
Time-Course Transcriptome Landscape of Bursa of Fabricius Development and Degeneration in Chickens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. Histological Observation
2.4. RNA Extraction, cDNA Library Preparation and RNA Sequencing (RNA-Seq)
2.5. Bioinformatics Analysis
2.6. Gene Expression Pattern Analysis
2.7. Functional Annotation
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Statistical Analysis
3. Results
3.1. Histomorphological Changes in the BF at Different Developmental Times
3.2. Sequencing Quality Analysis
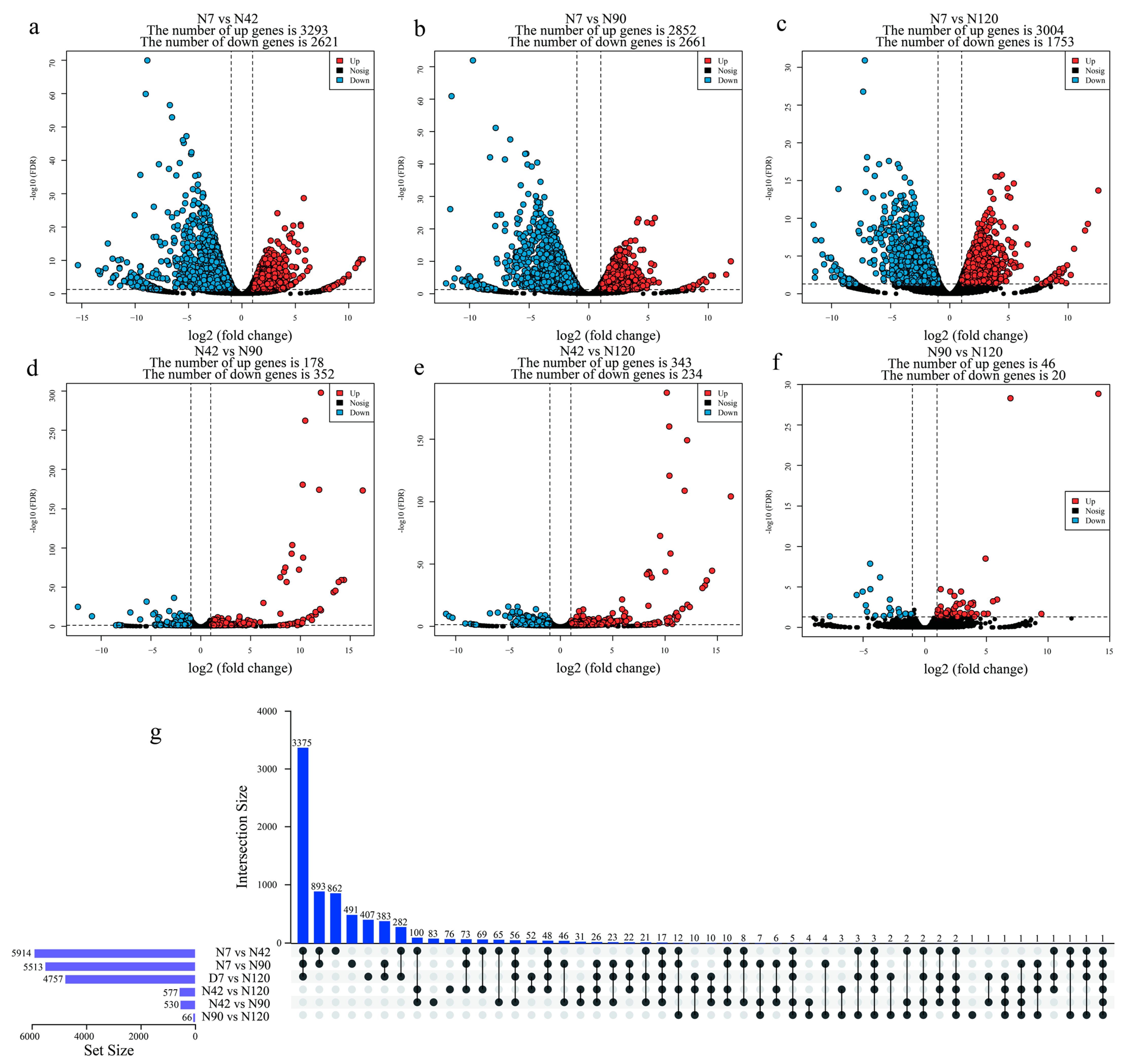
3.3. Identification and Functional Annotation of Differentially Expressed Genes (DEGs) during BF Development
3.4. Functional Analysis of DEGs Using the GO Database
3.5. Functional Analysis of DEGs Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) Database
3.6. Functional Analysis of DEGs Using the REACTOME Pathway
3.7. Gene Expression during Different BF Development Stages
3.8. Validation of DEGs by QRT-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Paramithiotis, E.; Ratcliffe, M.J. B cell emigration directly from the cortex of lymphoid follicles in the bursa of Fabricius. Eur. J. Immunol. 1994, 24, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Khan, M.Z.I.; Jahan, M.; Fujinaga, R.; Yanai, A.; Kokubu, K.; Shinoda, K. Histomorphological study on prenatal development of the lymphoid organs of native chickens of Bangladesh. Pak. Vet. J. 2012, 32, 175–178. [Google Scholar]
- Schat, K.A.; Kaspers, B.; Kaiser, P. Avian Immunology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Ribatti, D.; Porzionato, A.; Emmi, A.; de Caro, R. The bursa of Hieronymus Fabricius ab Aquapendente: From original iconography to most recent research. Rom. J. Morphol. Embryol. 2020, 61, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.; Chang, T.S.; Jaap, R.G. The Bursa of Fabricius and Antibody Production. Poult. Sci. 1956, 35, 224–225. [Google Scholar] [CrossRef]
- Monson, M.S.; van Goor, A.G.; Ashwell, C.M.; Persia, E.M.; Rothschild, M.F.; Schmidt, C.J.; Lamont, S.J. Immunomodulatory effects of heat stress and lipopolysaccharide on the bursal transcriptome in two distinct chicken lines. BMC Genom. 2018, 19, 643. [Google Scholar] [CrossRef]
- Kaspers, B.; Schat, K.; Göbel, T.; Vervelde, L. Avian Immunology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Cooper, M.D.; Raymond, D.A.; Peterson, R.D.; South, M.A.; Good, R.A. The functions of the thymus system and the bursa system in the chicken. J. Exp. Med. 1966, 123, 75–102. [Google Scholar] [CrossRef] [Green Version]
- Salvi, E.; Buffa, R.; Renda, T.G. Ontogeny, distribution and amine/peptide colocalization of chromogranin A- and B-immunoreactive cells in the chicken gizzard and antrum. Anat. Embryol. 1995, 192, 547–555. [Google Scholar] [CrossRef]
- Lassila, O.; Eskola, J.; Toivanen, P. Prebursal stem cells in the intraembryonic mesenchyme of the chick embryo at 7 days of incubation. J. Immunol. 1979, 123, 2091–2094. [Google Scholar]
- Otsubo, Y.; Chen, N.; Kajiwara, E.; Horiuchi, H.; Matsuda, H.; Furusawa, S. Role of bursin in the development of B lymphocytes in chicken embryonic Bursa of Fabricius. Dev. Comp. Immunol. 2001, 25, 485–593. [Google Scholar] [CrossRef]
- Le Douarin, N.M.; Houssaint, E.; Jotereau, F.V.; Belo, M. Origin of Hemopoietic Stem Cells in Embryonic Bursa of Fabricius and Bone Marrow Studied through Interspecific Chimeras. Proc. Natl. Acad. Sci. USA 1975, 72, 2701–2705. [Google Scholar] [CrossRef] [Green Version]
- Pike, K.A.; Baig, E.; Ratcliffe, M.J. The avian B-cell receptor complex: Distinct roles of Igalpha and Igbeta in B-cell development. Immunol. Rev. 2004, 197, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Omachi, R.; Suzuki, H.; Tomobe, K.; Kawashima, T.; Nakajima, T.; Kawashima-Ohya, Y. Apoptosis Occurs during Early Development of the Bursa of Fabricius in Chicken Embryos. Biol. Pharm. Bull. 2014, 37, 1982–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Fischer, D.S.; Theis, F.J.; Yosef, N. Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res. 2018, 46, e119. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4. 0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Tu, Y.; Chen, C.; Fan, G. Association between the expression of secreted phosphoprotein-related genes and prognosis of human cancer. BMC Cancer 2019, 19, 1230. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Thompson, D.E.; McConkey, J.; Murray, P.; Moorehead, R.A. Osteopontin regulates proliferation, apoptosis, and migration of murine claudin-low mammary tumor cells. BMC Cancer 2016, 16, 359. [Google Scholar] [CrossRef] [Green Version]
- Dalal, S.; Zha, Q.; Singh, M.; Singh, K. Osteopontin-stimulated apoptosis in cardiac myocytes involves oxidative stress and mitochondrial death pathway: Role of a pro-apoptotic protein BIK. Mol. Cell Biochem. 2016, 418, 22683–22690. [Google Scholar] [CrossRef] [Green Version]
- Ming, M.; Wang, S.; Wu, W.; Senyuk, V.; le Beau, M.M.; Nucifora, G.; Qian, Z. Activation of Wnt/β-catenin protein signaling induces mitochondria-mediated apoptosis in hematopoietic progenitor cells. J. Biol. Chem. 2012, 287, 22683–22690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grespi, F.; Soratroi, C.; Krumschnabel, G.; Sohm, B.; Ploner, C.; Geley, S.; Hengst, L.; Häcker, G.; Villunger, A. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010, 17, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Piñon, J.D.; Labi, V.; Egle, A.; Villunger, A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene 2008, 27, S41–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Lügering, N.; Pauels, H.G.; Schulze-Osthoff, K.; Domschke, W.; Kucharzik, T. IL-10 induces apoptosis in human monocytes involving the CD95 receptor/ligand pathway. Eur. J. Immunol. 2000, 30, 1769–1777. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Bapat, A.S.; Rayanade, R.J.; Dagtas, A.S.; Hoffmann, M.K. Interleukin-10 induces macrophage apoptosis and expression of CD16 (FcgammaRIII) whose engagement blocks the cell death programme and facilitates differentiation. Immunology 2001, 102, 331–337. [Google Scholar] [CrossRef]
- Sadeghi, I.; Behmanesh, M.; Chashmi, N.A.; Sharifi, M.; Soltani, B.M. 6-Methoxy Podophyllotoxin Induces Apoptosis via Inhibition of TUBB3 and TOPIIA Gene Expressions in 5637 and K562 Cancer Cell Lines. Cell J. 2015, 17, 502–509. [Google Scholar]
- Di Donato, N.; Timms, A.E.; Aldinger, K.A.; Mirzaa, G.M.; Bennett, J.T.; Collins, S.; Olds, C.; Mei, D.; Chiari, S.; Carvill, G.; et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet. Med. 2018, 20, 1354–1364. [Google Scholar] [CrossRef]
- Wu, I.; Shin, S.C.; Cao, Y.; Bender, I.K.; Jafari, N.; Feng, G.; Lin, S.; Cidlowski, J.A.; Schleimer, R.P.; Lu, N.Z. Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell Death Dis. 2013, 4, e453. [Google Scholar] [CrossRef]
- Zhao, C.-C.; Chen, J.; Zhang, L.-Y.; Liu, H.; Zhang, C.-G.; Liu, Y. Lamin B2 promotes the progression of triple negative breast cancer via mediating cell proliferation and apoptosis. Biosci. Rep. 2021, 41, BSR20203874. [Google Scholar] [CrossRef]
- Dong, C.-H.; Jiang, T.; Yin, H.; Song, H.; Zhang, Y.; Geng, H.; Shi, P.-C.; Xu, Y.-X.; Gao, H.; Liu, L.-Y.; et al. LMNB2 promotes the progression of colorectal cancer by silencing p21 expression. Cell Death Dis. 2021, 12, 331. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Dong, N.; Zhou, X.; Ma, L.; Xiang, R. Myosin light chain 9 promotes the proliferation, invasion, migration and angiogenesis of colorectal cancer cells by binding to Yes-associated protein 1 and regulating Hippo signaling. Bioengineered 2022, 13, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kuai, Y.; Lei, L.; Weng, Y.; Berberich-Siebelt, F.; Zhang, X.; Wang, J.; Zhou, Y.; Jiang, X.; Ren, G.; et al. The feedback loop of LITAF and BCL6 is involved in regulating apoptosis in B cell non-Hodgkin’s-lymphoma. Oncotarget 2016, 7, 77444–77456. [Google Scholar] [CrossRef] [Green Version]
- Alimperti, S.; Andreadis, S.T. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem. Cell Res. 2015, 14, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ying, J.; Li, H.; Zhang, Y.; Shu, X.; Fan, Y.; Tan, J.; Cao, Y.; Tsao, S.W.; Srivastava, G.; et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/β-catenin signaling and silenced in common carcinomas. Oncogene 2012, 31, 3901–3912. [Google Scholar] [CrossRef] [Green Version]
- Bolcun-Filas, E.; Bannister, L.A.; Barash, A.; Schimenti, K.J.; Hartford, S.A.; Eppig, J.J.; Handel, M.A.; Shen, L.; Schimenti, J.C. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 2011, 138, 3319–3330. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.E.; El-Deiry, W.S. Regulation of the human TRAIL gene. Cancer Biol. Ther. 2012, 13, 1143–1151. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef]
- Dunon, D.; Salomonsen, J.; Skjødt, K.; Kaufman, J.; Imhof, B.A. Ontogenic appearance of MHC class I (B-F) antigens during chicken embryogenesis. Dev. Immunol. 1990, 1, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Liu, X.; Mu, J.; Yan, W.; Wang, M.; Chai, H.; Sha, Y.; Jiang, S.; Wang, S.; Ren, Y.; et al. Intense Innate Immune Responses and Severe Metabolic Disorders in Chicken Embryonic Visceral Tissues Caused by Infection with Highly Virulent Newcastle Disease Virus Compared to the Avirulent Virus: A Bioinformatics Analysis. Viruses 2022, 14, 911. [Google Scholar] [CrossRef] [PubMed]
- Jax, E.; Wink, M.; Kraus, R.H.S. Avian transcriptomics: Opportunities and challenges. J. Ornithol. 2018, 159, 599–629. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.U.; Li, Y.; Ouyang, W.; Wang, Z.; Zuo, J.; Shi, S.; Yu, Q.; Lin, J.; Yang, Q. From nasal to basal: Single-cell sequencing of the bursa of Fabricius highlights the IBDV infection mechanism in chickens. Cell Biosci. 2021, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Sun, G.; Li, K.; Li, Z.; Su, A.; Liu, X.; Li, G.; Jiang, R.; Han, R.; et al. Transcriptome profile in bursa of Fabricius reveals potential mode for stress-influenced immune function in chicken stress model. BMC Genom. 2018, 19, 918. [Google Scholar] [CrossRef]
- Rai, M.F.; Cai, L.; Tycksen, E.D.; Chamberlain, A.; Keener, J. RNA-Seq analysis reveals sex-dependent transcriptomic profiles of human subacromial bursa stratified by tear etiology. J. Orthop. Res. 2022. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [Green Version]
- Khiem, D.; Cyster, J.G.; Schwarz, J.J.; Black, B.L. A p38 MAPK-MEF2C pathway regulates B-cell proliferation. Proc. Natl. Acad. Sci. USA 2008, 105, 17067–17072. [Google Scholar] [CrossRef] [Green Version]
- Kotelnikova, E.; Kiani, N.A.; Messinis, D.; Pertsovskaya, I.; Pliaka, V.; Bernardo-Faura, M.; Rinas, M.; Vila, G.; Zubizarreta, I.; Pulido-Valdeolivas, I.; et al. MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis. Proc. Natl. Acad. Sci. USA 2019, 116, 9671–9676. [Google Scholar] [CrossRef] [Green Version]
- McLaurin, J.D.; Weiner, O.D. Multiple sources of signal amplification within the B-cell Ras/MAPK pathway. Mol. Biol. Cell 2019, 30, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Quinn, W.J., 3rd; Salay, T.; Crowley, J.E.; Cancro, M.P.; Sen, J.M. Role of beta-catenin in B cell development and function. J. Immunol. 2008, 181, 3777–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.L.; Zierold, C.; Ranheim, E.A. Dysregulation of Frizzled 6 is a critical component of B-cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 2009, 113, 3031–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljungberg, J.K.; Kling, J.C.; Tran, T.T.; Blumenthal, A. Functions of the WNT Signaling Network in Shaping Host Responses to Infection. Front. Immunol. 2019, 10, 2521. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Ranheim, E.A.; Kwan, H.C.K.; Reya, T.; Wang, Y.-K.; Weissman, I.L.; Francke, U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood 2005, 105, 2487–2494. [Google Scholar] [CrossRef]
- Coffman, J.A. Cell Cycle Development. Dev. Cell 2004, 6, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Hagimoto, N.; Kuwano, K.; Kawasaki, M.; Yoshimi, M.; Kaneko, Y.; Kunitake, R.; Maeyama, T.; Tanaka, T.; Hara, N. Induction of interleukin-8 secretion and apoptosis in bronchiolar epithelial cells by Fas ligation. Am. J. Respir. Cell Mol. Biol. 1999, 21, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Kettritz, R.; Gaido, M.L.; Haller, H.; Luft, F.C.; Jennette, C.J.; Falk, R.J. Interleukin-8 delays spontaneous and tumor necrosis factor-alpha-mediated apoptosis of human neutrophils. Kidney Int. 1998, 53, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wu, X.; Pu, J.; Luo, P.; Ma, W.; Wang, J.; Wei, J.; Wang, Y.; Fei, Z. Lycium barbarum polysaccharide protects against oxygen glucose deprivation/reoxygenation-induced apoptosis and autophagic cell death via the PI3K/Akt/mTOR signaling pathway in primary cultured hippocampal neurons. Biochem. Biophys. Res. Commun. 2018, 495, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, W.J.; Wang, Y.Z.; Wang, Y.B.; Li, Y.L. Lobaplatin promotes (125)I-induced apoptosis and inhibition of proliferation in hepatocellular carcinoma by upregulating PERK-eIF2α-ATF4-CHOP pathway. Cell Death Dis. 2019, 10, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, L.; Chen, G.; Zhong, B.; Liu, F. Lycium barbarum polysaccharide induced apoptosis and inhibited proliferation in infantile hemangioma endothelial cells via down-regulation of PI3K/AKT signaling pathway. Biosci. Rep. 2019, 39, BSR20191182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef]
- Shan, M.; Yuan, X.; Song, L.Z.; Roberts, L.; Zarinkamar, N.; Seryshev, A.; Zhang, Y.; Hilsenbeck, S.; Chang, S.H.; Dong, C.; et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 2012, 4, 117ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, D.H.; Ndiaye, K.; Pate, J.L. Expression and regulation of secreted phosphoprotein 1 in the bovine corpus luteum and effects on T lymphocyte chemotaxis. Reproduction 2013, 146, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Ashkar, S.; Weber, G.F.; Panoutsakopoulou, V.; Sanchirico, M.E.; Jansson, M.; Zawaideh, S.; Rittling, S.R.; Denhardt, D.T.; Glimcher, M.J.; Cantor, H. Eta-1 (osteopontin): An early component of type-1 (cell-mediated) immunity. Science 2000, 287, 860–864. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef]
- Prince, C.W.; Oosawa, T.; Butler, W.T.; Tomana, M.; Bhown, A.S.; Bhown, M.; Schrohenloher, R.E. Isolation, characterization, and biosynthesis of a phosphorylated glycoprotein from rat bone. J. Biol. Chem. 1987, 262, 2900–2907. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Heinemeier, K.M.; Skovgaard, D.; Bayer, M.L.; Qvortrup, K.; Kjaer, A.; Kjaer, M.; Magnusson, S.P.; Kongsgaard, M. Uphill running improves rat Achilles tendon tissue mechanical properties and alters gene expression without inducing pathological changes. J. Appl. Physiol. 2012, 113, 827–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöblom, B.; Salmazo, A.; Djinović-Carugo, K. Alpha-actinin structure and regulation. Cell Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Hu, Y.; Guo, Q.; Chang, G.; Bai, H. Time-Course Transcriptome Landscape of Bursa of Fabricius Development and Degeneration in Chickens. Agriculture 2022, 12, 1194. https://doi.org/10.3390/agriculture12081194
Huang L, Hu Y, Guo Q, Chang G, Bai H. Time-Course Transcriptome Landscape of Bursa of Fabricius Development and Degeneration in Chickens. Agriculture. 2022; 12(8):1194. https://doi.org/10.3390/agriculture12081194
Chicago/Turabian StyleHuang, Lan, Yaodong Hu, Qixin Guo, Guobin Chang, and Hao Bai. 2022. "Time-Course Transcriptome Landscape of Bursa of Fabricius Development and Degeneration in Chickens" Agriculture 12, no. 8: 1194. https://doi.org/10.3390/agriculture12081194