
Accounting for Missing Pedigree Information with Single-Step Random Regression Test-Day Models


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.2. Models
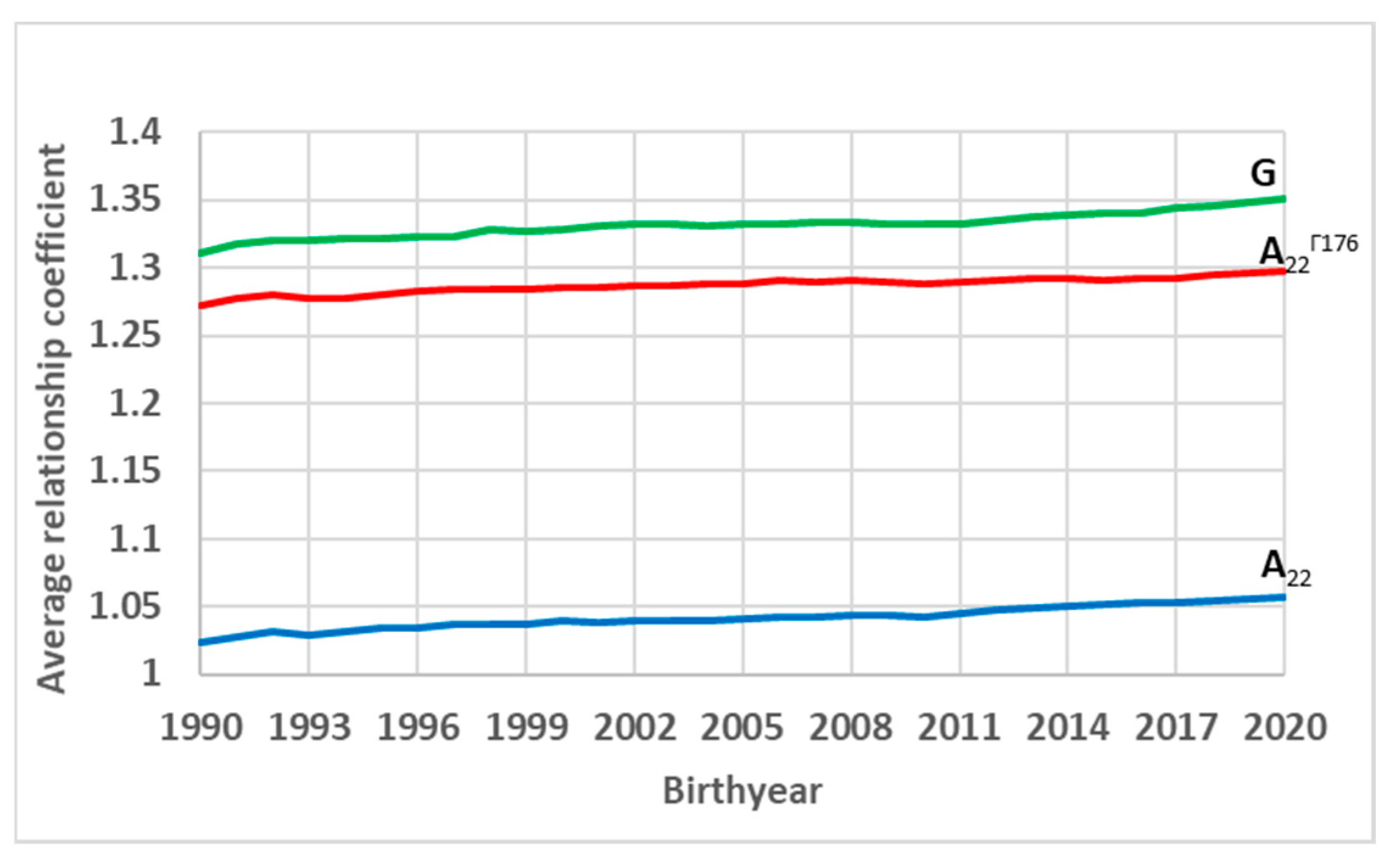
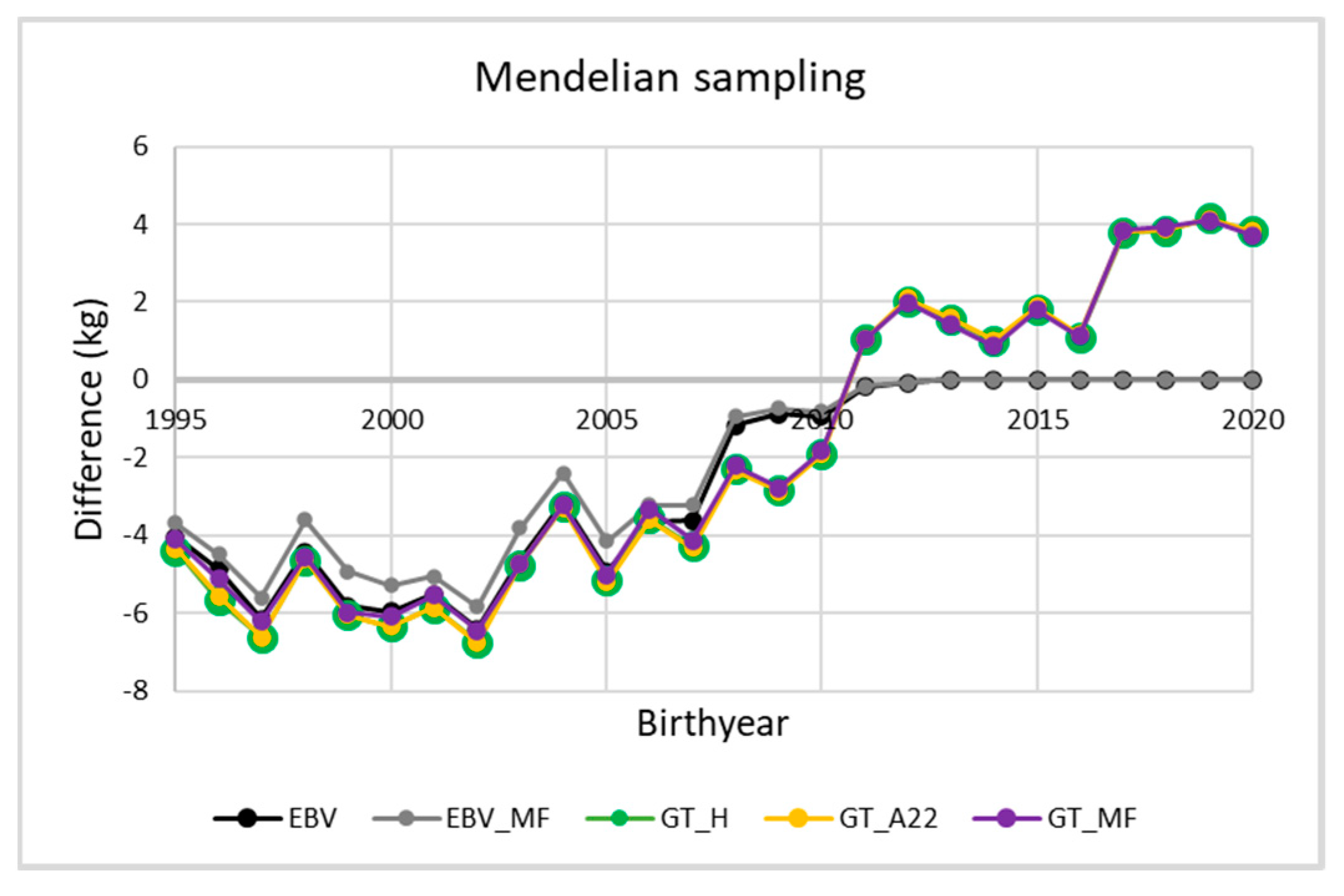
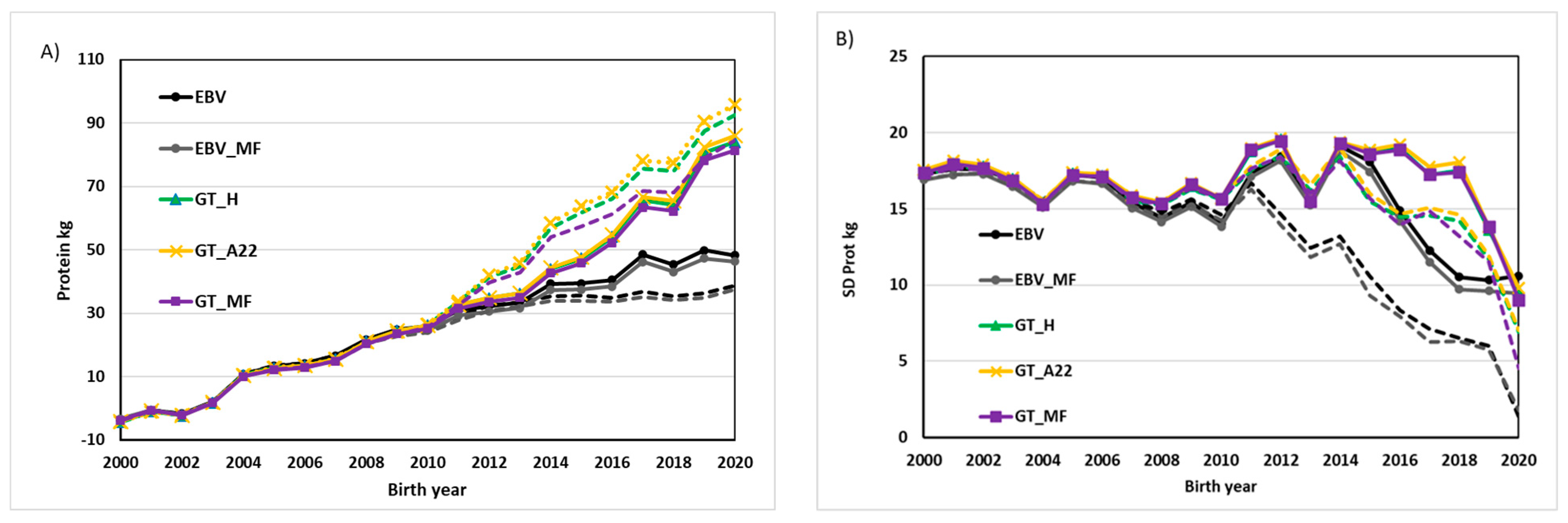
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Symposium review: How to implement genomic selection. J. Dairy Sci. 2020, 103, 5291–5301. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, I.; Misztal, I.; Johnson, D.-L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Christensen, O.F.; Lund, M.S. Genomic prediction when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäntysaari, E.A.; Koivula, M.; Strandén, I. Symposium review: Single-step genomic evaluations in dairy cattle. J. Dairy Sci. 2020, 103, 5314–5326. [Google Scholar] [CrossRef]
- Misztal, I.; Lourenco, D.; Legarra, A. Current status of genomic evaluation. J. Anim. Sci. 2020, 98, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Christensen, O.F. Compatibility of pedigree-based and marker-based relationship matrices for single-step genetic evaluation. Genet. Sel. Evol. 2012, 44, 37. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Vitezica, Z.G.; Aguilar, I.; Misztal, I.; Legarra, A. Bias in genomic predictions for populations under selection. Genet. Res. 2011, 93, 357–366. [Google Scholar] [CrossRef]
- Misztal, I.; Vitezica, Z.G.; Legarra, A.; Aguilar, I.; Swan, A.A. Unknown-parent groups in single-step genomic evaluation. J. Anim. Breed. Genet. 2013, 130, 252–258. [Google Scholar] [CrossRef]
- Thompson, R. The estimation of heritability with unbalanced data: II. Data available on more than two generations. Biometrics 1979, 33, 497–504. [Google Scholar] [CrossRef]
- Westell, R.A.; Quaas, R.L.; Van Vleck, L.D. Genetic groups in an animal model. J. Dairy Sci. 1988, 71, 1310–1318. [Google Scholar] [CrossRef]
- Quaas, R.L.; Pollak, E.J. Modified equations for sire models with groups. J. Dairy Sci. 1981, 64, 1868–1872. [Google Scholar] [CrossRef]
- Matilainen, K.; Strandén, I.; Aamand, G.P.; Mäntysaari, E.A. Single step genomic evaluation for female fertility in Nordic Red dairy cattle. J. Anim. Breed. Genet. 2018, 135, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Koivula, M.; Strandén, I.; Aamand, G.P.; Mäntysaari, E.A. Practical implementation of genetic groups in single-step genomic evaluations with Woodbury matrix identity based genomic relationship inverse. J. Dairy Sci. 2021, 104, 10049–10058. [Google Scholar] [CrossRef]
- Tsuruta, S.; Lourenco, D.A.L.; Masuda, Y.; Misztal, I.; Lawlor, T.J. Controlling bias in genomic breeding values for young genotyped bulls. J. Dairy Sci. 2019, 102, 9956–9970. [Google Scholar] [CrossRef] [PubMed]
- Cesarani, A.; Masuda, Y.; Tsuruta, S.; Nicolazzi, E.K.; VanRaden, P.M.; Lourenco, D.; Misztal, I. Genomic predictions for yield traits in US Holsteins with unknown parent groups. J. Dairy Sci. 2021, 104, 5843–5853. [Google Scholar] [CrossRef]
- Masuda, Y.; Tsuruta, S.; Bermann, M.; Bradford, H.L.; Misztal, I. Comparison of models for missing pedigree in single-step genomic prediction. J. Anim. Sci. 2021, 99, 1–10. [Google Scholar] [CrossRef]
- Legarra, A.; Christensen, O.F.; Vitezica, Z.G.; Aguilar, I.; Misztal, I. Ancestral relationships using metafounders: Finite ancestral populations and across population relationships. Genetics 2015, 200, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Baccino, C.; Legarra, A.A.; Christensen, O.F.; Misztal, I.; Pocrnic, I.; Vitezica, Z.G.; Cantet, R.J. Metafounders are related to Fst fixation indices and reduce bias in single-step genomic evaluations. Genet. Sel. Evol. 2017, 49, 34. [Google Scholar] [CrossRef] [Green Version]
- Kudinov, A.A.; Mäntysaari, E.A.; Aamand, G.P.; Uimari, P.; Strandén, I. Metafounder approach for single-step genomic evaluations of Red Dairy cattle. J. Dairy Sci. 2020, 103, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Hill, W.G.; Thompson, R. Estimating the covariance structure of traits during growth and ageing, illustrated with lactation in dairy cattle. Genet. Res. 1994, 64, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudinov, A.A.; Koivula, M.; Strandén, I.; Aamand, G.P.; Mäntysaari, E.A. Single-step genomic predictions of a minor breed concurrently with a main breed large national genomic evaluation. Interbull Bull. 2021, 56, 174–179. [Google Scholar]
- EuroGenomics. A European Network for a Reliable Cattle Breeding. 2022. Available online: https://www.eurogenomics.com/?rub=88&unce_contenus_webclient=0&view=afficher_elasticsearch_results&submitted=1&q=A+European+Network+for+a+Reliable+Cattle+Breeding.+2022 (accessed on 10 January 2022).
- Sargolzaei, M.; Chesnais, I.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [Green Version]
- Mäntysaari, E.A.; Evans, R.D.; Strandén, I. Efficient single-step genomic evaluation for a multibreed beef cattle population having many genotyped animals. J. Anim. Sci. 2017, 95, 4728–4737. [Google Scholar] [CrossRef] [Green Version]
- Lidauer, M.; Pösö, J.; Pederson, J.; Lassen, J.; Madsen, P.; Mäntysaari, E.A.; Nielsen, U.; Eriksson, K.-Å.; Johansson, K.; Pitkänen, T.; et al. Across-country test-day model evaluations for Nordic Holstein, Red Cattle and Jersey. J. Dairy Sci. 2015, 98, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Legarra, A.; Reverter, A. Semi-parametric estimates of population accuracy and bias of predictions of breeding values and future phenotypes using the LR method. Genet. Sel. Evol. 2018, 50, 53. [Google Scholar] [CrossRef] [Green Version]
- Stranden, I.; Lidauer, M.; Mäntysaari, E.A.; Pösö, J. Calculation of Interbull weighting factors for the Finnish test day model. Interbull Bull. 2001, 26, 78–79. [Google Scholar]
- Strandén, I.; Lidauer, M. Solving large mixed models using preconditioned conjugate gradient iteration. J. Dairy Sci. 1999, 82, 2779–2787. [Google Scholar] [CrossRef]
- Strandén, I.; Matilainen, K.; Aamand, G.P.; Mäntysaari, E.A. Solving efficiently large single-step genomic best linear unbiased prediction models. J. Anim. Breed. Genet. 2017, 134, 264–274. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M.; Olson, K.M.; Wiggans, G.R.; Cole, J.B.; Tooker, M.E. Genomic inbreeding and relationships among Holsteins, Jerseys, and Brown Swiss. J. Dairy Sci. 2011, 94, 5673–5682. [Google Scholar] [CrossRef] [PubMed]
- Tyrisevä, A.-M.; Mäntysaari, E.A.; Jakobsen, J.; Aamand, G.P.; Dürr, J.; Fikse, W.F.; Lidauer, M.H. Detection of evaluation bias caused by genomic preselection. J. Dairy Sci. 2018, 101, 3155–3163. [Google Scholar] [CrossRef]
- Masuda, Y.; VanRaden, P.M.; Misztal, I.; Lawlor, T.J. Differing genetic trend estimates from traditional and genomic evaluations of genotyped animals as evidence of preselection bias in US Holsteins. J. Dairy Sci. 2018, 101, 5194–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi-Arpanahi, R.; Lourenco, D.; Misztal, I. Detecting effective starting point of genomic selection by divergent trends from best linear unbiased prediction and single-step genomic best linear unbiased prediction in pigs, beef cattle, and broilers. J. Anim. Sci. 2021, 99, 1–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EBV | EBV_MF | GT_H | GT_A22 | GT_MF | |
|---|---|---|---|---|---|
| Building T matrix time (h) | 23 | 20 | 20 | ||
| Peak memory GB | 208 | 207 | 207 | ||
| Solving | |||||
| Iterations | 1227 | 1264 | 1019 | 1051 | 1307 |
| Seconds/PCG round | 101 | 125 | 173 | 178 | 125 |
| Time (h) | 34 | 44 | 49 | 52 | 45 |
| Peak memory GB | 14.9 | 15.0 | 114.5 | 114.3 | 114.5 |
| Total computing time (h) | 34 | 44 | 72 | 72 | 65 |
| Model | b0 | b1 | R2 | |
|---|---|---|---|---|
| Milk | EBV | −101.7 | 0.84 | 0.32 |
| EBV_MF | −141.36 | 0.89 | 0.35 | |
| GT_H | −319.8 | 0.87 | 0.67 | |
| GT_A22 | −315.2 | 0.87 | 0.67 | |
| GT_MF | −272.3 | 0.89 | 0.68 | |
| Protein | EBV | 0.80 | 0.74 | 0.24 |
| EBV_MF | 0.58 | 0.82 | 0.27 | |
| GT_H | −11.10 | 0.81 | 0.63 | |
| GT_A22 | −10.99 | 0.81 | 0.62 | |
| GT_MF | −9.71 | 0.83 | 0.64 | |
| Fat | EBV | −2.18 | 0.73 | 0.23 |
| EBV_MF | −2.24 | 0.80 | 0.26 | |
| GT_H | −16.16 | 0.82 | 0.64 | |
| GT_A22 | −15.61 | 0.81 | 0.63 | |
| GT_MF | −14.67 | 0.85 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koivula, M.; Strandén, I.; Aamand, G.P.; Mäntysaari, E.A. Accounting for Missing Pedigree Information with Single-Step Random Regression Test-Day Models. Agriculture 2022, 12, 388. https://doi.org/10.3390/agriculture12030388
Koivula M, Strandén I, Aamand GP, Mäntysaari EA. Accounting for Missing Pedigree Information with Single-Step Random Regression Test-Day Models. Agriculture. 2022; 12(3):388. https://doi.org/10.3390/agriculture12030388
Chicago/Turabian StyleKoivula, Minna, Ismo Strandén, Gert P. Aamand, and Esa A. Mäntysaari. 2022. "Accounting for Missing Pedigree Information with Single-Step Random Regression Test-Day Models" Agriculture 12, no. 3: 388. https://doi.org/10.3390/agriculture12030388