Therapeutic Potential of Heme Oxygenase-1 in Aneurysmal Diseases

 , , , , and
, , , , and 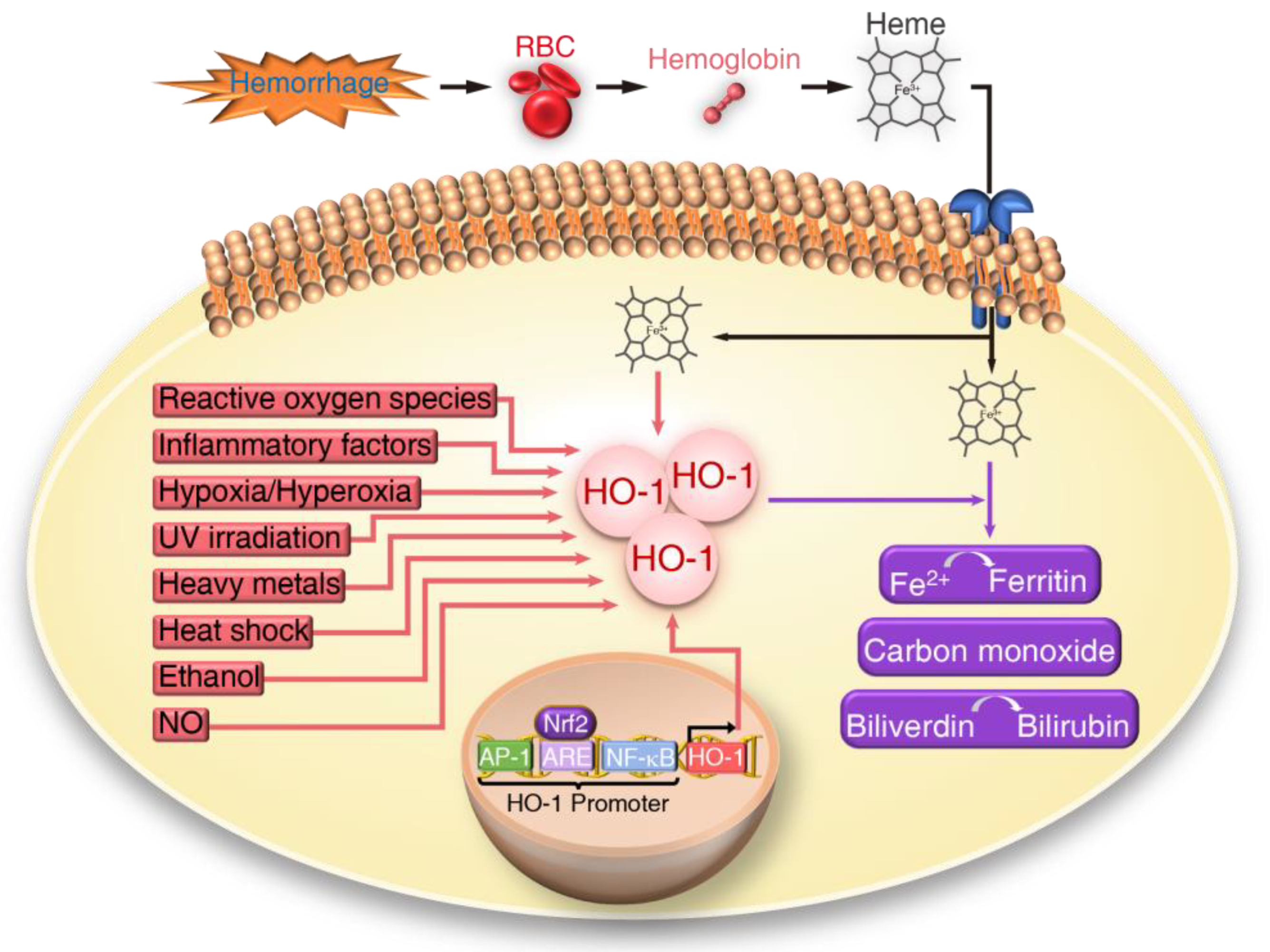{kind=link}
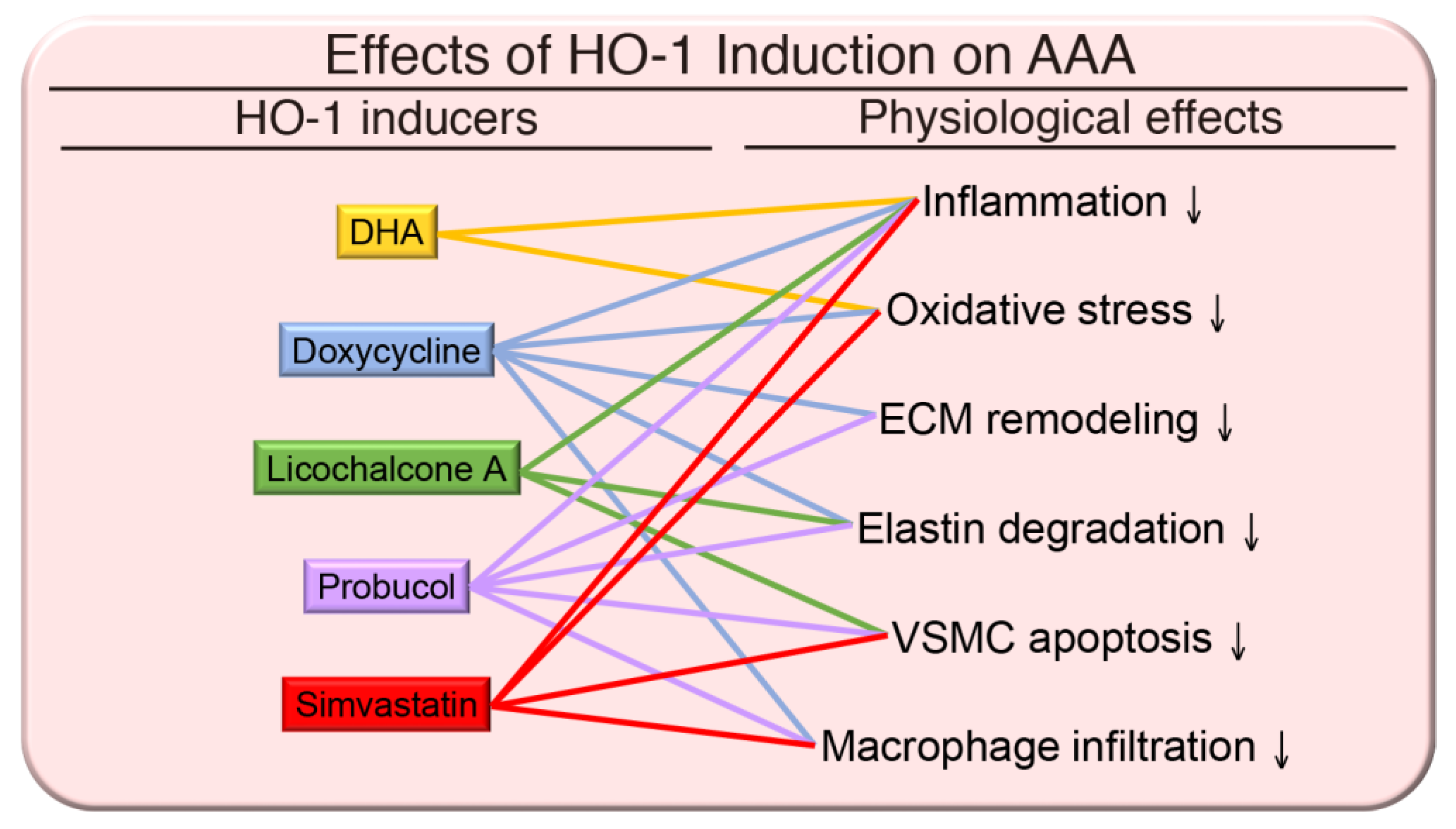{kind=link}
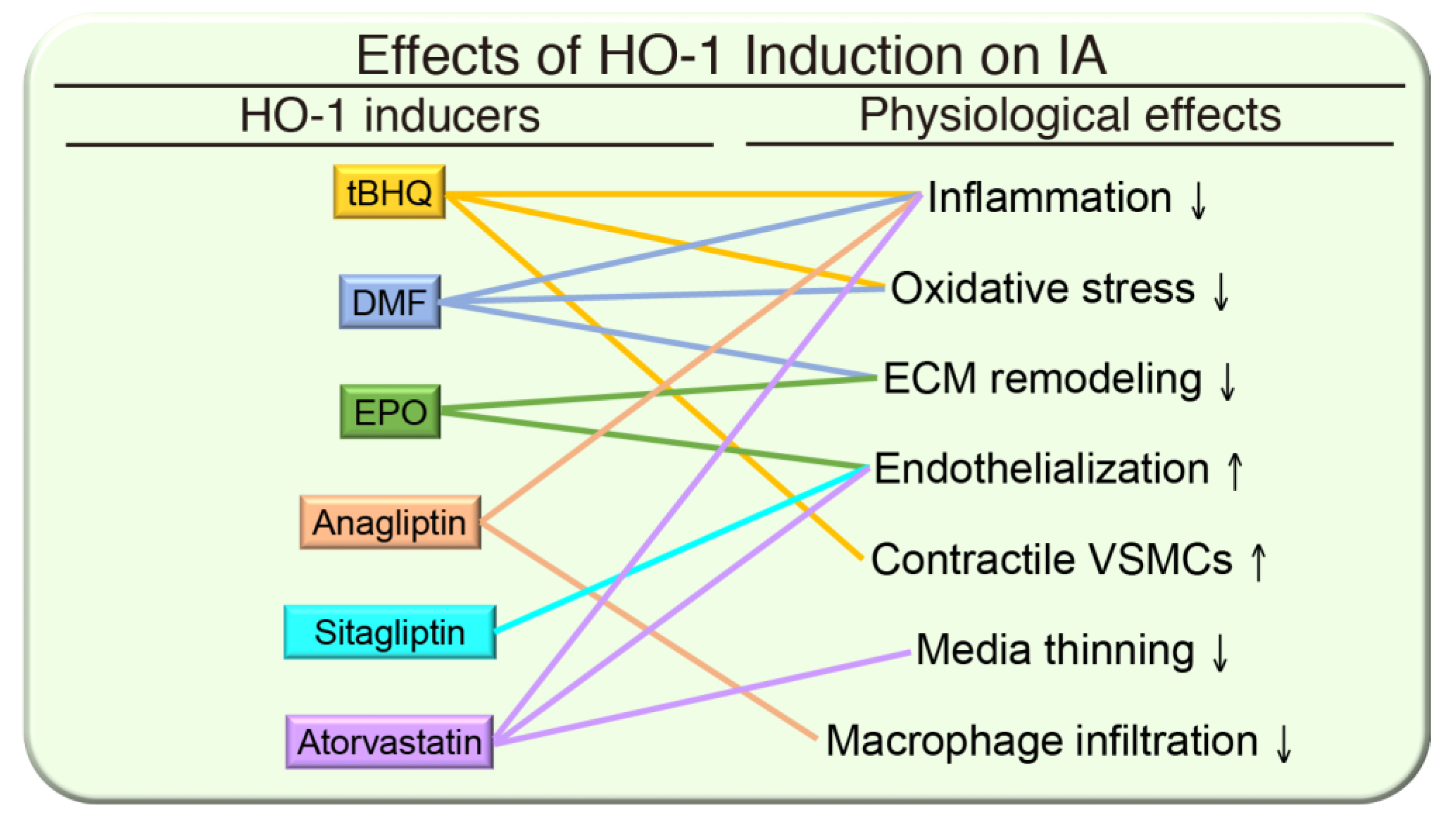{kind=link}
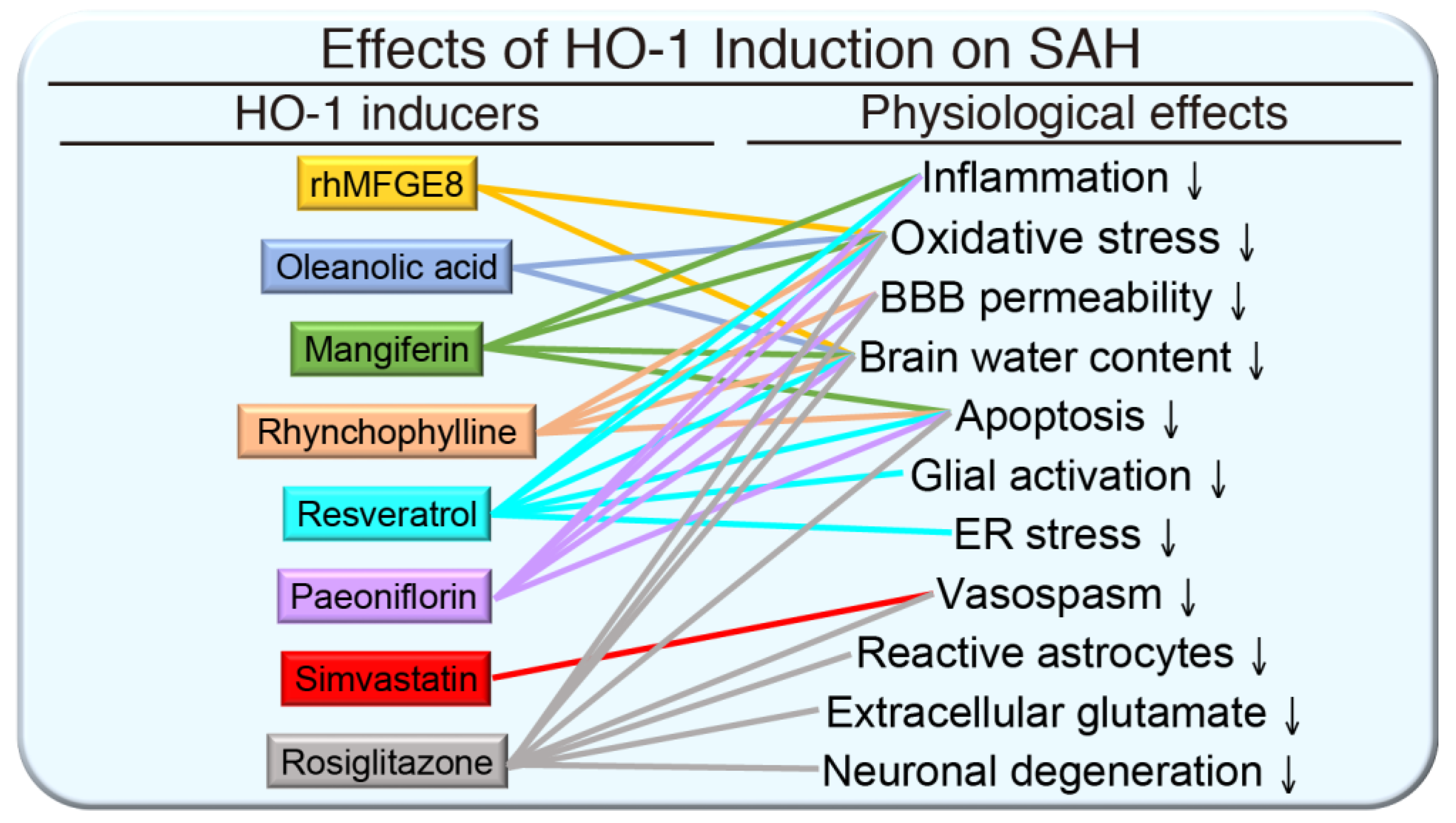{kind=link}
{kind=link}
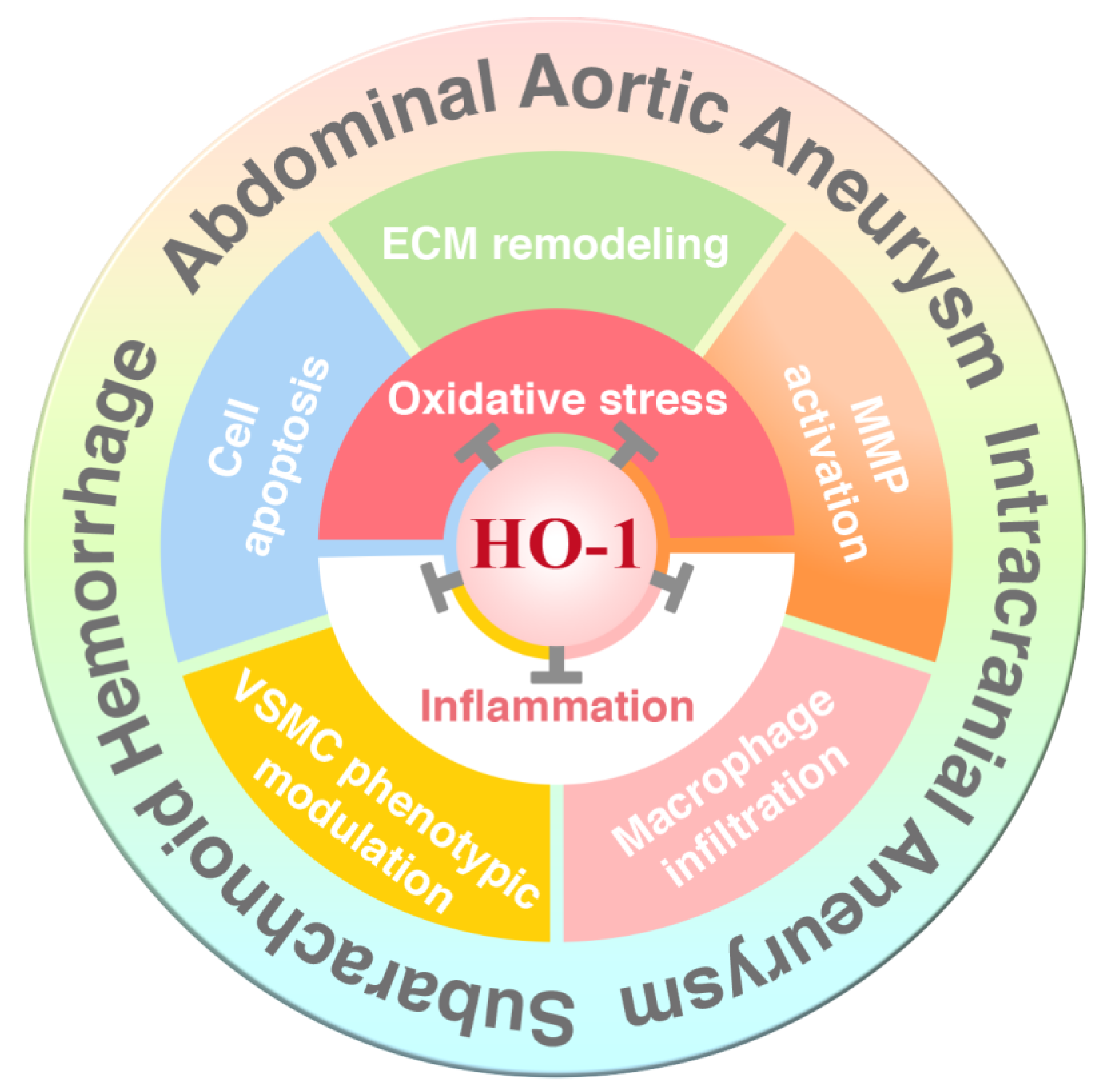
Abstract
:1. Introduction
1.1. Aneurysmal Diseases
1.2. Current Treatments for Aneurysmal Diseases
1.3. Opportunity of Heme Oxygenase-1 in Aneurysmal Diseases
2. HO-1 in Abdominal Aortic Aneurysm
3. HO-1 in Intracranial Aneurysm
4. HO-1 in Subarachnoid Hemorrhage
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2605–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, U.K.; Norman, P.E.; Fowkes, F.G.R.; Aboyans, V.; Song, Y., Jr.; Forouzanfar, M.H.; Naghavi, M.; Denenberg, J.O.; McDermott, M.M.; Criqui, M.H.; et al. Estimation of Global and Regional Incidence and Prevalence of Abdominal Aortic Aneurysms 1990 to 2010. Glob. Hear. 2014, 9, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sampson, U.K.; Norman, P.E.; Fowkes, F.G.R.; Aboyans, V.; Song, Y., Jr.; Forouzanfar, M.H.; Naghavi, M.; Denenberg, J.O.; McDermott, M.M.; Criqui, M.H.; et al. Global and Regional Burden of Aortic Dissection and Aneurysms: Mortality Trends in 21 World Regions, 1990 to 2010. Glob. Hear. 2014, 9, 171–180.e10. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.J.; Gelijns, A.C.; Greco, G. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altobelli, E.; Rapacchietta, L.; Profeta, V.F.; Fagnano, R. Risk Factors for Abdominal Aortic Aneurysm in Population-Based Studies: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Heal. 2018, 15, 2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UN World Population Ageing 2017—Highlights; United Nations, Department of Economic and Social Affairs, Population Division: New York, NY, USA, 2017.
- Best, V.A.; Price, J.F.; Fowkes, F.G.R. Persistent increase in the incidence of abdominal aortic aneurysm in Scotland, 1981–2000. BJS 2003, 90, 1510–1515. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; White, P.M. The detection and management of unruptured intracranial aneurysms. Brain 2000, 123, 205–221. [Google Scholar] [CrossRef] [Green Version]
- Wiebers, D.O.; Torner, J.; Meissner, I. Impact of unruptured intracranial aneurysms on public health in the United States. Stroke 1992, 23, 1416–1419. [Google Scholar] [CrossRef] [Green Version]
- Tanweer, O.; Wilson, T.A.; Metaxa, E.; Riina, H.A.; Meng, H. A comparative review of the hemodynamics and pathogenesis of cerebral and abdominal aortic aneurysms: Lessons to learn from each other. J. Cerebrovasc. Endovasc. Neurosurg. 2014, 16, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Vlak, M.H.; Algra, A.; Brandenburg, R.; Rinkel, G.J. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: A systematic review and meta-analysis. Lancet Neurol. 2011, 10, 626–636. [Google Scholar] [CrossRef]
- Dharmadhikari, S.; Atchaneeyasakul, K.; Ambekar, S.; Saini, V.; Haussen, D.C.; Yavagal, D.R. Association of Menopausal Age with Unruptured Intracranial Aneurysm Morphology. Interv. Neurol. 2019, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.T.; Vates, G.E. Subarachnoid Hemorrhage. N. Engl. J. Med. 2017, 377, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Rincon, F.; Rossenwasser, R.H.; Dumont, A. The Epidemiology of Admissions of Nontraumatic Subarachnoid Hemorrhage in the United States. Neurosurgery 2013, 73, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, P.J.; Dippel, D.W.; Vermeulen, M.; Lindsay, K.W.; Hasan, D.; Van Gijn, J. Amount of blood on computed tomography as an independent predictor after aneurysm rupture. Stroke 1993, 24, 809–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gijn, J.; Rinkel, G.J.E. Subarachnoid haemorrhage: Diagnosis, causes and management. Brain 2001, 124, 249–278. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Johnston, S.C.; Selvin, S.; Gress, D.R. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology 1998, 50, 1413–1418. [Google Scholar] [CrossRef]
- Mayer, S.A.; Kreiter, K.T.; Copeland, D.; Bernardini, G.L.; Bates, J.E.; Peery, S.; Claassen, J.; Du, Y.E.; Connolly, E.S. Global and domain-specific cognitive impairment and outcome after subarachnoid hemorrhage. Neurology 2002, 59, 1750–1758. [Google Scholar] [CrossRef]
- English, S.W. Long-Term Outcome and Economic Burden of Aneurysmal Subarachnoid Hemorrhage: Are we Only Seeing the Tip of the Iceberg? Neurocrit. Care 2020, 33, 37–38. [Google Scholar] [CrossRef]
- Bor, A.S.E.; Koffijberg, H.; Wermer, M.J.; Rinkel, G.J. Optimal screening strategy for familial intracranial aneurysms: A cost-effectiveness analysis. Neurology 2010, 74, 1671–1679. [Google Scholar] [CrossRef]
- Lall, R.R.; Eddleman, C.S.; Bendok, B.R.; Batjer, H.H. Unruptured intracranial aneurysms and the assessment of rupture risk based on anatomical and morphological factors: Sifting through the sands of data. Neurosurg. Focus 2009, 26, E2. [Google Scholar] [CrossRef] [PubMed]
- Chaikof, E.L.; Brewster, D.C.; Dalman, R.L.; Makaroun, M.S.; Illig, K.A.; Sicard, G.A.; Timaran, C.H.; Upchurch, G.R.; Veith, F.J. The care of patients with an abdominal aortic aneurysm: The Society for Vascular Surgery practice guidelines. J. Vasc. Surg. 2009, 50, S2–S49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlösser, F.J.; Tangelder, M.J.; Verhagen, H.J.; Van Der Heijden, G.J.; Muhs, B.E.; Van Der Graaf, Y.; Moll, F.L. Growth predictors and prognosis of small abdominal aortic aneurysms. J. Vasc. Surg. 2008, 47, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Calero, A.; Illig, K.A. Overview of aortic aneurysm management in the endovascular era. Semin. Vasc. Surg. 2016, 29, 3–17. [Google Scholar] [CrossRef]
- Wiebers, D.O. Unruptured intracranial aneurysms: Natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet 2003, 362, 103–110. [Google Scholar] [CrossRef]
- Ries, T.; Groden, C. Endovascular Treatment of Intracranial Aneurysms: Long-Term Stability, Risk Factors for Recurrences, Retreatment and Follow-Up. Clin. Neuroradiol. 2009, 19, 62–72. [Google Scholar] [CrossRef]
- Connolly, E.S.; Rabinstein, A.A.; Carhuapoma, J.R.; Derdeyn, C.P.; Dion, J.; Higashida, R.T.; Hoh, B.L.; Kirkness, C.J.; Naidech, A.M.; Ogilvy, C.S.; et al. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage. Stroke 2012, 43, 1711–1737. [Google Scholar] [CrossRef] [Green Version]
- Baxter, B.T. Invited commentary: Abdominal aortic aneurysm regression by medical treatment: Possibility or pipe dream? J. Vasc. Surg. 2006, 43, 1068–1069. [Google Scholar] [CrossRef] [Green Version]
- Bergoeing, M.P.; Thompson, R.W.; Curci, J. Pharmacological targets in the treatment of abdominal aortic aneurysms. Expert Opin. Ther. Targets 2006, 10, 547–559. [Google Scholar] [CrossRef]
- Chalouhi, N.; Hoh, B.L.; Hasan, D. Review of Cerebral Aneurysm Formation, Growth, and Rupture. Stroke 2013, 44, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting Heme Oxygenase-1 in the Arterial Response to Injury and Disease. Antioxidants 2020, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Hinds, J.T.D.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Fernández-Fierro, A.; Covián, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef]
- Pennell, R.C.; Hollier, L.H.; Lie, J.T.; Bernatz, P.E.; Joyce, J.W.; Pairolero, P.C.; Cherry, K.J.; Hallett, J.W. Inflammatory abdominal aortic aneurysms: A thirty-year review. J. Vasc. Surg. 1985, 2, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Dua, M.M.; Dalman, R.L. Hemodynamic Influences on abdominal aortic aneurysm disease: Application of biomechanics to aneurysm pathophysiology. Vasc. Pharmacol. 2010, 53, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsdahl, S.H.; Singh, K.; Solberg, S.; Jacobsen, B.K. Risk Factors for Abdominal Aortic Aneurysms. Circulation 2009, 119, 2202–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, D.P.J.; Banerjee, A.; Fairhead, J.F.; Handa, A.; Silver, L.E.; Rothwell, P.M. The Oxford Vascular Study Population-Based Study of Incidence of Acute Abdominal Aortic Aneurysms with Projected Impact of Screening Strategy. J. Am. Hear. Assoc. 2015, 4, e001926. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.M.; Rateri, D.L.; Daugherty, A. Abdominal aortic aneurysm. Curr. Opin. Cardiol. 2015, 30, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golledge, J.; Norman, P.E. Atherosclerosis and Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2010, 30, 1075–1077. [Google Scholar] [CrossRef] [Green Version]
- Biros, E.; Gäbel, G.; Moran, C.S.; Schreurs, C.; Lindeman, J.H.N.; Walker, P.J.; Nataatmadja, M.; West, M.; Holdt, L.M.; Hinterseher, I.; et al. Differential gene expression in human abdominal aortic aneurysm and aortic occlusive disease. Oncotarget 2015, 6, 12984–12996. [Google Scholar] [CrossRef] [Green Version]
- Folsom, A.R.; Yao, L.; Alonso, A.; Lutsey, P.L.; Missov, E.; Lederle, F.A.; Ballantyne, C.M.; Tang, W. Circulating Biomarkers and Abdominal Aortic Aneurysm Incidence. Circulation 2015, 132, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.T.; Tromp, G.; Kuivaniemi, H.; Gretarsdottir, S.; Baas, A.F.; Giusti, B.; Strauss, E.; Hof, F.N.G.V.; Webb, T.R.; Erdman, R.; et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ. Res. 2017, 120, 341–353. [Google Scholar] [CrossRef]
- Klarin, D.; Verma, S.S.; Judy, R.; Dikilitas, O.; Wolford, B.N.; Paranjpe, I.; Levin, M.G.; Pan, C.; Tcheandjieu, C.; Spin, J.M.; et al. Genetic Architecture of Abdominal Aortic Aneurysm in the Million Veteran Program. Circulation 2020, 142, 1633–1646. [Google Scholar] [CrossRef]
- Schillinger, M.; Exner, M.; Mlekusch, W.; Ahmadi, R.; Rumpold, H.; Mannhalter, C.; Wagner, O.; Minar, E. Heme oxygenase-1 genotype is a vascular anti-inflammatory factor following balloon angioplasty. J. Endovasc. Ther. 2002, 9, 385–394. [Google Scholar] [CrossRef]
- Schillinger, M.; Exner, M.; Mlekusch, W.; Domanovits, H.; Huber, K.; Mannhalter, C.; Wagner, O.; Minar, E. Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aortic aneurysm. Thromb. Res. 2002, 106, 131–136. [Google Scholar] [CrossRef]
- Dean, J. Marfan syndrome: Clinical diagnosis and management. Eur. J. Hum. Genet. 2007, 15, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.R.; Bridges, A.B.; West, R.R.; McLeish, L.; Stuart, A.G.; Dean, J.; Porteous, M.; Boxer, M.; Davies, S.J. Life expectancy in British Marfan syndrome populations. Clin. Genet. 2008, 54, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, C.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nat. Cell Biol. 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Mizuguchi, T.; Collod-Beroud, G.; Akiyama, T.; Abifadel, M.; Harada, N.; Morisaki, T.; Allard, D.; Varret, M.; Claustres, M.; Morisaki, H.; et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 2004, 36, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Adès, L.C.; Sullivan, K.; Biggin, A.; Haan, E.A.; Brett, M.; Holman, K.J.; Dixon, J.; Robertson, S.; Holmes, A.D.; Rogers, J.; et al. FBN1,TGFBR1, and the Marfan-craniosynostosis/mental retardation disorders revisited. Am. J. Med. Genet. Part A 2006, 140, 1047–1058. [Google Scholar] [CrossRef]
- Singh, K.K.; Rommel, K.; Mishra, A.; Karck, M.; Haverich, A.; Schmidtke, J.; Arslan-Kirchner, M. TGFBR1andTGFBR2mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum. Mutat. 2006, 27, 770–777. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Matsumura, J.S. Pharmacologic Management of Aneurysms. Circ. Res. 2019, 124, 631–646. [Google Scholar] [CrossRef]
- Wu, M.-L.; Ho, Y.-C.; Lin, C.-Y.; Yet, S.-F. Heme oxygenase-1 in inflammation and cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 150–158. [Google Scholar]
- Nakahashi, T.K.; Hoshina, K.; Tsao, P.S.; Sho, E.; Sho, M.; Karwowski, J.K.; Yeh, C.; Yang, R.-B.; Topper, J.N.; Dalman, R.L. Flow Loading Induces Macrophage Antioxidative Gene Expression in Experimental Aneurysms. Arter. Thromb. Vasc. Biol. 2002, 22, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Azuma, J.; Wong, R.J.; Morisawa, T.; Hsu, M.; Maegdefessel, L.; Zhao, H.; Kalish, F.; Kayama, Y.; Wallenstein, M.B.; Deng, A.C.; et al. Heme Oxygenase-1 Expression Affects Murine Abdominal Aortic Aneurysm Progression. PLoS ONE 2016, 11, e0149288. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Wu, M.-L.; Gung, P.-Y.; Chen, C.-H.; Kuo, C.-C.; Yet, S.-F. Heme oxygenase-1 deficiency exacerbates angiotensin II-induced aortic aneurysm in mice. Oncotarget 2016, 7, 67760–67776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meital, L.T.; Windsor, M.T.; Perissiou, M.; Schulze, K.; Magee, R.; Kuballa, A.; Golledge, J.; Bailey, T.G.; Askew, C.D.; Russell, F.D. Omega-3 fatty acids decrease oxidative stress and inflammation in macrophages from patients with small abdominal aortic aneurysm. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sapadin, A.N.; Fleischmajer, R. Tetracyclines: Nonantibiotic properties and their clinical implications. J. Am. Acad. Dermatol. 2006, 54, 258–265. [Google Scholar] [CrossRef]
- Manning, M.W.; Cassis, L.A.; Daugherty, A. Differential Effects of Doxycycline, a Broad-Spectrum Matrix Metalloproteinase Inhibitor, on Angiotensin II–Induced Atherosclerosis and Abdominal Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2003, 23, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Chen, C.; Cao, Y.; Qi, R. Inhibitory effects of doxycycline on the onset and progression of abdominal aortic aneurysm and its related mechanisms. Eur. J. Pharmacol. 2017, 811, 101–109. [Google Scholar] [CrossRef]
- Mosorin, M.; Juvonen, J.; Biancari, F.; Satta, J.; Surcel, H.-M.; Leinonen, M.; Saikku, P.; Juvonen, T. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: A randomized, double-blind, placebo-controlled pilot study. J. Vasc. Surg. 2001, 34, 606–610. [Google Scholar] [CrossRef] [Green Version]
- Baxter, B.; Pearce, W.H.; Waltke, E.A.; Littooy, F.N.; Hallett, J.W.; Kent, K.; Upchurch, G.R.; Chaikof, E.L.; Mills, J.L.; Fleckten, B.; et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: Report of a prospective (Phase II) multicenter study. J. Vasc. Surg. 2002, 36, 1–12. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Abdul-Hussien, H.; Van Bockel, J.H.; Wolterbeek, R.; Kleemann, R. Clinical Trial of Doxycycline for Matrix Metalloproteinase-9 Inhibition in Patients With an Abdominal Aneurysm. Circulation 2009, 119, 2209–2216. [Google Scholar] [CrossRef]
- Hou, X.; Yang, S.; Zheng, Y. Licochalcone A attenuates abdominal aortic aneurysm induced by angiotensin II via regulating the miR-181b/SIRT1/HO-1 signaling. J. Cell. Physiol. 2019, 234, 7560–7568. [Google Scholar] [CrossRef]
- Deng, Y.-M.; Wu, B.J.; Witting, P.K.; Stocker, R. Probucol Protects Against Smooth Muscle Cell Proliferation by Upregulating Heme Oxygenase-1. Circulation 2004, 110, 1855–1860. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, Y.; Cao, Y.; Wang, Q.; Anwaier, G.; Zhang, Q.; Qi, R. Mechanisms underlying the inhibitory effects of probucol on elastase-induced abdominal aortic aneurysm in mice. Br. J. Pharmacol. 2019, 177, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Goraca, A.; Demyanets, S.; Mittlboeck, M.; Domenig, C.; Neumayer, C.; Wojta, J.; Nanobachvili, J.; Huk, I.; Klinger, M. Simvastatin Decreases Free Radicals Formation in the Human Abdominal Aortic Aneurysm Wall via NF-κB. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, E.F.; Buckley, C.; Shames, M.L.; Ennis, T.L.; VanVickle-Chavez, S.J.; Mao, D.; Goeddel, L.A.; Hawkins, C.J.; Thompson, R.W. Treatment with Simvastatin Suppresses the Development of Experimental Abdominal Aortic Aneurysms in Normal and Hypercholesterolemic Mice. Ann. Surg. 2005, 241, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Schouten, O.; Van Laanen, J.; Boersma, E.; Vidakovic, R.; Feringa, H.; Dunkelgrün, M.; Bax, J.; Koning, J.; Van Urk, H.; Poldermans, D. Statins are Associated with a Reduced Infrarenal Abdominal Aortic Aneurysm Growth. Eur. J. Vasc. Endovasc. Surg. 2006, 32, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechota-Polańczyk, A.; Ejózkowicz, A.; Nowak, W.N.; Eeilenberg, W.; Eneumayer, C.; Emalinski, T.; Ehuk, I.; Brostjan, C. The Abdominal Aortic Aneurysm and Intraluminal Thrombus: Current Concepts of Development and Treatment. Front. Cardiovasc. Med. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salata, K.; Syed, M.; Hussain, M.A.; De Mestral, C.; Greco, E.; Mamdani, M.; Tu, J.V.; Forbes, T.L.; Bhatt, D.L.; Verma, S.; et al. Statins Reduce Abdominal Aortic Aneurysm Growth, Rupture, and Perioperative Mortality: A Systematic Review and Meta-Analysis. J. Am. Hear. Assoc. 2018, 7, e008657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechota-Polańczyk, A.; Kopacz, A.; Kloska, D.; Zagrapan, B.; Neumayer, C.; Grochot-Przeczek, A.; Huk, I.; Brostjan, C.; Dulak, J.; Józkowicz, A. Simvastatin Treatment Upregulates HO-1 in Patients with Abdominal Aortic Aneurysm but Independently of Nrf2. Oxidative Med. Cell. Longev. 2018, 2018, 2028936. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, K.; Taneda, M.; Asai, T.; Kinoshita, A.; Ito, M.; Kuroda, R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke 1999, 30, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Li, S.; Song, Y.; Liu, P.; Yang, Z.; Liu, Y.; Quan, K.; Yu, G.; Fan, Z.; Zhu, W. Nrf-2 signaling inhibits intracranial aneurysm formation and progression by modulating vascular smooth muscle cell phenotype and function. J. Neuroinflamm. 2019, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pascale, C.L.; Martinez, A.N.; Carr, C.; Sawyer, D.M.; Ribeiro-Alves, M.; Chen, M.; O’Donnell, D.B.; Guidry, J.J.; Amenta, P.S.; Dumont, A.S. Treatment with dimethyl fumarate reduces the formation and rupture of intracranial aneurysms: Role of Nrf2 activation. Br. J. Pharmacol. 2019, 40, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, R.L.E.; Bartelds, B.; Wagener, F.A.D.T.G.; Affara, N.; Mohaupt, S.; Wijnberg, H.; Pennings, S.W.C.; Takens, J.; Berger, R.M.F. Erythropoietin Attenuates Pulmonary Vascular Remodeling in Experimental Pulmonary Arterial Hypertension through Interplay between Endothelial Progenitor Cells and Heme Oxygenase. Front. Pediatr. 2015, 3, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Tian, Y.; Wei, H.-J.; Chen, J.; Dong, J.-F.; Zacharek, A.; Zhang, J. Erythropoietin increases circulating endothelial progenitor cells and reduces the formation and progression of cerebral aneurysm in rats. Neuroscience 2011, 181, 292–299. [Google Scholar] [CrossRef]
- Choi, S.-H.; Park, S.; Oh, C.J.; Leem, J.; Park, K.-G.; Lee, I.K. Dipeptidyl peptidase-4 inhibition by gemigliptin prevents abnormal vascular remodeling via NF-E2-related factor 2 activation. Vasc. Pharmacol. 2015, 73, 11–19. [Google Scholar] [CrossRef]
- Si, J.; Meng, R.; Gao, P.; Hui, F.; Li, Y.; Liu, X.; Yang, B. Linagliptin protects rat carotid artery from balloon injury and activates the NRF2 antioxidant pathway. Exp. Anim. 2019, 68, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Ikedo, T.; Minami, M.; Kataoka, H.; Hayashi, K.; Nagata, M.; Fujikawa, R.; Higuchi, S.; Yasui, M.; Aoki, T.; Fukuda, M.; et al. Dipeptidyl Peptidase-4 Inhibitor Anagliptin Prevents Intracranial Aneurysm Growth by Suppressing Macrophage Infiltration and Activation. J. Am. Hear. Assoc. 2017, 6, 6. [Google Scholar] [CrossRef]
- Wei, H.; Yang, M.; Yu, K.; Dong, W.; Liang, W.; Wang, Z.; Jiang, R.; Zhang, J. Atorvastatin Protects Against Cerebral Aneurysmal Degenerative Pathology by Promoting Endothelial Progenitor Cells (EPC) Mobilization and Attenuating Vascular Deterioration in a Rat Model. Med. Sci. Monit. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Hashimoto, N. Simvastatin Suppresses the Progression of Experimentally Induced Cerebral Aneurysms in Rats. Stroke 2008, 39, 1276–1285. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nakagami, H.; Nozaki, K.; Morishita, R.; Hashimoto, N. Pitavastatin Suppresses Formation and Progression of Cerebral Aneurysms Through Inhibition of the Nuclear Factor κB Pathway. Neurosurgery 2009, 64, 357–366. [Google Scholar] [CrossRef]
- Kimura, N.; Shimizu, H.; Eldawoody, H.; Nakayama, T.; Saito, A.; Tominaga, T.; Takahashi, A. Effect of olmesartan and pravastatin on experimental cerebral aneurysms in rats. Brain Res. 2010, 1322, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Kitazato, K.T.; Yagi, K.; Shimada, K.; Matsushita, N.; Kinouchi, T.; Kanematsu, Y.; Satomi, J.; Kageji, T.; Nagahiro, S. Statins Promote the Growth of Experimentally Induced Cerebral Aneurysms in Estrogen-Deficient Rats. Stroke 2011, 42, 2286–2293. [Google Scholar] [CrossRef] [PubMed]
- Marbacher, S.; Schläppi, J.-A.; Fung, C.; Hüsler, J.; Beck, J.; Raabe, A. Do statins reduce the risk of aneurysm development: A case-control study. J. Neurosurg. 2012, 116, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, K.; Salem, M.M.; Alturki, A.Y.; Thomas, A.J.; Ogilvy, C.; Moore, J.M. Endothelialization following Flow Diversion for Intracranial Aneurysms: A Systematic Review. Am. J. Neuroradiol. 2019, 40, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, M.B.; Purdy, P.D.; Burns, D.; Bellotto, D. Scanning electron microscopic findings in a basilar tip aneurysm embolized with Guglielmi detachable coils. Am. J. Neuroradiol. 1997, 18, 688–690. [Google Scholar]
- Tenjin, H.; Fushiki, S.; Nakahara, Y.; Masaki, H.; Matsuo, T.; Johnson, C.M.; Ueda, S. Effect of Guglielmi Detachable Coils on Experimental Carotid Artery Aneurysms in Primates. Stroke 1995, 26, 2075–2080. [Google Scholar] [CrossRef]
- Brinjikji, W.; Kallmes, D.F.; Kadirvel, R. Mechanisms of Healing in Coiled Intracranial Aneurysms: A Review of the Literature. Am. J. Neuroradiol. 2015, 36, 1216–1222. [Google Scholar] [CrossRef]
- Urbich, C.; Dimmeler, S. Endothelial Progenitor Cells. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.H.; Chen, Y.H.; Yet, S.F.; Chau, L.Y. After vascular injury, heme oxygenase-1/carbon monoxide enhances re-endothelialization via promoting mobilization of circulating endothelial progenitor cells. J. Thromb. Haemost. 2009, 7, 1401–1408. [Google Scholar] [CrossRef]
- Wu, B.J.; Midwinter, R.G.; Cassano, C.; Beck, K.; Wang, Y.; Changsiri, D.; Gamble, J.R.; Stocker, R. Heme Oxygenase-1 Increases Endothelial Progenitor Cells. Arter. Thromb. Vasc. Biol. 2009, 29, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Liu, P.; Shi, Y.; Li, S.; Liu, Y.; Zhu, W. Sitagliptin stimulates endothelial progenitor cells to induce endothelialization in aneurysm necks through the SDF-1/CXCR4/NRF2 signaling pathway. Front. Endocrinol. 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhou, Y.; An, Q.; Song, Y.; Chen, X.; Yang, G.-Y.; Zhu, W. Erythropoietin Stimulates Endothelial Progenitor Cells to Induce Endothelialization in an Aneurysm Neck After Coil Embolization by Modulating Vascular Endothelial Growth Factor. STEM CELLS Transl. Med. 2016, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; An, Q.; Chen, X.; Huang, J.; Yang, G.-Y.; Zhu, W. Rosuvastatin for enhancement of aneurysm neck endothelialization after coil embolization: Promotion of endothelial progenitor cells in a rodent model. J. Neurosurg. 2016, 124, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Hong, D.Y.; Dai, D.; Schroeder, D.; Kallmes, D.F.; Kadirvel, R. Statins are not associated with short-term improved aneurysm healing in a rabbit model of unruptured aneurysms. J. NeuroInterv. Surg. 2016, 9, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Shahi, V.; Cloft, H.J.; Lanzino, G.; Kallmes, D.F.; Kadirvel, R. Could Statin Use Be Associated with Reduced Recurrence Rates following Coiling in Ruptured Intracranial Aneurysms? Am. J. Neuroradiol. 2015, 36, 2104–2107. [Google Scholar] [CrossRef] [Green Version]
- Brinjikji, W.; Cloft, H.; Cekirge, S.; Fiorella, D.; Hanel, R.; Jabbour, P.; Lylyk, P.; McDougall, C.G.; Moran, C.J.; Siddiqui, A.H.; et al. Lack of Association between Statin Use and Angiographic and Clinical Outcomes after Pipeline Embolization for Intracranial Aneurysms. Am. J. Neuroradiol. 2017, 38, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.M.; Maragkos, G.A.; Enriquez-Marulanda, A.; Ascanio, L.; Ravindran, K.; Alturki, A.Y.; Ogilvy, C.S.; Thomas, A.J.; Moore, J.M. Statin Therapy and Diabetes Do Not Affect Aneurysm Occlusion or Clinical Outcomes After Pipeline Embolization Device Treatment: A Preliminary Study. World Neurosurg. 2018, 120, e525–e532. [Google Scholar] [CrossRef]
- Beitzke, M.; Enzinger, C.; Wünsch, G.; Asslaber, M.; Gattringer, T.; Fazekas, F. Contribution of Convexal Subarachnoid Hemorrhage to Disease Progression in Cerebral Amyloid Angiopathy. Stroke 2015, 46, 1533–1540. [Google Scholar] [CrossRef]
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160. [Google Scholar] [CrossRef]
- Regan, R.F.; Panter, S. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci. Lett. 1993, 153, 219–222. [Google Scholar] [CrossRef]
- Jaremko, K.M.; Chen-Roetling, J.; Chen, L.; Regan, R.F. Accelerated hemolysis and neurotoxicity in neuron-glia-blood clot co-cultures. J. Neurochem. 2010, 114, 1063–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.-C.; Yen, B.L.; Lee, F.-K.; Lai, T.-H.; Chen, Y.-C.; Chan, S.-H.; Huang, H.-I. In Vitro Differentiation of Human Placenta-Derived Multipotent Cells into Hepatocyte-Like Cells. STEM CELLS 2006, 24, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; Brough, D.; Robinson, E.M.; Girard, S.; Rothwell, N.J.; Allan, S.M. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis. Model. Mech. 2012, 5, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Reyes, S.; Guzmán-Beltrán, S.; Medina-Campos, O.N.; Pedraza-Chaverri, J. Curcumin Pretreatment Induces Nrf2 and an Antioxidant Response and Prevents Hemin-Induced Toxicity in Primary Cultures of Cerebellar Granule Neurons of Rats. Oxidative Med. Cell. Longev. 2013, 2013, 801418. [Google Scholar] [CrossRef]
- Matz, P.; Turner, C.; Weinstein, P.R.; Massa, S.M.; Panter, S.; Sharp, F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Schallner, N.; Pandit, R.; Leblanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, S.; Frase, S.; Selzner, L.; Lieberum, J.-L.; Wollborn, J.; Niesen, W.-D.; Foit, N.A.; Heiland, D.H.; Schallner, N. Neuroprotection after Hemorrhagic Stroke Depends on Cerebral Heme Oxygenase-1. Antioxidants 2019, 8, 496. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Fang, Q.; Zhang, J.; Zhou, D.; Wang, Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 2011, 89, 515–523. [Google Scholar] [CrossRef]
- Wang, Z.; Ji, C.; Wu, L.; Qiu, J.; Li, Q.; Shao, Z.; Chen, G. Tert-Butylhydroquinone Alleviates Early Brain Injury and Cognitive Dysfunction after Experimental Subarachnoid Hemorrhage: Role of Keap1/Nrf2/ARE Pathway. PLoS ONE 2014, 9, e97685. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qiu, J.; Wang, Z.; You, W.; Wu, L.; Ji, C.; Chengyuan, J. Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system. J. Neurosurg. 2015, 123, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Morgan, L.; Hawe, E.; Palmén, J.; Montgomery, H.; Humphries, S.E.; Kitchen, N. Polymorphism of the heme oxygenase-1 gene and cerebral aneurysms. Br. J. Neurosurg. 2005, 19, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hu, Q.; Li, B.; Manaenko, A.; Chen, Y.; Tang, J.; Guo, Z.; Tang, J.; Zhang, J.H. Recombinant milk fat globule-EGF factor-8 reduces oxidative stress via integrin β3/nuclear factor erythroid 2-related factor 2/heme oxygenase pathway in subarachnoid hemorrhage rats. Stroke 2014, 45, 3691–3697. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Koh, E.S.; Shin, S.J.; Choi, B.S.; Park, C.W.; Chang, Y.S.; Chung, S. Delayed treatment with oleanolic acid attenuates tubulointerstitial fibrosis in chronic cyclosporine nephropathy through Nrf2/HO-1 signaling. J. Transl. Med. 2014, 12, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, R.; Carvalho-Tavares, J.; Hernández, M.; Arnés, M.; Ruiz-Gutiérrez, V.; Nieto, M.L. Beneficial actions of oleanolic acid in an experimental model of multiple sclerosis: A potential therapeutic role. Biochem. Pharmacol. 2010, 79, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-W.; Liu, X.-J.; Zhao, Y.; Li, X. Role of Oleanolic acid in maintaining BBB integrity by targeting p38MAPK/VEGF/Src signaling pathway in rat model of subarachnoid hemorrhage. Eur. J. Pharmacol. 2018, 839, 12–20. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, S.; Wang, J.; Shen, Y.; Zhang, J.; Wu, Q. Nrf2/HO-1 mediates the neuroprotective effect of mangiferin on early brain injury after subarachnoid hemorrhage by attenuating mitochondria-related apoptosis and neuroinflammation. Sci. Rep. 2017, 7, 11883. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, L.; Jiang, R.; Ye, M.; Yin, X.; Wan, J. Anti-inflammatory effects of mangiferin on sepsis-induced lung injury in mice via up-regulation of heme oxygenase-1. J. Nutr. Biochem. 2013, 24, 1173–1181. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Zhu, S.; Xu, T.; Lu, J.; Han, H.; Zhou, C.; Yan, J. The role of rhynchophylline in alleviating early brain injury following subarachnoid hemorrhage in rats. Brain Res. 2016, 1631, 92–100. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2016, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol Pretreatment Attenuates Cerebral Ischemic Injury by Upregulating Expression of Transcription Factor Nrf2 and HO-1 in Rats. Neurochem. Res. 2011, 36, 2352–2362. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Zhou, M.; Zhang, X.-S.; Zhuang, Z.; Li, T.; Shi, J.-X.; Zhang, X. Resveratrol prevents neuronal apoptosis in an early brain injury model. J. Surg. Res. 2014, 189, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-C.; Wang, Z.; Xie, Y.-K.; Zhou, X.; Yuan, H.-T.; Qiu, J.; Xin, D.-Q.; Chu, X.-L. Resveratrol reduces brain injury after subarachnoid hemorrhage by inhibiting oxidative stress and endoplasmic reticulum stress. Neural Regen. Res. 2019, 14, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Saito, K.; Ohmi, Y.; Fujie, N.; Ohtsuka, K. Paeoniflorin, a novel heat shock protein–inducing compound. Cell Stress Chaperon 2004, 9, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Xu, L.; Gao, L.; Zhao, L.; Liu, X.-H.; Chang, Y.-Y.; Liu, Y.-L. Paeoniflorin attenuates early brain injury through reducing oxidative stress and neuronal apoptosis after subarachnoid hemorrhage in rats. Metab. Brain Dis. 2020, 35, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.-L.; Yang, C.-C.; Tseng, H.-C.; Hsiao, L.-D.; Lin, C.-C.; Yang, C.-M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.-F.; Kuo, C.-Y.; Wen, L.-L.; Chen, C.-M.; Chang, Y.-F.; Wong, C.-S.; Cherng, C.-H.; Chuang, M.-Y.; Wu, Z.-F. Rosiglitazone Attenuates Cerebral Vasospasm and Provides Neuroprotection in an Experimental Rat Model of Subarachnoid Hemorrhage. Neurocritic. Care 2014, 21, 316–331. [Google Scholar] [CrossRef]
- Gu, C.; Wang, Y.; Li, J.; Chen, J.; Yan, F.; Chen, G.; Chen, G. Rosiglitazone attenuates early brain injury after experimental subarachnoid hemorrhage in rats. Brain Res. 2015, 1624, 199–207. [Google Scholar] [CrossRef]
- Zolnourian, A.; Galea, I.; Bulters, D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxidative Med. Cell. Longev. 2019, 2019, 6218239. [Google Scholar] [CrossRef]
- Tseng, M.-Y.; Hutchinson, P.; Richards, H.K.; Czosnyka, M.; Pickard, J.D.; Erber, W.N.; Brown, S.J.; Kirkpatrick, P.J. Acute systemic erythropoietin therapy to reduce delayed ischemic deficits following aneurysmal subarachnoid hemorrhage: A Phase II randomized, double-blind, placebo-controlled trial. J. Neurosurg. 2009, 111, 171–180. [Google Scholar] [CrossRef]
- Helbok, R.; Shaker, E.; Beer, R.; Chemelli, A.; Sojer, M.; Sohm, F.; Broessner, G.; Lackner, P.; Beck, M.; Zangerle, A.; et al. High dose Erythropoietin increases Brain Tissue Oxygen Tension in Severe Vasospasm after Subarachnoid Hemorrhage. BMC Neurol. 2012, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Springborg, J.B.; Møller, C.; Gideon, P.; Jørgensen, O.S.; Juhler, M.; Olsen, N.V. Erythropoietin in patients with aneurysmal subarachnoid haemorrhage: A double blind randomised clinical trial. Acta Neurochir. 2007, 149, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; McGirt, M.; Laskowitz, D.T.; Friedman, A.H.; Alexander, M.J. Simvastatin Reduces Vasospasm after Aneurysmal Subarachnoid Hemorrhage: Results of a Pilot Randomized Clinical Trial. Neurosurgery 2005, 57, 420. [Google Scholar] [CrossRef]
- Kirkpatrick, P.J.; Turner, C.L.; Smith, C.; Hutchinson, P.J.; Murray, F.R.S.C.G.D. Simvastatin in aneurysmal subarachnoid haemorrhage (STASH): A multicentre randomised phase 3 trial. Lancet Neurol. 2014, 13, 666–675. [Google Scholar] [CrossRef]
- Tseng, M.-Y.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Effects of Acute Treatment With Pravastatin on Cerebral Vasospasm, Autoregulation, and Delayed Ischemic Deficits After Aneurysmal Subarachnoid Hemorrhage. Stroke 2005, 36, 1627–1632. [Google Scholar] [CrossRef]
- Sanchez-Peña, P.; Nouet, A.; Clarençon, F.; Colonne, C.; Jean, B.; Le Jean, L.; Fonfrede, M.; Aout, M.; Vicaut, E.; Puybasset, L. Atorvastatin decreases computed tomography and S100-assessed brain ischemia after subarachnoid aneurysmal hemorrhage. Crit. Care Med. 2012, 40, 594–602. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Murakami, Y.; Saitoh, M.; Yokoi, T.; Aoki, T.; Miura, K.; Ueshima, H.; Nozaki, K. Statin Use and Risk of Cerebral Aneurysm Rupture: A Hospital-based Case–control Study in Japan. J. Stroke Cerebrovasc. Dis. 2014, 23, 343–348. [Google Scholar] [CrossRef]
- Chen, W.-J.; Chen, Y.-H.; Lai, Y.-J.; Hsu, Y.-J.; Yeh, Y.-H.; Tsai, C.-S.; Lin, C.-Y. GT-repeat length polymorphism in heme oxygenase-1 promoter determines the effect of cilostazol on vascular smooth muscle cells. Int. J. Cardiol. 2016, 222, 407–415. [Google Scholar] [CrossRef]
- Niu, P.-P.; Yang, G.; Xing, Y.; Guo, Z.-N.; Yang, Y. Effect of cilostazol in patients with aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. J. Neurol. Sci. 2014, 336, 146–151. [Google Scholar] [CrossRef]
- Saber, H.; Desai, A.; Palla, M.; Mohamed, W.; Seraji-Bozorgzad, N.; Ibrahim, M. Efficacy of Cilostazol in Prevention of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage: A Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2018, 27, 2979–2985. [Google Scholar] [CrossRef]
- Shan, T.; Zhang, T.; Qian, W.; Ma, L.; Li, H.; You, C.; Xie, X. Effectiveness and feasibility of cilostazol in patients with aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. J. Neurol. 2019, 267, 1577–1584. [Google Scholar] [CrossRef]
- Sugimoto, K.; Nomura, S.; Shirao, S.; Inoue, T.; Ishihara, H.; Kawano, R.; Kawano, A.; Oka, F.; Suehiro, E.; Sadahiro, H.; et al. Cilostazol decreases duration of spreading depolarization and spreading ischemia after aneurysmal subarachnoid hemorrhage. Ann. Neurol. 2018, 84, 873–885. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.-C.; Chen, C.-M.; Hamdin, C.D.; Orekhov, A.N.; Sobenin, I.A.; Layne, M.D.; Yet, S.-F. Therapeutic Potential of Heme Oxygenase-1 in Aneurysmal Diseases. Antioxidants 2020, 9, 1150. https://doi.org/10.3390/antiox9111150
Jiang W-C, Chen C-M, Hamdin CD, Orekhov AN, Sobenin IA, Layne MD, Yet S-F. Therapeutic Potential of Heme Oxygenase-1 in Aneurysmal Diseases. Antioxidants. 2020; 9(11):1150. https://doi.org/10.3390/antiox9111150
Chicago/Turabian StyleJiang, Wei-Cheng, Chen-Mei Chen, Candra D. Hamdin, Alexander N. Orekhov, Igor A. Sobenin, Matthew D. Layne, and Shaw-Fang Yet. 2020. "Therapeutic Potential of Heme Oxygenase-1 in Aneurysmal Diseases" Antioxidants 9, no. 11: 1150. https://doi.org/10.3390/antiox9111150