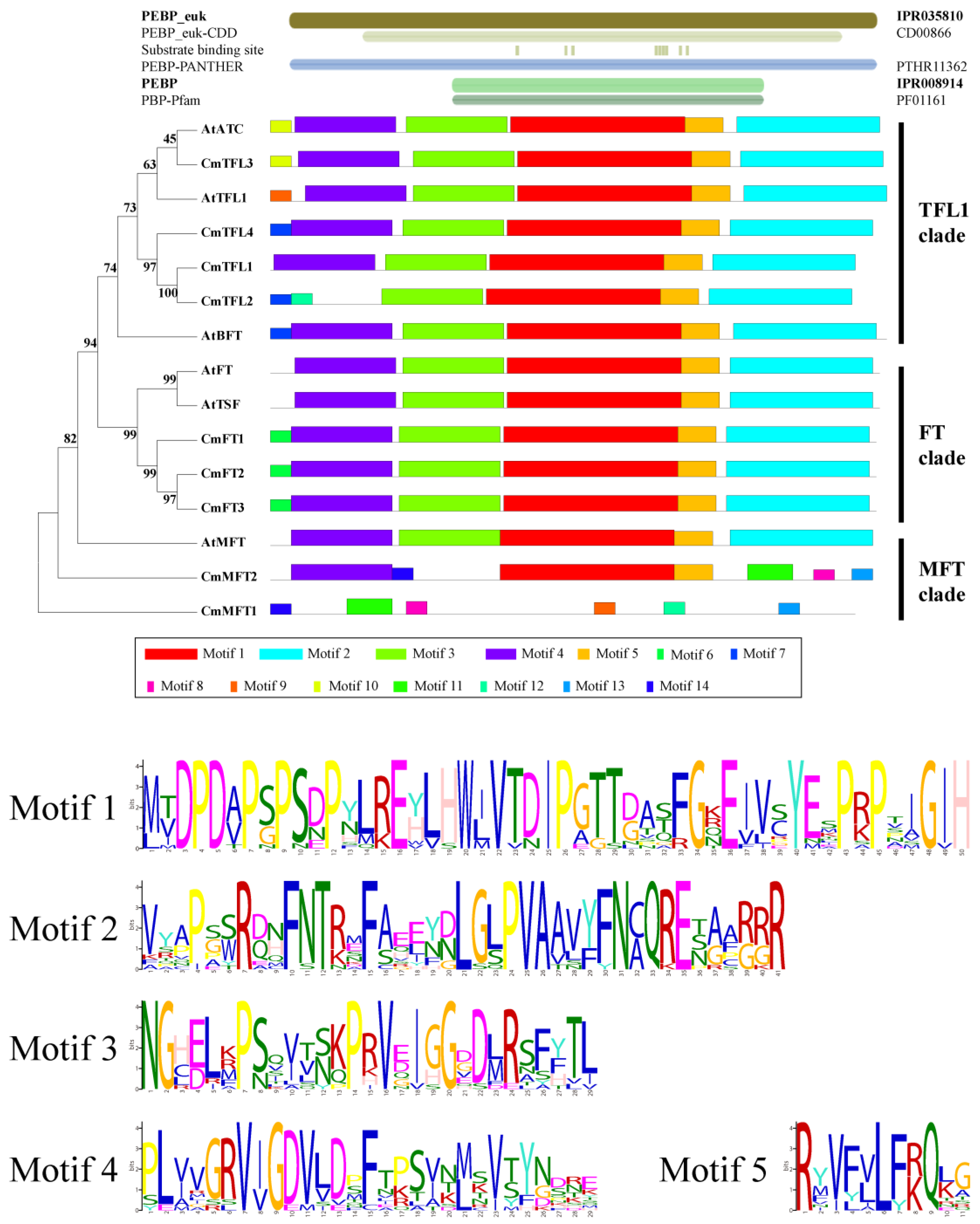
3.2. Similarity and Phylogenetic Gene Structure Analysis
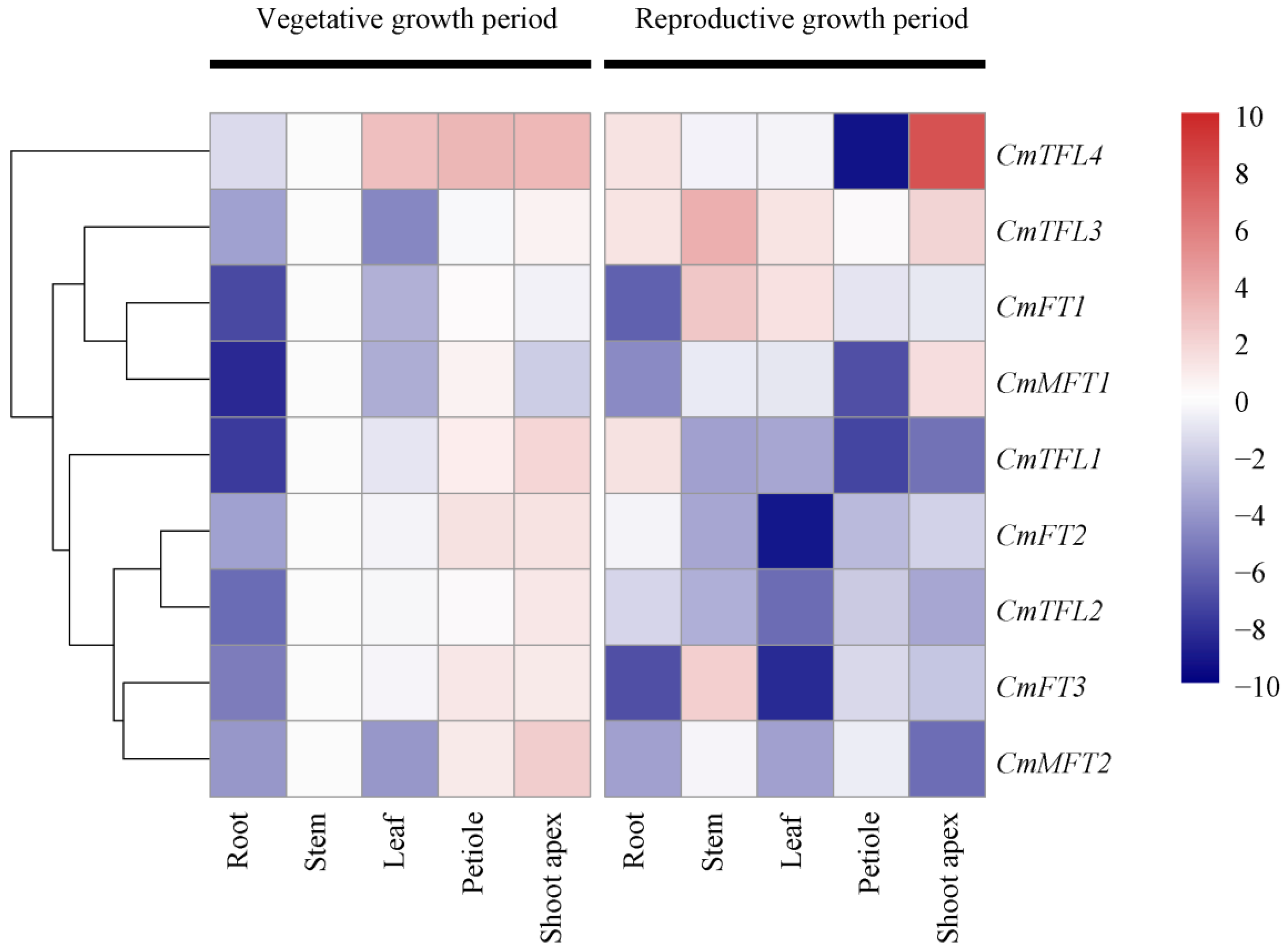
3.3. Tissue-Specific CmPEBP Gene Expression
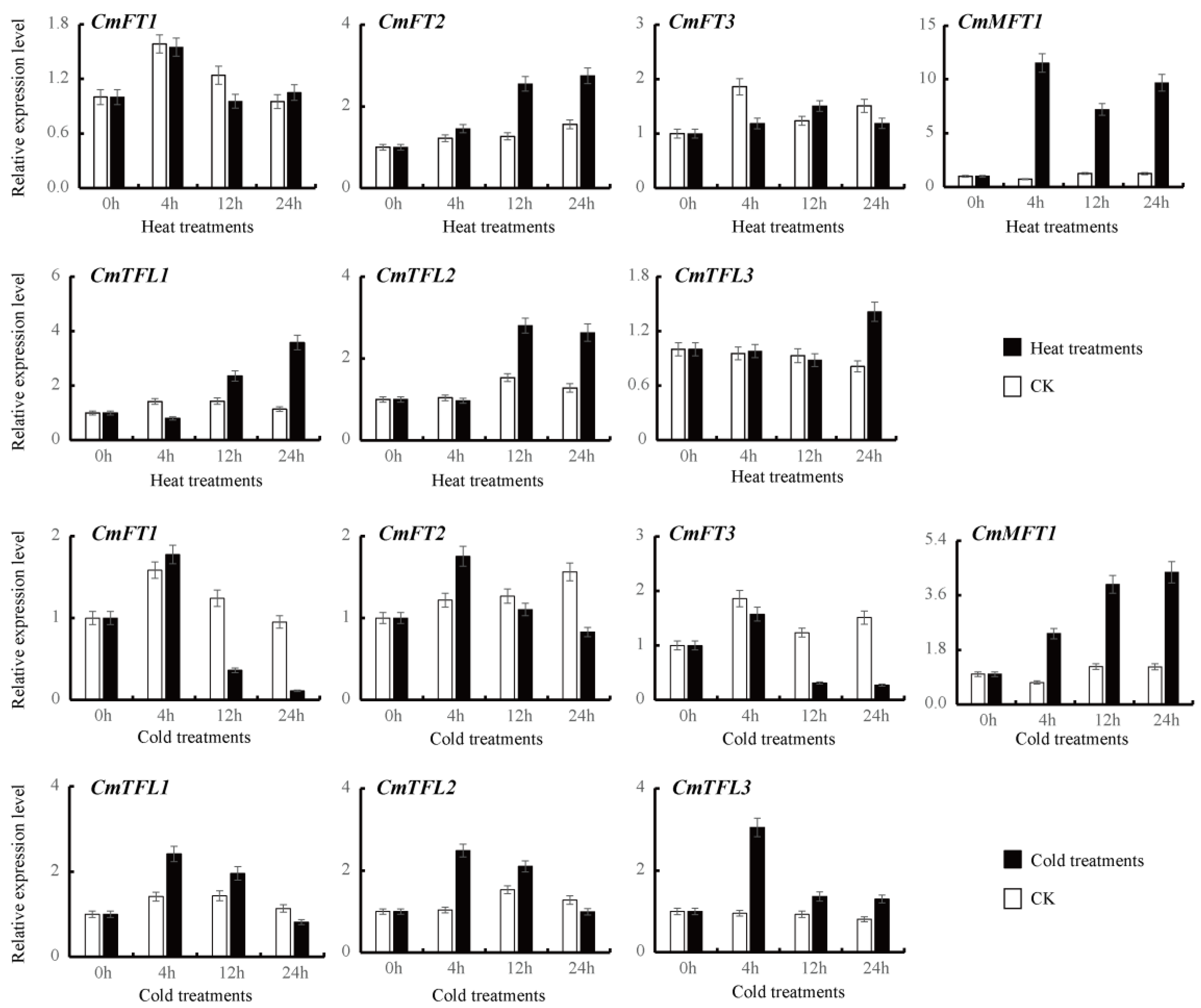
3.4. Differential CmPEBP Gene Expression under High and Low Temperature Treatments
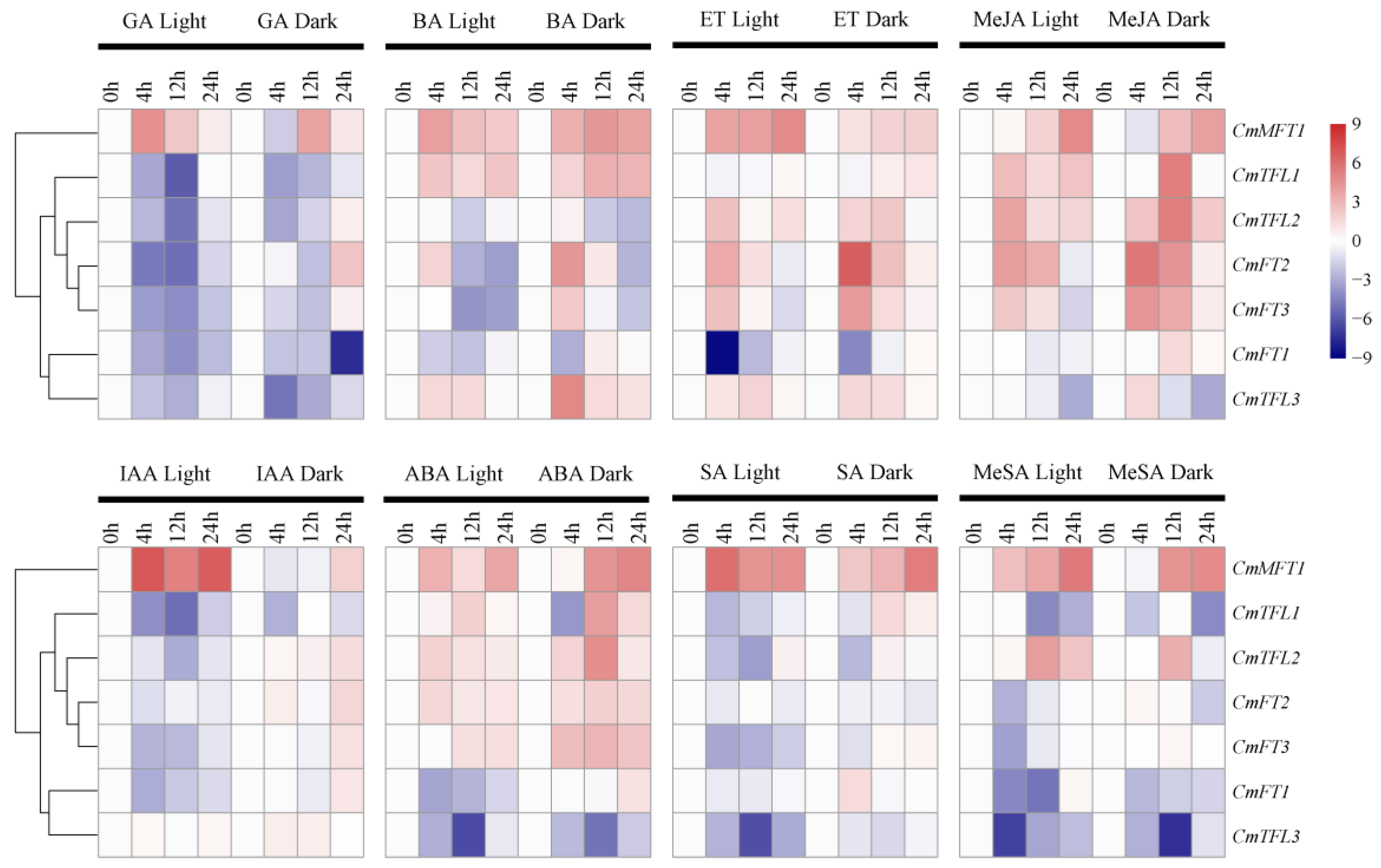
3.5. Differential CmPEBP Gene Expression under Hormone Treatments
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eshed, Y.; Lippman, Z.B. Revolutions in agriculture chart a course for targeted breeding of old and new crops. Science 2019, 366, eaax0025. [Google Scholar] [CrossRef] [PubMed]
- Khanouki, M.A.; Rezanejad, F.; Millar, A.A. Sequence and functional analysis of a TERMINAL FLOWER 1 homolog from Brassica juncea: A putative biotechnological tool for flowering time adjustment. GM Crops Food 2020, 11, 79–92. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, Z.; Liu, Y.; Liu, T.; Li, Q.; Ji, Y.; Li, C.; Fang, C.; Wang, M.; Wu, M.; et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and Arabidopsis. Plant Cell 2015, 27, 323–336. [Google Scholar] [CrossRef]
- Zheng, X.; Wu, F.; Zhang, X.; Lin, Q.; Wang, J.; Guo, X.; Lei, C.; Cheng, Z.; Zou, C.; Wan, J. Evolution of the PEBP gene family and selective signature on FT-like clade. J. Syst. Evol. 2016, 54, 502–510. [Google Scholar] [CrossRef]
- Schoentgen, F.; Jollès, P. From structure to function: Possible biological roles of a new widespread protein family binding hydrophobic ligands and displaying a nucleotide binding site. FEBS Lett. 1995, 369, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Argiriou, A.; Michailidis, G.; Tsaftaris, A.S. Characterization and expression analysis of TERMINAL FLOWER1 homologs from cultivated alloteraploid cotton (Gossypium hirsutum) and its diploid progenitors. J. Plant Physiol. 2008, 165, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Danilevskaya, O.N.; Meng, X.; Hou, Z.; Ananiev, E.V.; Simmons, C.R. A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 2008, 146, 250–264. [Google Scholar] [CrossRef]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef]
- Tamaki, S.; Matsuo, S.; Wong, H.L.; Yokoi, S.; Shimamoto, K. Hd3a protein is a mobile flowering signal in rice. Science 2007, 316, 1033–1036. [Google Scholar] [CrossRef]
- Lifschitz, E.; Eviatar, T.; Rozman, A.; Shalit, A.; Goldshmidt, A.; Amsellem, Z.; Alvarez, J.P.; Eshed, Y. The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. Proc. Natl. Acad. Sci. USA 2006, 103, 6398–6403. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.J.; Chung, K.S.; Jung, S.H.; Yoo, S.Y.; Lee, J.S.; Ahn, J.H. BROTHER OF FT AND TFL1 (BFT) has TFL1-like activity and functions redundantly with TFL1 in inflorescence meristem development in Arabidopsis. Plant J. 2010, 63, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Hanano, S.; Goto, K. Arabidopsis TERMINAL FLOWER1 is involved in the regulation of flowering time and inflorescence development through transcriptional repression. Plant Cell 2011, 23, 3172–3184. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Ratcliffe, O.; Vincent, C.; Carpenter, R.; Coen, E. Inflorescence commitment and architecture in Arabidopsis. Science 1997, 275, 80–83. [Google Scholar] [CrossRef]
- Bradley, D.; Carpenter, R.; Copsey, L.; Vincent, C.; Rothstein, S.; Coen, E. Control of inflorescence architecture in Antirrhinum. Nature 1996, 379, 791–797. [Google Scholar] [CrossRef]
- Yano, M.; Kojima, S.; Takahashi, Y.; Lin, H.; Sasaki, T. Genetic control of flowering time in rice, a short-day plant. Plant Physiol. 2001, 127, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Kralemann, L.E.M.; Scalone, R.; Andersson, L.; Hennig, L. North European invasion by common ragweed is associated with early flowering and dominant changes in FT/TFL1 expression. J. Exp. Botany 2018, 69, 2647–2658. [Google Scholar] [CrossRef]
- Tao, Y.B.; Luo, L.; He, L.L.; Ni, J.; Xu, Z.F. A promoter analysis of MOTHER OF FT AND TFL1 1 (JcMFT1), a seed-preferential gene from the biofuel plant Jatropha curcas. J. Plant Res. 2014, 127, 513–524. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, L.; Kohnen, M.V.; Xiong, B.; Zhen, X.; Liao, J.; Oka, Y.; Zhu, Q.; Gu, L.; Linet, C.; et al. Identification and characterization of the PEBP family genes in moso bamboo (Phyllostachys heterocycla). Sci. Rep. 2019, 9, 14998. [Google Scholar] [CrossRef]
- Hedman, H.; Kallman, T.; Lagercrantz, U. Early evolution of the MFT-like gene family in plants. Plant Mol. Biol. 2009, 70, 359–369. [Google Scholar] [CrossRef]
- Wang, M.; Tan, Y.; Cai, C.; Zhang, B. Identification and expression analysis of phosphatidy ethanolamine-binding protein (PEBP) gene family in cotton. Genomics 2019, 111, 1373–1380. [Google Scholar] [CrossRef]
- Kardailsky, I.; Shukla, V.K.; Ahn, J.H.; Dagenais, N.; Christensen, S.K.; Nguyen, J.T.; Chory, J.; Harrison, M.J.; Weigel, D. Activation tagging of the floral inducer FT. Science 1999, 286, 1962–1965. [Google Scholar] [CrossRef]
- Cao, K.; Cui, L.; Zhou, X.; Ye, L.; Zou, Z.; Deng, S. Four tomato FLOWERING LOCUS T-Like proteins act antagonistically to regulate floral initiation. Front Plant Sci. 2016, 6, 1213. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Pang, C.; Wei, H.; Wang, H.; Song, M.; Fan, S.; Yu, S. Characterization and Functional Analysis of PEBP Family Genes in Upland Cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0161080. [Google Scholar] [CrossRef]
- Sato, H.; Heang, D.; Sassa, H.; Koba, T. Identification and characterization of FT/TFL1 gene family in cucumber. Breed. Sci. 2009, 59, 3–11. [Google Scholar] [CrossRef]
- Mohamed, R.; Wang, C.-T.; Ma, C.; Shevchenko, O.; Dye, S.J.; Puzey, J.R.; Etherington, E.; Sheng, X.; Meilan, R.; Strauss, S.H.; et al. Populus CEN/TFL1 regulates first onset of flowering, axillary meristem identity and dormancy release in Populus. Plant J. 2010, 62, 674–688. [Google Scholar] [CrossRef]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Chardon, F.; Damerval, C. Phylogenomic analysis of the PEBP gene family in cereals. J. Mol. Evolution 2005, 61, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Yang, C. Functional analysis of FT and TFL1 orthologs from orchid (Oncidium Gower Ramsey) that regulate the vegetative to reproductive transition. Plant Cell Physiol. 2009, 50, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Schiessl, S.V.; Huettel, B.; Kuehn, D.; Reinhardt, R.; Snowdon, R.J. Flowering time gene variation in Brassica species shows evolutionary principles. Front Plant Sci. 2017, 8, 1742. [Google Scholar] [CrossRef]
- Muhammad, N.; Khurram, S.; Saddam, S.; Asim, S.; Nasrullah; Muhammad, Y.; Muhammad, I.A. The Solanum melongena COP1LIKE manipulates fruit ripening and flowering time in tomato (Solanum lycopersicum). Plant Growth Regul. 2022, 96, 369–382. [Google Scholar]
- Karlgren, A.; Gyllenstrand, N.; Källman, T.; Sundström, J.F.; Moore, D.; Lascoux, M.; Lagercrantz, U. Evolution of the PEBP gene family in plants: Functional diversification in seed plant evolution. Plant Physiol. 2011, 156, 1967–1977. [Google Scholar] [CrossRef] [Green Version]
- Banfield, M.J.; Brady, R.L. The structure of Antirrhinum centroradialis protein (CEN) suggests a role as a kinase regulator. J. Mol. Biol. 2000, 297, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Harper, J.F.; Palanivelu, R. A fruitful journey: Pollen tube navigation from germination to fertilization. Annu Rev. Plant Biol. 2019, 70, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Hanzawa, Y.; Money, T.; Bradley, D. A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. USA. 2005, 102, 7748–7753. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Miller, D.; Winter, V.J.; Banfield, M.J.; Lee, J.H.; Yoo, S.Y.; Henz, S.R.; Brady, R.L.; Weigel, D. A divergent external loop confers antagonistic activity on floral regulators FT and TFL1. EMBO J. 2006, 25, 605–614. [Google Scholar] [CrossRef]
- Pin, P.A.; Benlloch, R.; Bonnet, D.; Wremerth-Weich, E.; Kraft, T.; Gielen, J.J.L.; Nilssonet, O. An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 2010, 330, 1397–1400. [Google Scholar] [CrossRef]
- Okada, M. Classification of chrysanthemum varieties in view of their enivronmental responses to flowering. J. Jpn. Soc. Hortic. Sci. 1957, 26, 59–72. [Google Scholar] [CrossRef]
- Amasino, R. Seasonal and developmental timing of flowering. Plant J. 2010, 61, 1001–1013. [Google Scholar] [CrossRef]
- Kazan, K.; Lyons, R. The link between flowering time and stress tolerance. J. Exp. Botany 2016, 67, 47–60. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Smith, S.M. Hormone Metabolism and Signaling in Plants. J. Exp. Botany 2016, 67, 47–60. [Google Scholar]
- Santner, A.; Estelle, M. Recent advances and emerging trends in plant hormone signalling. Nature 2009, 459, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Wolters, H.; Jürgens, G. Survival of the flexible: Hormonal growth control and adaptation in plant development. Nat. Rev. Genet. 2009, 10, 305–317. [Google Scholar] [CrossRef]
- Conti, L. Hormonal control of the floral transition: Can one catch them all? Dev. Biol. 2017, 430, 288–301. [Google Scholar] [CrossRef]
- Bao, S.; Hua, C.; Shen, L.; Yu, H. New insights into gibberellin signaling in regulating flowering in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 118–131. [Google Scholar] [CrossRef]
- Piotrowska-Niczyporuk, A.; Bajguz, A.; Kotowska, U.; Zambrzycka-Szelewa, E.; Sienkiewicz, A. Auxins and cytokinins regulate phytohormone homeostasis and thiol-mediated detoxification in the green alga acutodesmus obliquus exposed to lead stress. Sci. Rep. 2020, 10, 10193. [Google Scholar] [CrossRef] [PubMed]
- Kesawat, M.S.; Kherawat, B.S.; Singh, A.; Dey, P.; Routray, S.; Mohapatra, C.; Saha, D.; Ram, C.; Siddique, K.H.M.; Kumar, A.; et al. Genome-wide analysis and characterization of the proline-rich extensin-like receptor kinases (perks) gene family reveals their role in different developmental stages and stress conditions in wheat (Triticum aestivum L.). Plants 2022, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Qiao, L.; Guo, H.; Guo, L.; Ren, F.; Bai, J.; Wang, Y. Genome-wide identification of wheat WRKY gene family reveals that TaWRKY75-A is referred to drought and salt resistances. Front. Plant Sci. 2021, 12, 663118. [Google Scholar] [CrossRef]
- Kesawat, M.S.; Kherawat, B.S.; Singh, A.; Dey, P.; Kabi, M.; Debnath, D.; Saha, D.; Khandual, A.; Rout, S.; Manorama; et al. Genome-wide identification and characterization of the brassinazole-resistant (BZR) gene family and its expression in the various developmental stage and stress conditions in wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2021, 22, 8743. [Google Scholar]
- Zhu, T.; Liu, Y.; Ma, L.; Wang, X.; Zhang, D.; Han, Y.; Ding, Q.; Ma, L. Genome-wide identification, phylogeny and expression analysis of the SPL gene family in wheat. BMC Plant Biol. 2020, 20, 420. [Google Scholar] [CrossRef]
- Li, S.; Liu, Z.; Guo, L.; Li, H.; Nie, X.; Chai, S.; Zheng, W. Genome-wide identification of wheat ZIP gene family and functional characterization of the TaZIP13-B in plants. Front. Plant Sci. 2021, 12, 748146. [Google Scholar] [CrossRef]
- Shao, W.; Chen, W.; Zhu, X.; Zhou, X.; Jin, Y.; Zhan, C.; Liu, G.; Liu, X.; Ma, D.; Qiao, Y. Genome-wide identification and characterization of wheat 14-3-3 genes unravels the role of TaGRF6-A in salt stress tolerance by binding MYB transcription factor. Int. J. Mol. Sci. 2021, 22, 1904. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, W. Chrysanthemum—Flower Production Technology and Its Application Series; China Forestry Publishing House: Beijing, China, 2001. [Google Scholar]
- Li, H.; Huang, W.; Wang, G.; Wu, Z.; Zhuang, J. Expression profile analysis of ascorbic acid-related genes in response to temperature stress in the tea plant, Camellia sinensis (L.) O. Kuntze. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, S.; Chen, F.; Teng, N.; Fang, W.; Guan, Z. Anatomical structure and gravitropic response of the creeping shoots of ground-cover chrysanthemum ‘Yuhuajinhua’. Plant Growth Regul. 2008, 56, 141. [Google Scholar] [CrossRef]
- Pan, C.; Tian, K.; Ban, Q.; Wang, L.; Sun, Q.; He, Y.; Yang, Y.; Pan, Y.; Li, Y.; Jiang, J.; et al. Genome-wide analysis of the biosynthesis and deactivation of gibberellin-dioxygenases gene family in Camellia sinensis (L.) O. Kuntze. Genes 2017, 8, 235. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Yue, C.; Cao, H.; Wang, L.; Zhou, Y.; Hao, X.; Zeng, J.; Wang, X.; Yang, Y. Molecular cloning and expression analysis of tea plant aquaporin (AQP) gene family. Plant Physiol. Biochem. 2014, 83, 65–76. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012, 40, 302–305. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protocols 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 725–2729. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.E.; Nobleet, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zaman, W.; Lu, J.; Niu, Q.; Zhang, X.; Ayaz, A.; Saqib, S.; Yang, B.; Zhang, J.; Zhao, H.; et al. Natural lupeol level variation among castor accessions and the upregulation of lupeol synthesis in response to light. Ind. Crop Prod. 2023, 192, 116090. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liao, W.Y.; Lin, L.F.; Lin, M.D.; Hsieh, S.C.; Li, A.Y.S.; Tsay, Y.S.; Chou, M.L. Overexpression of Lilium formosanumMADS-box (LFMADS) Causing Floral Defects While Promoting Flowering in Arabidopsis thaliana, Whereas only affecting floral transition time in Nicotiana tabacum. Int. J. Mol. Sci. 2018, 19, 2217. [Google Scholar] [CrossRef]
- Cho, L.H.; Yoon, J.; An, G. The control of flowering time by environmental factors. Plant J. 2017, 90, 708–719. [Google Scholar] [CrossRef]
- Campos-Rivero, G.; Osorio-Montalvo, P.; Sánchez-Borges, R.; Us-Camas, R.; Duarte-Aké, F.; De-la-Peña, C. Plant hormone signaling in flowering: An epigenetic point of view. J. Plant Physiol. 2017, 214, 16–27. [Google Scholar] [CrossRef]
- Yan, X.; Liu, J.; Wu, K.X.; Yang, N.; Pan, L.B.; Song, Y.; Liu, Y.; Tang, Z.H. Comparative analysis of endogenous hormones and metabolite profiles in early-spring flowering plants and unflowered plants revealing the strategy of blossom. J. Plant Growth Regul. 2022, 41, 2421–2434. [Google Scholar] [CrossRef]
- Wilson, R.N.; Heckman, J.W.; Somerville, C.R. Gibberellin is required for flowering in arabidopsis thaliana under short days. Plant Physiol. 1992, 100, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Xue, J.; Xue, Y.; Yang, R.; Wang, S.; Zhang, X. Effect of exogenous GA_3 on flowering quality, endogenous hormones, and hormone- and flowering-associated gene expression in forcing-cultured tree peony (Paeonia suffruticosa). J. Integr. Agric. 2019, 18, 1295–1311. [Google Scholar] [CrossRef]
- Liang, N.; Cheng, D.; Liu, Q.; Cui, J.; Luo, C. Difference of proteomics vernalization-induced in bolting and flowering transitions of Beta vulgaris. Plant Physiol. Biochem. 2018, 123, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Ye, T.; Lu, Y.; Chen, X.; Wu, Y. The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J. Exp. Botany 2013, 64, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Riboni, M.; Test, A.; Galbiati, M.; Tonelli, C.; Conti, L. ABA-dependent control of GIGANTEA signalling enables drought escape via up-regulation of FLOWERING LOCUS T in Arabidopsis thaliana. J. Exp. Botany 2016, 67, 6309–6322. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, F.; Zhao, X. Salicylic acid-mediated signal transduction pathway associated with stress resistance of plants. Chin. Agric. Sci. Bulletin. 2005, 21, 227–229. [Google Scholar]
- Böhlenius, H.; Huang, T.; Charbonnel-Campaa, L.; Brunner, A.M.; Jansson, S.; Strauss, S.H.; Nilssonet, O. CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science 2006, 312, 1040–1043. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | Accession Nos | Open Reading Frame (bp) | Number of Deduced Amino Acid (aa) | Molecular Weight (kDa) | Subcellular Location (WoLF PSORT/TargetP) | Theoretical pI | Groups |
|---|---|---|---|---|---|---|---|
| CmFT1 | OP617277 | 525 | 174 | 19.26 | chlo/cyto/nucl | 6.83 | Group A |
| CmFT2 | OP617278 | 525 | 174 | 19.79 | cyto/chlo/nucl/mito | 8.64 | Group A |
| CmFT3 | OP617279 | 525 | 174 | 19.52 | cyto/nucl/plas/chlo | 6.82 | Group A |
| CmTFL1 | OP617271 | 507 | 168 | 19.04 | cyto/nucl | 8.96 | Group B |
| CmTFL2 | OP617272 | 504 | 167 | 19.14 | cyto/nucl/extr | 9.39 | Group B |
| CmTFL3 | OP617273 | 531 | 176 | 19.87 | cyto/chlo/vacu | 8.86 | Group B |
| CmTFL4 | OP617274 | 522 | 173 | 19.44 | cyto | 8.93 | Group B |
| CmMFT1 | OP617275 | 507 | 168 | 18.45 | cyto/cysk | 4.71 | Group C |
| CmMFT2 | OP617276 | 522 | 173 | 18.99 | cyto/chlo | 6.83 | Group C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, C.; He, X.; Song, S.; Zou, L.; Wang, M.; Han, B. Identification and Analysis of Phosphatidylethanolamine-Binding Protein Family Genes in the Hangzhou White Chrysanthemum (Chrysanthemum morifolium Ramat). Agriculture 2023, 13, 374. https://doi.org/10.3390/agriculture13020374
Pan C, He X, Song S, Zou L, Wang M, Han B. Identification and Analysis of Phosphatidylethanolamine-Binding Protein Family Genes in the Hangzhou White Chrysanthemum (Chrysanthemum morifolium Ramat). Agriculture. 2023; 13(2):374. https://doi.org/10.3390/agriculture13020374
Chicago/Turabian StylePan, Cheng, Xueyi He, Shiyue Song, Liping Zou, Mengxin Wang, and Baoyu Han. 2023. "Identification and Analysis of Phosphatidylethanolamine-Binding Protein Family Genes in the Hangzhou White Chrysanthemum (Chrysanthemum morifolium Ramat)" Agriculture 13, no. 2: 374. https://doi.org/10.3390/agriculture13020374