
Positive Selection and Adaptive Introgression of Haplotypes from Bos indicus Improve the Modern Bos taurus Cattle


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing and SNP Discovery
2.2. Runs of Homozygosity
2.3. QTL Enrichment
2.4. Detection of Selection Signatures
2.5. Association Mapping of Haplotypes from the ROH Hotspot Containing the CYP19A1 Gene
3. Results
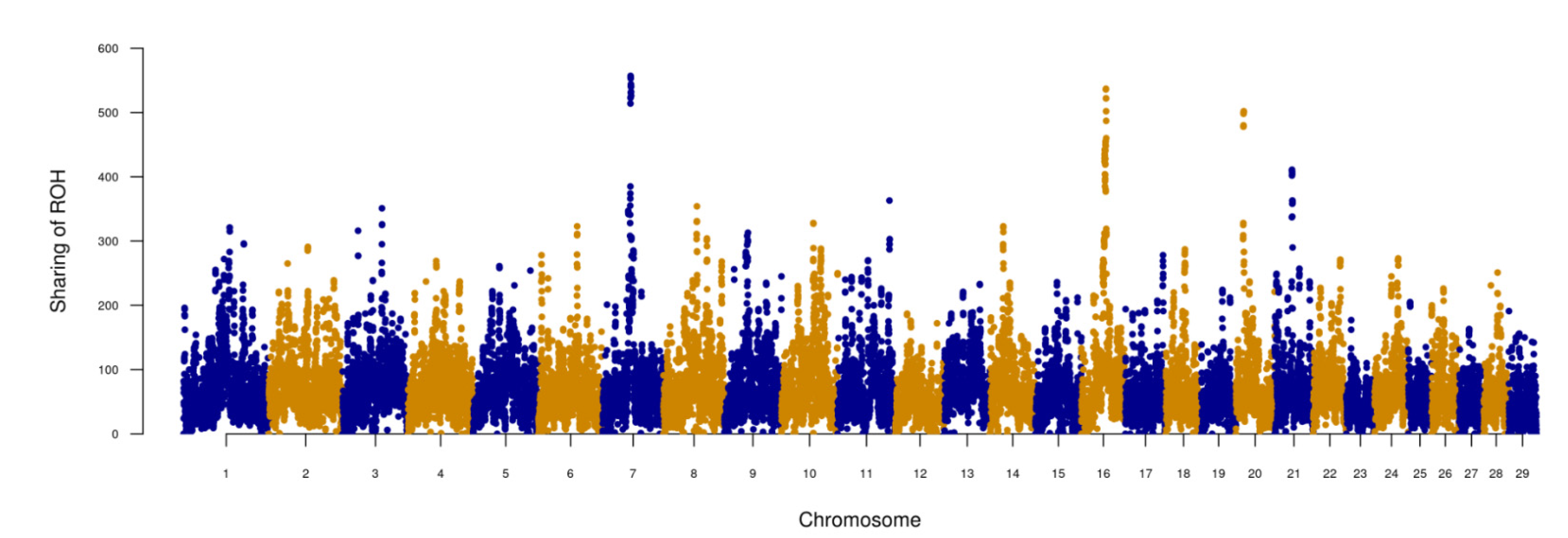
3.1. The Distribution of ROH on Genomes Shaped by Demography
3.2. Effect of Positive Selection on ROH Occurrence
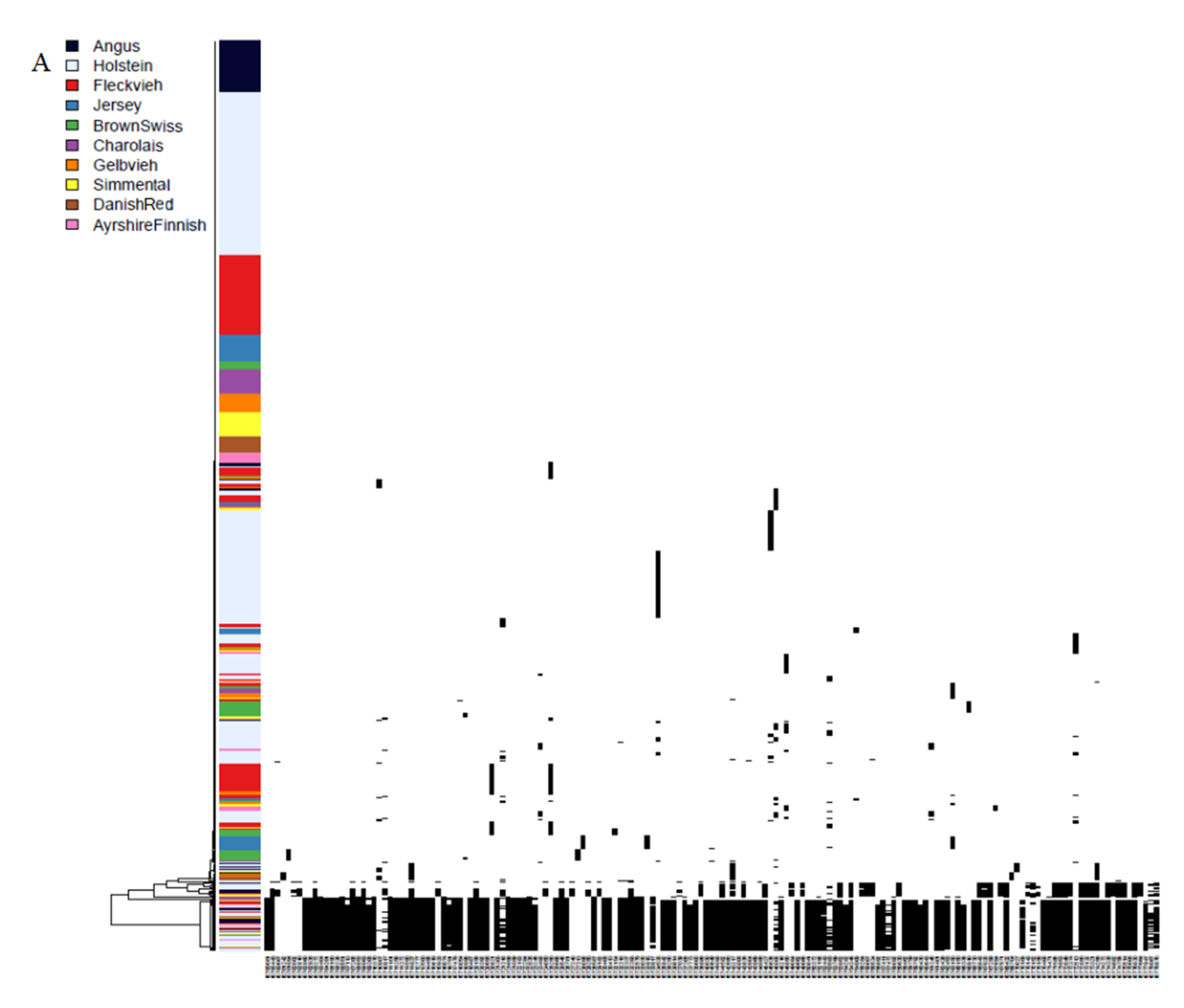
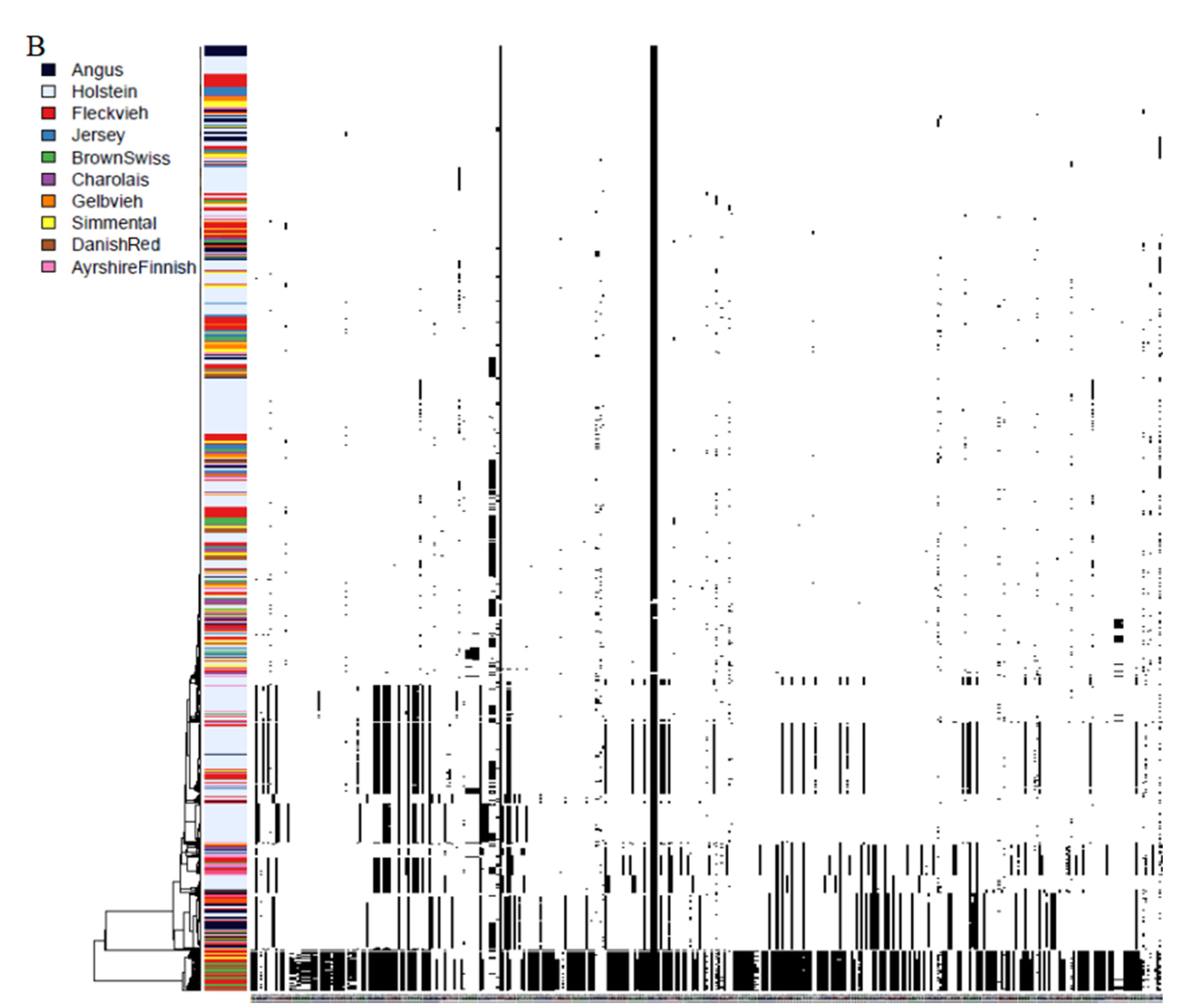
3.3. Phylogenies and Genealogies in ROH Hotspots
3.4. Effect of Haplotype in the ROH Hotspot around the CYP19A1 Gene on Milk Yield
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Calus, M.P.L.; Bosse, M.; Sahana, G.; Lund, M.S.; Guldbrandtsen, B. Human-Mediated Introgression of Haplotypes in a Modern Dairy Cattle Breed. Genetics 2018, 209, 1305–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiró, H.V.; Li, G.; Trindade, F.J.; Assis, J.; Pais, F.; Fernandes, G.; Santos, S.H.D.; Hughes, G.M.; Komissarov, A.; Antunes, A.; et al. Genome-wide signatures of complex introgression and adaptive evolution in the big cats. Sci. Adv. 2017, 3, e1700299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, E.R.; Gonzalez-Fortes, G.; Connell, S.; Siska, V.; Eriksson, A.; Martiniano, R.; McLaughlin, R.; Llorente, M.G.; Cassidy, L.M.; Gamba, C.; et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Commun. 2015, 6, 8912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, R.D.; Amato, R.; Auburn, S.; Miotto, O.; Almagro-Garcia, J.; Amaratunga, C.; Suon, S.; Mao, S.; Noviyanti, R.; Trimarsanto, H.; et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 2016, 48, 959–964. [Google Scholar] [CrossRef]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Pool, J.E.; Hellmann, I.; Jensen, J.D.; Nielsen, R. Population genetic inference from genomic sequence variation. Genome Res. 2010, 20, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Bersaglieri, T.; Sabeti, P.C.; Patterson, N.; Vanderploeg, T.; Schaffner, S.F.; Drake, J.A.; Rhodes, M.; Reich, D.E.; Hirschhorn, J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004, 74, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Voight, B.F.; Kudaravalli, S.; Wen, X.Q.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, 446–458. [Google Scholar]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Ellegren, H.; Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Loftus, R.T.; MacHugh, D.E.; Bradley, D.G.; Sharp, P.M.; Cunningham, P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA 1994, 91, 2757–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollongino, R.; Burger, J.; Powell, A.; Mashkour, M.; Vigne, J.-D.; Thomas, M. Modern Taurine Cattle Descended from Small Number of Near-Eastern Founders. Mol. Biol. Evol. 2012, 29, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Götherström, A.; Anderung, C.; Hellborg, L.; Galil, R.; Smith, E.C.; Bradley, D.G.; Ellegren, H. Cattle domestication in the Near East was followed by hybridization with aurochs bulls in Europe. Proc. R. Soc. B Boil. Sci. 2005, 272, 2660. [Google Scholar] [CrossRef] [Green Version]
- Flori, L.; Fritz, S.; Jaffrézic, F.; Boussaha, M.; Gut, I.; Heath, S.; Foulley, J.-L.; Gautier, M. The Genome Response to Artificial Selection: A Case Study in Dairy Cattle. PLoS ONE 2009, 4, e6595. [Google Scholar] [CrossRef] [Green Version]
- Pryce, J.E.; Daetwyler, H.D. Designing dairy cattle breeding schemes under genomic selection: A review of international research. Anim. Prod. Sci. 2012, 52, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.; Bowman, P.; Chamberlain, A.; Goddard, M. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Berglund, B. Genetic improvement of dairy cow reproductive performance. Reprod. Domest. Anim. 2008, 43, 89–95. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ding, X.-D.; Wang, S.; Wójcik, J.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.-S.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [Green Version]
- Boitard, S.; Boussaha, M.; Capitan, A.; Rocha, D.; Servin, B. Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds. Genetics 2016, 203, 433–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc GPD. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosse, M.; Megens, H.-J.; Madsen, O.; Paudel, Y.; Frantz, L.A.F.; Schook, L.B.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Regions of Homozygosity in the Porcine Genome: Consequence of Demography and the Recombination Landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res. 2016, 44, D827–D833. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Marnetto, D.; Huerta-Sánchez, E. Haplostrips: Revealing population structure through haplotype visualization. Methods Ecol. Evol. 2017, 8, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.; Billerey, C.; Samson, F.; Boichard, D.; Boussaha, M. Identification of the putative ancestral allele of bovine single-nucleotide polymorphisms. J. Anim. Breed. Genet. 2014, 131, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Uyttendaele, H.; Soriano, J.V.; Montesano, R.; Kitajewski, J. Notch4 and Wnt-1 proteins function to regulate branching morphogenesis of mammary epithelial cells in an opposing fashion. Dev. Biol. 1998, 196, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Gil, B.; Arranz, J.J.; Wiener, P. An interpretive review of selective sweep studies in Bos taurus cattle populations: Identification of unique and shared selection signals across breeds. Front. Genet. 2015, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Pausch, H.; Flisikowski, K.; Jung, S.; Emmerling, R.; Edel, C.; Götz, K.-U.; Fries, R. Genome-Wide Association Study Identifies Two Major Loci Affecting Calving Ease and Growth-Related Traits in Cattle. Genetics 2011, 187, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Karim, L.; Takeda, H.; Lin, L.; Druet, T.; Arias, J.A.C.; Baurain, D.; Cambisano, N.; Davis, S.R.; Farnir, F.; Grisart, B.; et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 2011, 43, 405–413. [Google Scholar] [CrossRef]
- Hou, L.; Panthier, J.-J.; Arnheiter, H. Signaling and transcriptional regulation in the neural crest-derived melanocyte lineage: Interactions between KIT and MITF. Development 2000, 127, 5379–5389. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic Runs of Homozygosity Record Population History and Consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Holdt, B.M.; Pollinger, J.P.; Earl, D.A.; Knowles, J.C.; Boyko, A.R.; Parker, H.; Geffen, E.; Pilot, M.; Jedrzejewski, W.; Jedrzejewska, B.; et al. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 2011, 21, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferenčaković, M.; Hamzic, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Fregeau, B.; Kim, B.J.; Hernandez-Garcia, A.; Jordan, V.K.; Cho, M.T.; Schnur, R.E.; Monaghan, K.G.; Juusola, J.; Rosenfeld, J.A.; Bhoj, E.; et al. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am. J. Hum. Genet. 2016, 98, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Scott, D.A. Mouse Model Reveals the Role of RERE in Cerebellar Foliation and the Migration and Maturation of Purkinje Cells. PLoS ONE 2014, 9, e87518. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, F.; Chen, Y.; Liang, X.; Hirschy, A.; Stenbit, A.E.; Gu, Y.; Dalton, N.D.; Yajima, T.; Lu, Y.C.; Knowlton, K.U.; et al. Alpha-E-Catenin inactivation disrupts the cardiomyocyte adherens junction, resulting in cardiomyopathy and susceptibility to wall rupture. Circulation 2006, 114, 1046–1055. [Google Scholar] [CrossRef]
- Cochran, S.D.; Cole, J.B.; Null, D.J.; Hansen, P.J. Discovery of single nucleotide polymorphisms in candidate genes associated with fertility and production traits in Holstein cattle. BMC Genet. 2013, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, M.S.; Denicol, A.C.; Cole, J.B.; Null, D.J.; Taylor, J.F.; Schnabel, R.D.; Hansen, P.J. Association of single nucleotide polymorphisms in candidate genes previously related to genetic variation in fertility with phenotypic measurements of reproductive function in Holstein cows. J. Dairy Sci. 2017, 100, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Pryce, J.; Chamberlain, A.J.; Bowman, P.J.; Goddard, M.E. Genetic Architecture of Complex Traits and Accuracy of Genomic Prediction: Coat Colour, Milk-Fat Percentage, and Type in Holstein Cattle as Contrasting Model Traits. PLoS Genet. 2010, 6, e1001139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofidou, P.; Nelson, C.; Nikpay, M.; Qu, L.; Li, M.; Loley, C.; Debiec, R.; Braund, P.S.; Denniff, M.; Charchar, F.; et al. Runs of Homozygosity: Association with Coronary Artery Disease and Gene Expression in Monocytes and Macrophages. Am. J. Hum. Genet. 2015, 97, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Ghani, M.; Reitz, C.; Cheng, R.; Vardarajan, B.N.; Jun, G.; Sato, C.; Naj, A.C.; Rajbhandary, R.; Wang, L.-S.; Valladares, O.; et al. Association of Long Runs of Homozygosity with Alzheimer Disease Among African American Individuals. JAMA Neurol. 2015, 72, 1313–1323. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. A Fuller Theory of Junctions in Inbreeding. Heredity 1954, 8, 187–197. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–143. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Effect Size | Standard Error | Significance (p Value) of Substitution Effects between Haplotypes |
|---|---|---|---|
| 11,111 | 0.629 | 0.730 | 0.010 * |
| 12,111 | 0.135 | 0.984 | |
| 22,211 | 0.081 | 0.971 | |
| 22,222 | −0.846 | 0.740 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Schönherz, A.A.; Lund, M.S.; Guldbrandtsen, B. Positive Selection and Adaptive Introgression of Haplotypes from Bos indicus Improve the Modern Bos taurus Cattle. Agriculture 2022, 12, 844. https://doi.org/10.3390/agriculture12060844
Zhang Q, Schönherz AA, Lund MS, Guldbrandtsen B. Positive Selection and Adaptive Introgression of Haplotypes from Bos indicus Improve the Modern Bos taurus Cattle. Agriculture. 2022; 12(6):844. https://doi.org/10.3390/agriculture12060844
Chicago/Turabian StyleZhang, Qianqian, Anna Amanda Schönherz, Mogens Sandø Lund, and Bernt Guldbrandtsen. 2022. "Positive Selection and Adaptive Introgression of Haplotypes from Bos indicus Improve the Modern Bos taurus Cattle" Agriculture 12, no. 6: 844. https://doi.org/10.3390/agriculture12060844