

Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions
 ,
,  , , , , ,
, , , , ,
Abstract
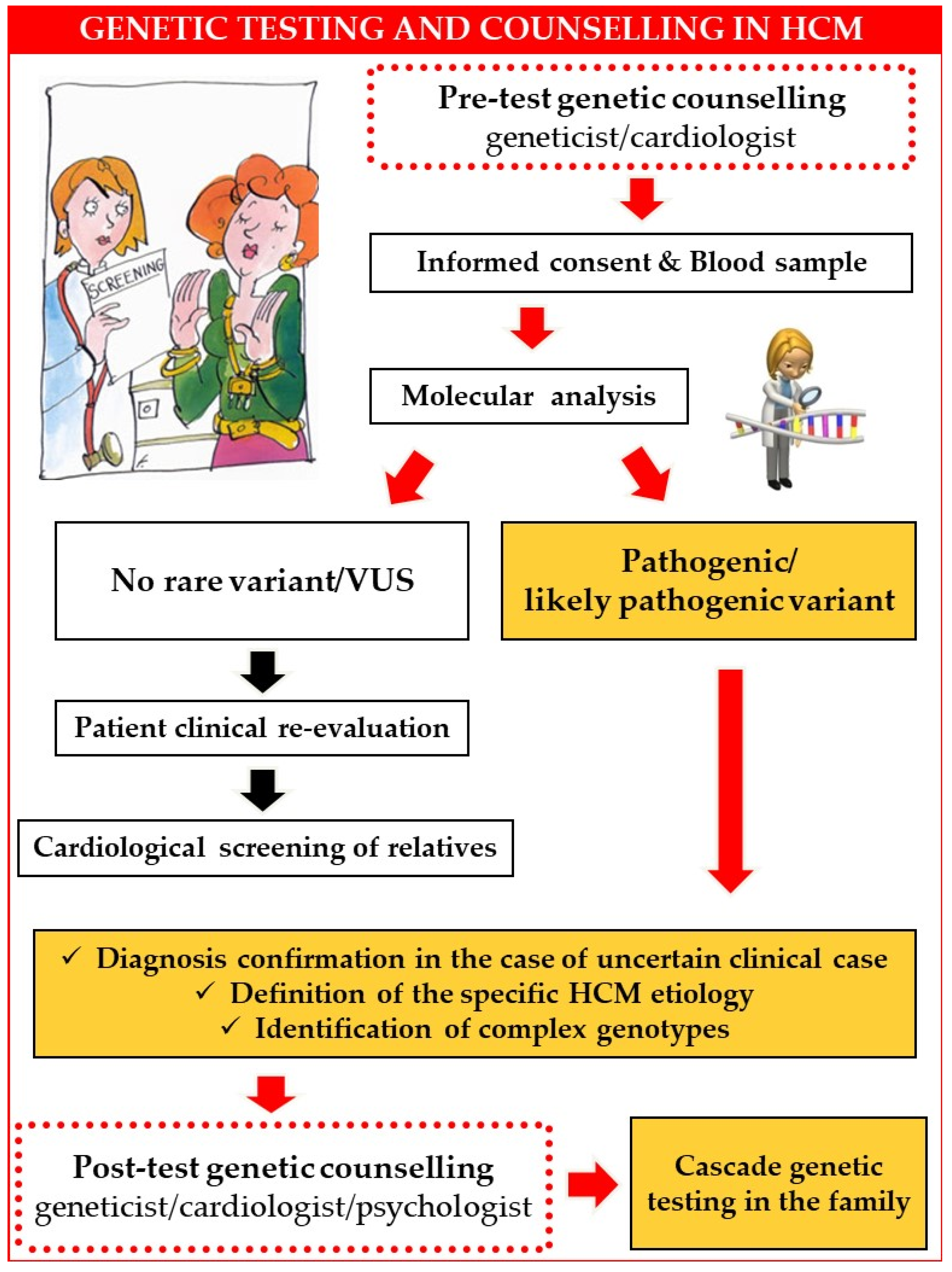
:1. Introduction
2. Genetic Counselling
3. Patients’ Questions, Real-World Answers
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Dearani, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Management of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 390–414. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Braunwald, E.; Lambrew, C.T.; Rockoff, S.D.; Ross, J., Jr.; Morrow, A.G. Idiopathic Hypertrophic Subaortic Stenosis. I. A Description of the Disease Based Upon an Analysis of 64 Patients. Circulation 1964, 30 (Suppl. S4), 3–119. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2020, 76, 3022–3055. [Google Scholar] [CrossRef]
- Jarcho, J.A.; McKenna, W.; Pare, J.A.; Solomon, S.D.; Holcombe, R.F.; Dickie, S.; Levi, T.; Donis-Keller, H.; Seidman, J.G.; Seidman, C.E. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 1989, 321, 1372–1378. [Google Scholar] [CrossRef] [Green Version]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. JACC 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Semsarian, C.; Semsarian, C.R. Variable Penetrance in Hypertrophic Cardiomyopathy: In Search of the Holy Grail. JACC 2020, 76, 560–562. [Google Scholar] [CrossRef]
- Ho, C.Y.; Charron, P.; Richard, P.; Girolami, F.; Van Spaendonck-Zwarts, K.Y.; Pinto, Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 2015, 105, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo. Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y.; et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009, 301, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banankhah, P.; Fishbein, G.A.; Dota, A.; Ardehali, R. Cardiac manifestations of PRKAG2 mutation. BMC Med. Genet. 2018, 19, 1. [Google Scholar] [CrossRef] [Green Version]
- Arends, M.; Wijburg, F.A.; Wanner, C.; Vaz, F.M.; van Kuilenburg, A.B.P.; Hughes, D.A.; Biegstraaten, M.; Mehta, A.; Hollak, C.E.M.; Langeveld, M. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol. Genet. Metab. 2017, 121, 157–161. [Google Scholar] [CrossRef]
- Gagliardi, C.; Perfetto, F.; Lorenzini, M.; Ferlini, A.; Salvi, F.; Milandri, A.; Quarta, C.C.; Taborchi, G.; Bartolini, S.; Frusconi, S.; et al. Phenotypic profile of Ile68Leu transthyretin amyloidosis: An underdiagnosed cause of heart failure. Eur. J. Heart Fail. 2018, 20, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Girolami, F.; Vergaro, G.; Pieroni, M.; Passantino, S.; Giannotti, G.; Grippo, G.; Canale, M.L.; Favilli, S.; Cappelli, F.; Olivotto, I.; et al. Clinical pathway for cardiomyopathies: A genetic testing strategy proposed by ANMCO in Tuscany. G. Ital. Cardiol. 2020, 21, 926–934. [Google Scholar]
- Mazzarotto, F.; Girolami, F.; Boschi, B.; Barlocco, F.; Tomberli, A.; Baldini, K.; Coppini, R.; Tanini, I.; Bardi, S.; Contini, E.; et al. Defining the diagnostic effectiveness of genes for inclusion in panels: The experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet. Med. 2019, 21, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Girolami, F.; Iascone, M.; Pezzoli, L.; Passantino, S.; Limongelli, G.; Monda, E.; Rubino, M.; Adorisio, R.; Lombardi, M.; Ragni, L.; et al. Clinical pathway on pediatric cardiomyopathies: A genetic testing strategy proposed by the Italian Society of Pediatric Cardiology. G. Ital. Cardiol. 2022, 23, 505–515. [Google Scholar]
- Girolami, F.; Frisso, G.; Benelli, M.; Crotti, L.; Iascone, M.; Mango, R.; Mazzaccara, C.; Pilichou, K.; Arbustini, E.; Tomberli, B.; et al. Contemporary genetic testing in inherited cardiac disease: Tools, ethical issues, and clinical applications. J. Cardiovasc. Med. 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbustini, E.; Behr, E.R.; Carrier, L.; van Duijn, C.; Evans, P.; Favalli, V.; van der Harst, P.; Haugaa, K.H.; Jondeau, G.; Kääb, S.; et al. Interpretation and actionability of genetic variants in cardiomyopathies: A position statement from the European Society of Cardiology Council on cardiovascular genomics. Eur. Heart J. 2022, 43, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Charron, P.; Héron, D.; Gargiulo, M.; Richard, P.; Dubourg, O.; Desnos, M.; Bouhour, J.B.; Feingold, J.; Carrier, L.; Hainque, B.; et al. Genetic testing and genetic counselling in hypertrophic cardiomyopathy: The French experience. J. Med. Genet. 2002, 39, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Cirino, A.L.; Seidman, C.E.; Ho, C.Y. Genetic Testing and Counselling for Hypertrophic Cardiomyopathy. Cardiol. Clin. 2019, 37, 35–43. [Google Scholar] [CrossRef]
- Skrzynia, C.; Demo, E.M.; Baxter, S.M. Genetic counselling and testing for hypertrophic cardiomyopathy: An adult perspective. J. Cardiovasc. Transl. Res. 2009, 2, 493–499. [Google Scholar] [CrossRef]
- Cirino, A.L.; Harris, S.L.; Murad, A.M.; Hansen, B.; Malinowski, J.; Natoli, J.L.; Kelly, M.A.; Christian, S. The uptake and utility of genetic testing and genetic counselling for hypertrophic cardiomyopathy—A systematic review and meta-analysis. J. Genet. Couns. 2022, 31, 1290–1305. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, F.; Munkhsaikhan, U.; Boyle, C.; Borcky, T.; Zhao, W.; Purevjav, E.; Towbin, J.A.; Liao, F.; Williams, R.W.; et al. Identifying modifier genes for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2020, 144, 119–126. [Google Scholar] [CrossRef]
- Torricelli, F.; Girolami, F.; Olivotto, I.; Passerini, I.; Frusconi, S.; Vargiu, D.; Richard, P.; Cecchi, F. Prevalence and clinical profile of troponin T mutations among patients with hypertrophic cardiomyopathy in Tuscany. Am. J. Cardiol. 2003, 92, 1358–1362. [Google Scholar] [CrossRef]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Torricelli, F.; Yeates, L.; Cecchi, F.; Ackerman, M.J.; et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurizi, N.; Michels, M.; Rowin, E.J.; Semsarian, C.; Girolami, F.; Tomberli, B.; Cecchi, F.; Maron, M.S.; Olivotto, I.; Maron, B.J. Clinical Course and Significance of Hypertrophic Cardiomyopathy Without Left Ventricular Hypertrophy. Circulation 2019, 139, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Monda, E.; Vritz, O.; Albert, D.C.; Rubino, M.; Verrillo, F.; Caiazza, M.; Lioncino, M.; Amodio, F.; Guarnaccia, N.; et al. Medical treatment of patients with hypertrophic cardiomyopathy: An overview of current and emerging therapy. Arch. Cardiovasc. Dis. 2022, 115, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Helms, A.S.; Thompson, A.D.; Day, S.M. Translation of New and Emerging Therapies for Genetic Cardiomyopathies. JACC 2021, 7, 70–83. [Google Scholar] [CrossRef]
- Investor Portal. Tenaya Therapeutics Receives Orphan Drug Designation and Presents Pre-Clinical Data for Its Most Advanced Gene Therapy Product Candidate for Genetic Hypertrophic Cardiomyopathy. Available online: https://investors.tenayatherapeutics.com/news-releases/news-release-details/tenaya-therapeutics-receives-orphan-drug-designation-and (accessed on 11 February 2023).
- Globe Newswire. LEXEO Therapeutics Expands Cardiac Gene Therapy Pipeline with Acquisition of Stelios Therapeutics and its Gene Therapy Programs for Rare Cardiovascular Diseases. Available online: https://www.globenewswire.com/news-release/2021/07/21/2266478/0/en/LEXEO-Therapeutics-Expands-Cardiac-Gene-Therapy-Pipeline-with-Acquisition-of-Stelios-Therapeutics-and-its-Gene-Therapy-Programs-for-Rare-Cardiovascular-Diseases.html (accessed on 11 February 2023).
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Baldi, M.; Girolami, F. A new type of “tracing” for cardiologists? G. Ital. Cardiol. 2009, 10, 266. [Google Scholar]
- Semsarian, C.; Gray, B.; Haugaa, K.H.; Lampert, R.; Sharma, S.; Kovacic, J.C. Athletic Activity for Patients with Hypertrophic Cardiomyopathy and Other Inherited Cardiovascular Diseases: JACC Focus Seminar ¾. JACC 2022, 80, 1268–1283. [Google Scholar] [CrossRef]
- GPDP. Autorizzazione Generale al Trattamento Dei Dati Genetici. Autorizzazione n. 8/2016, 15 Dicembre 2016. Available online: https://www.garanteprivacy.it/home/docweb/-/docweb-display/docweb/5803688 (accessed on 11 February 2023).
- Kassem, H.S.; Girolami, F.; Sanoudou, D. Molecular genetics made simple. Glob. Cardiol. Sci. Pract. 2012, 2012, 6. [Google Scholar] [CrossRef]
- Semsarian, C.; Ho, C.Y. Screening children at risk for hypertrophic cardiomyopathy: Balancing benefits and harms. Eur. Heart J. 2019, 40, 3682–3684. [Google Scholar] [CrossRef]
- Mital, S.; Ommen, S. To Screen or Not to Screen, That Is the Question. Circulation 2019, 140, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, D.; Bale, S.; Bick, J.; Das, W.W.; Gastier-Foster, M.; Grody, E.; Hegde, E.; Lyon, K.; Spector, H.L.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Barajas-Martinez, H.S.; Behr, E.R.; Breckpot, B.J.; Charron, P.; Chockalingam, P.; et al. N EHRA/HRS/APHRS/LAHRS. Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace 2022, 24, 1307–1367. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Genetic Testing in HCM: Tips for the Cardiologist |
| It is important to inform the patient that genetic testing: |
| Usually needs a long time (up to three months) before results are ready. |
| Has a chance of finding a causative mutation ranging from 30 to 60%, depending on the cardiomyopathy and its presence in other family members. |
| Can be extended to other family members ONLY in the presence of a pathogenic/likely pathogenic variant in the proband. |
| It is undesirable to suggest: |
| Performing the test as a matter of urgency. |
| Waiting for the test result to start treatment. |
| Submitting the entire family to genetic testing before determining the proband’s mutation. |
| Advising a prenatal diagnosis before genetic counselling. |
| Waiting for test results to decide on physical and sport activity. |
| Performing genetic testing for individuals in the family without phenotypic evidence of HCM, including relatives of sudden cardiac death victims. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girolami, F.; Gozzini, A.; Pálinkás, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. https://doi.org/10.3390/jcm12072489
Girolami F, Gozzini A, Pálinkás ED, Ballerini A, Tomberli A, Baldini K, Marchi A, Zampieri M, Passantino S, Porcedda G, et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. Journal of Clinical Medicine. 2023; 12(7):2489. https://doi.org/10.3390/jcm12072489
Chicago/Turabian StyleGirolami, Francesca, Alessia Gozzini, Eszter Dalma Pálinkás, Adelaide Ballerini, Alessia Tomberli, Katia Baldini, Alberto Marchi, Mattia Zampieri, Silvia Passantino, Giulio Porcedda, and et al. 2023. "Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions" Journal of Clinical Medicine 12, no. 7: 2489. https://doi.org/10.3390/jcm12072489