
Comparison of Methods to Select Candidates for High-Density Genotyping; Practical Observations in a Cattle Breeding Program

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
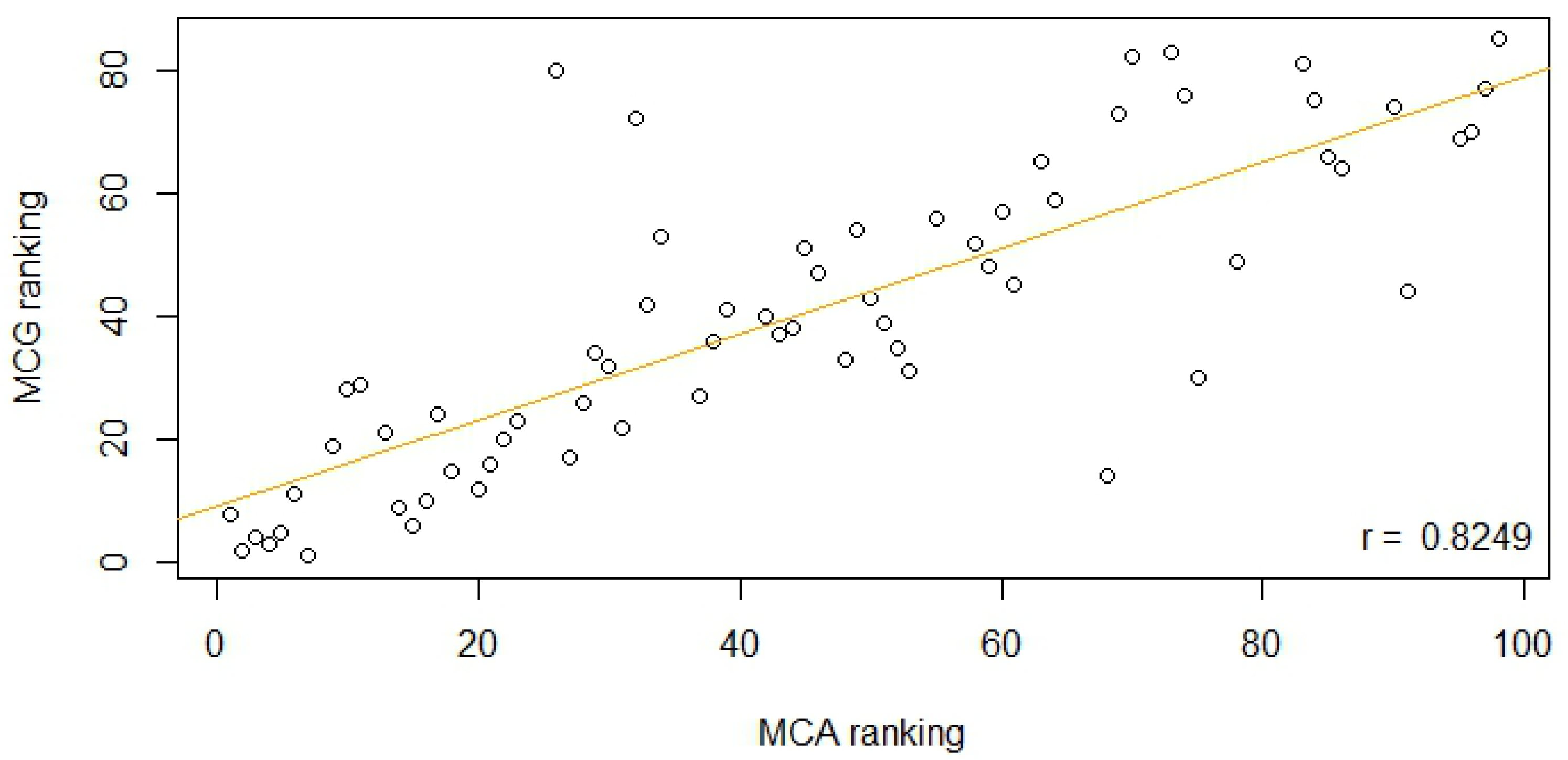
3.1. Overlap between Chosen Candidates
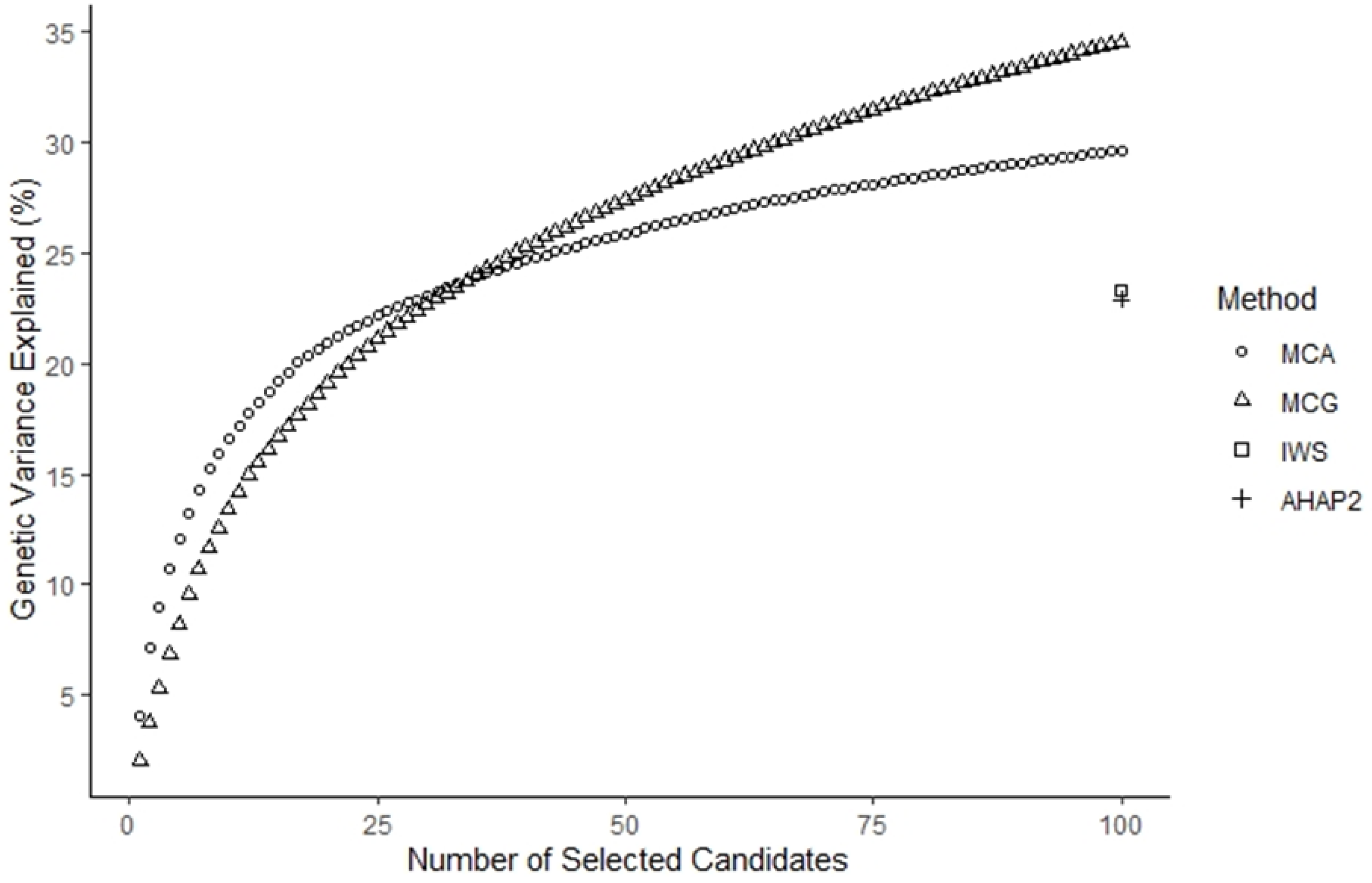
3.2. Percentage of Genetic Variance Explained
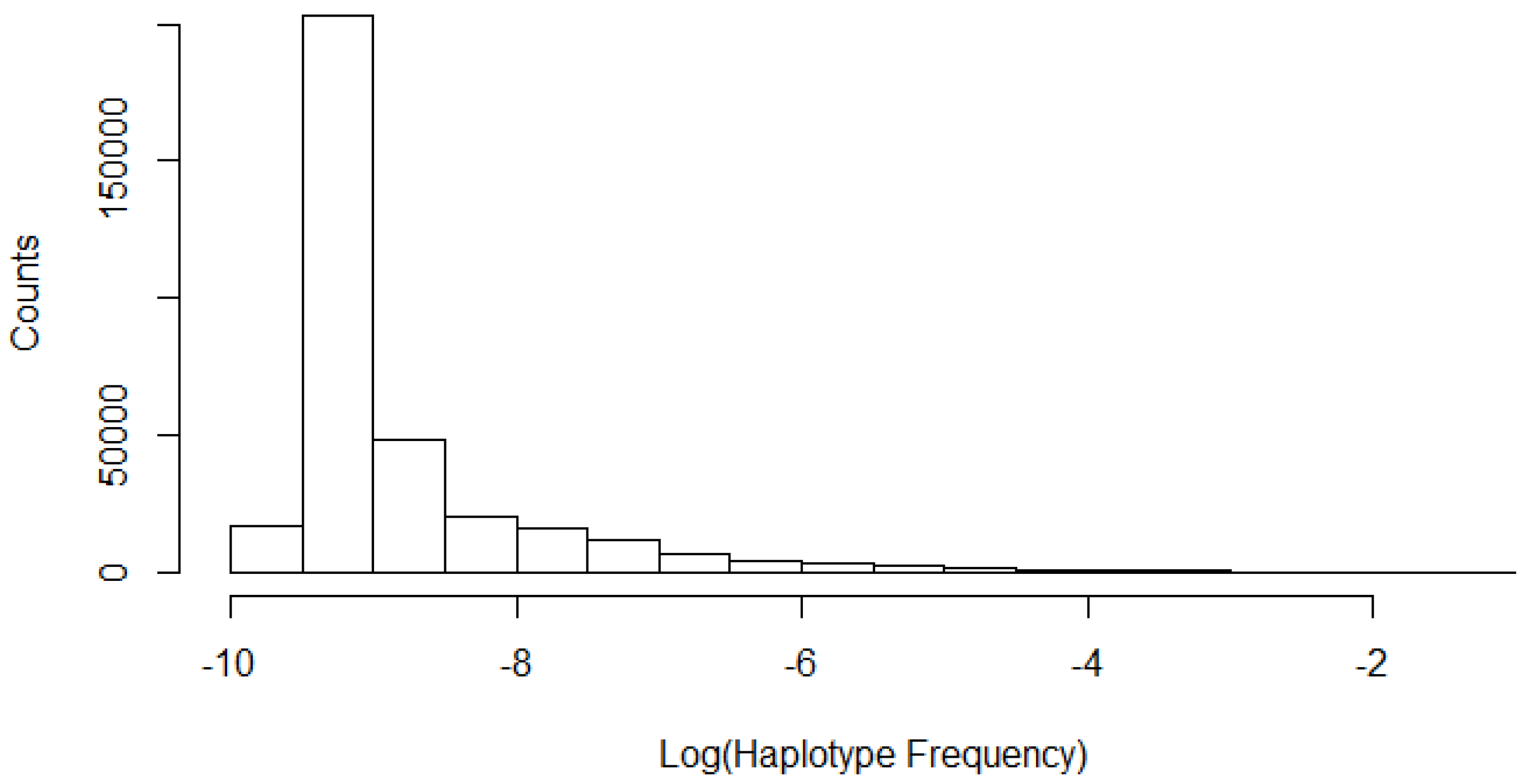
3.3. Number of Unique Haplotypes Accounted for
4. Discussion
4.1. Comparison of Relationship Matrix Methods
4.2. Comparison of Haplotype Block Methods
4.3. Practical Considerations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.; Goddard, M. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.M.; Dang, C.G.; Alam, M.Z.; Kim, Y.S.; Cho, K.H.; Park, K.D.; Kim, J.J. The effectiveness of genomic selection for milk production traits of Holstein dairy cattle. Asian-Australas. J. Anim. Sci. 2020, 33, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisart, B.; Coppieters, W.; Farnir, F.; Karim, L.; Ford, C.; Berzi, P.; Cambisano, N.; Mni, M.; Reid, S.; Simon, P. Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002, 12, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatkar, M.S.; Moser, G.; Hayes, B.J.; Raadsma, H.W. Strategies and utility of imputed SNP genotypes for genomic analysis in dairy cattle. BMC Genom. 2012, 13, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, S.; Matsuda, H.; Taniguchi, Y.; Watanabe, T.; Kitamura, Y.; Tabuchi, I.; Sugimoto, Y.; Iwaisaki, H. Genomic prediction for carcass traits in Japanese Black cattle using single nucleotide polymorphism markers of different densities. Anim. Prod. Sci. 2017, 57, 1631–1636. [Google Scholar] [CrossRef]
- Goddard, M. Genomic selection: Prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Georges, M. Towards sequence-based genomic selection of cattle. Nat. Genet. 2009, 46, 807. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; Tooker, M.E.; O’connell, J.R.; Cole, J.B.; Bickhart, D.M. Selecting sequence variants to improve genomic predictions for dairy cattle. Genet. Sel. Evol. 2017, 49, 32. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Woolliams, J.A.; Meuwissen, T.H. Prioritizing animals for dense genotyping in order to impute missing genotypes of sparsely genotyped animals. Genet. Sel. Evol. 2014, 46, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickhart, D.; Hutchison, J.; Null, D.; VanRaden, P.; Cole, J. Reducing animal sequencing redundancy by preferentially selecting animals with low-frequency haplotypes. J. Dairy Sci. 2016, 99, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Vazquez, A.I. Pedigreemm: Pedigree-Based Mixed-Effects Models. R Package Version 0.0-3. Available online: https://CRAN.R-project.org/package=pedigreemm (accessed on 2 March 2021).
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Sargolzaei, M.; Chesnais, J.; Schenkel, F. FImpute-An efficient imputation algorithm for dairy cattle populations. J. Dairy Sci. 2011, 94, 421. [Google Scholar]
- Druet, T.; Macleod, I.; Hayes, B. Toward genomic prediction from whole-genome sequence data: Impact of sequencing design on genotype imputation and accuracy of predictions. Heredity 2014, 112, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.D.; Banks, R. Genetic diversity and trends of Australian Japanese Black cattle. Proc. Assoc. Adv. Anim. Breed. Genet. 2021, 24, 451–454. [Google Scholar]
- Butty, A.M.; Sargolzaei, M.; Miglior, F.; Stothard, P.; Schenkel, F.S.; Gredler-Grandl, B.; Baes, C.F. Optimizing selection of the reference population for genotype imputation from array to sequence variants. Front. Genet. 2019, 10, 510. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858. [Google Scholar] [CrossRef]
- Gonen, S.; Ros-Freixedes, R.; Battagin, M.; Gorjanc, G.; Hickey, J.M. A method for the allocation of sequencing resources in genotyped livestock populations. Genet. Sel. Evol. 2017, 49, 47. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Gonen, S.; Gorjanc, G.; Hickey, J.M. A method for allocating low-coverage sequencing resources by targeting haplotypes rather than individuals. Genet. Sel. Evol. 2017, 49, 78. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Whalen, A.; Gorjanc, G.; Mileham, A.J.; Hickey, J.M. Evaluation of sequencing strategies for whole-genome imputation with hybrid peeling. Genet. Sel. Evol. 2020, 52, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, O.F.; Lund, M.S. Genomic prediction when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| MCA | MCG | IWS | AHAP2 | PROG | |
|---|---|---|---|---|---|
| MCA | 100 | ||||
| MCG | 70 | 100 | |||
| IWS | 5 | 7 | 100 | ||
| AHAP2 | 2 | 4 | 61 | 100 | |
| PROG | 80 | 78 | 7 | 4 |
| Method | Common | Uncommon | Rare | Total |
|---|---|---|---|---|
| ≥5% | 1%–<5% | 0.1%–<1% | ||
| MCA A | - | - | - | - |
| MCG | 588 | 3557 | 8175 | 12,320 |
| IWS | 588 | 3507 | 6492 | 10,587 |
| AHAP2 | 588 | 3524 | 5137 | 9249 |
| Max # B | 588 | 3666 | 16,600 | 20,854 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McEwin, R.A.; Hebart, M.L.; Oakey, H.; Tearle, R.; Grose, J.; Popplewell, G.; Pitchford, W.S. Comparison of Methods to Select Candidates for High-Density Genotyping; Practical Observations in a Cattle Breeding Program. Agriculture 2022, 12, 276. https://doi.org/10.3390/agriculture12020276
McEwin RA, Hebart ML, Oakey H, Tearle R, Grose J, Popplewell G, Pitchford WS. Comparison of Methods to Select Candidates for High-Density Genotyping; Practical Observations in a Cattle Breeding Program. Agriculture. 2022; 12(2):276. https://doi.org/10.3390/agriculture12020276
Chicago/Turabian StyleMcEwin, Rudi A., Michelle L. Hebart, Helena Oakey, Rick Tearle, Joe Grose, Greg Popplewell, and Wayne S. Pitchford. 2022. "Comparison of Methods to Select Candidates for High-Density Genotyping; Practical Observations in a Cattle Breeding Program" Agriculture 12, no. 2: 276. https://doi.org/10.3390/agriculture12020276