Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders

Abstract
:1. Introduction




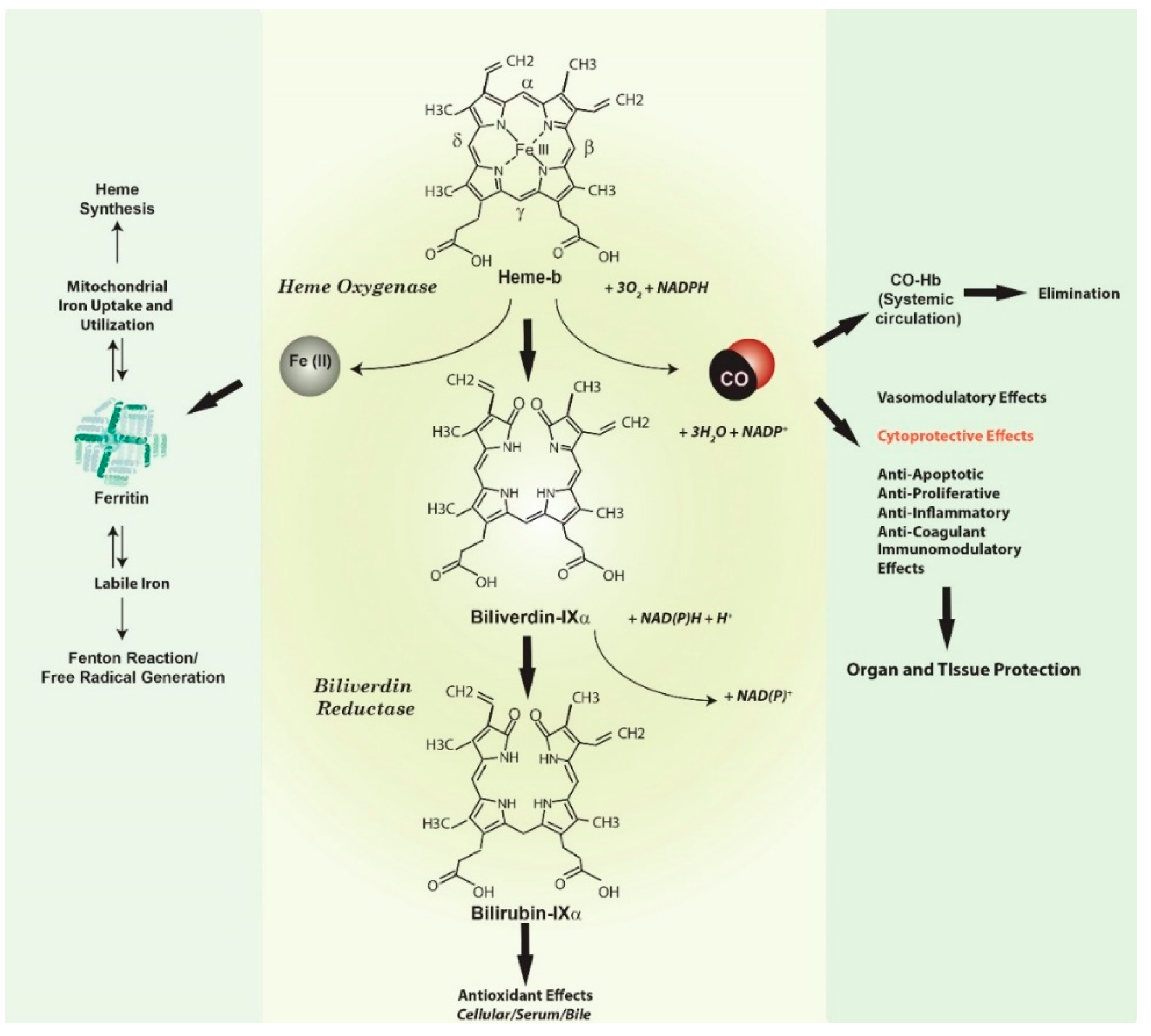{kind=link}
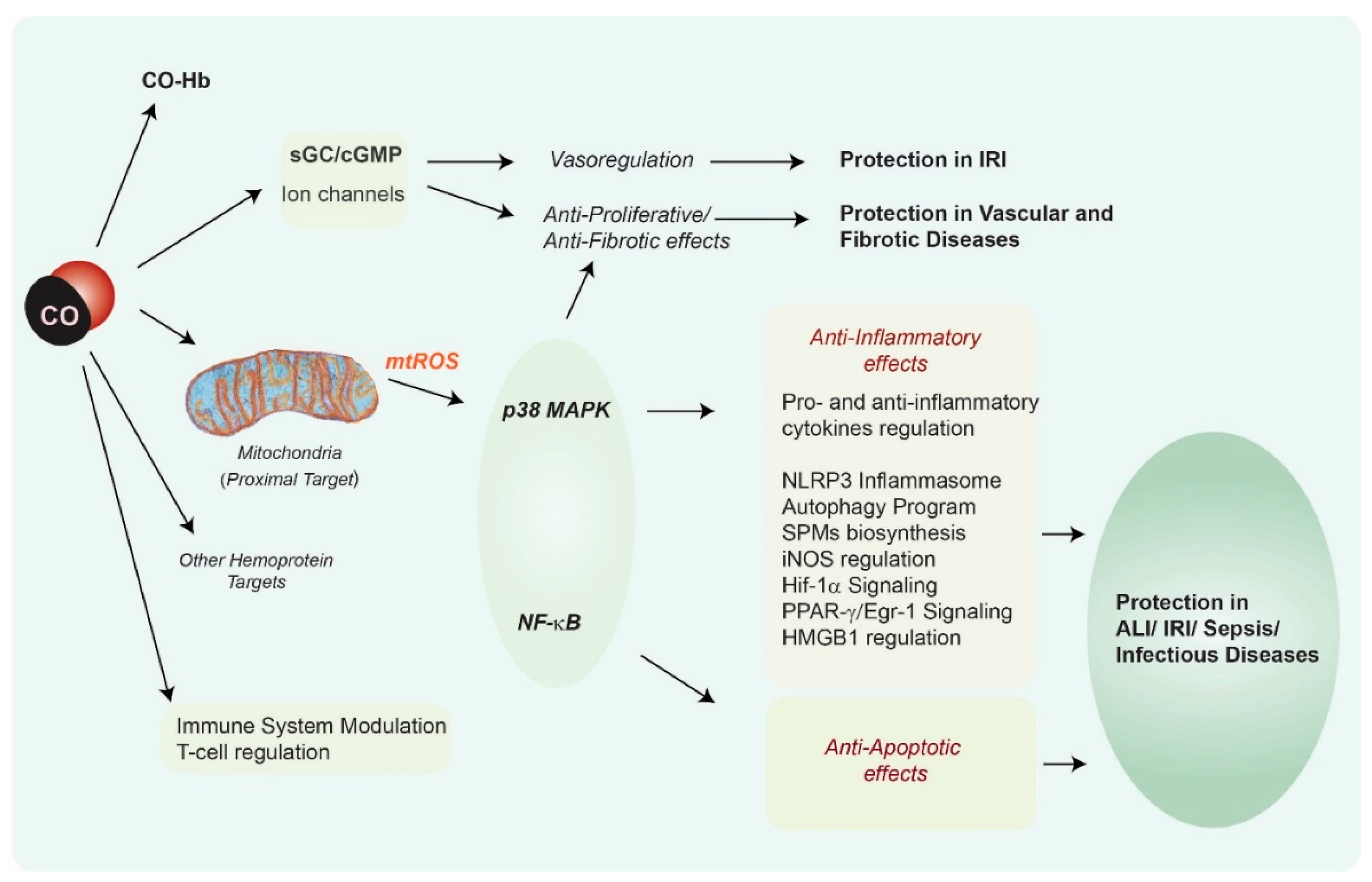{kind=link}
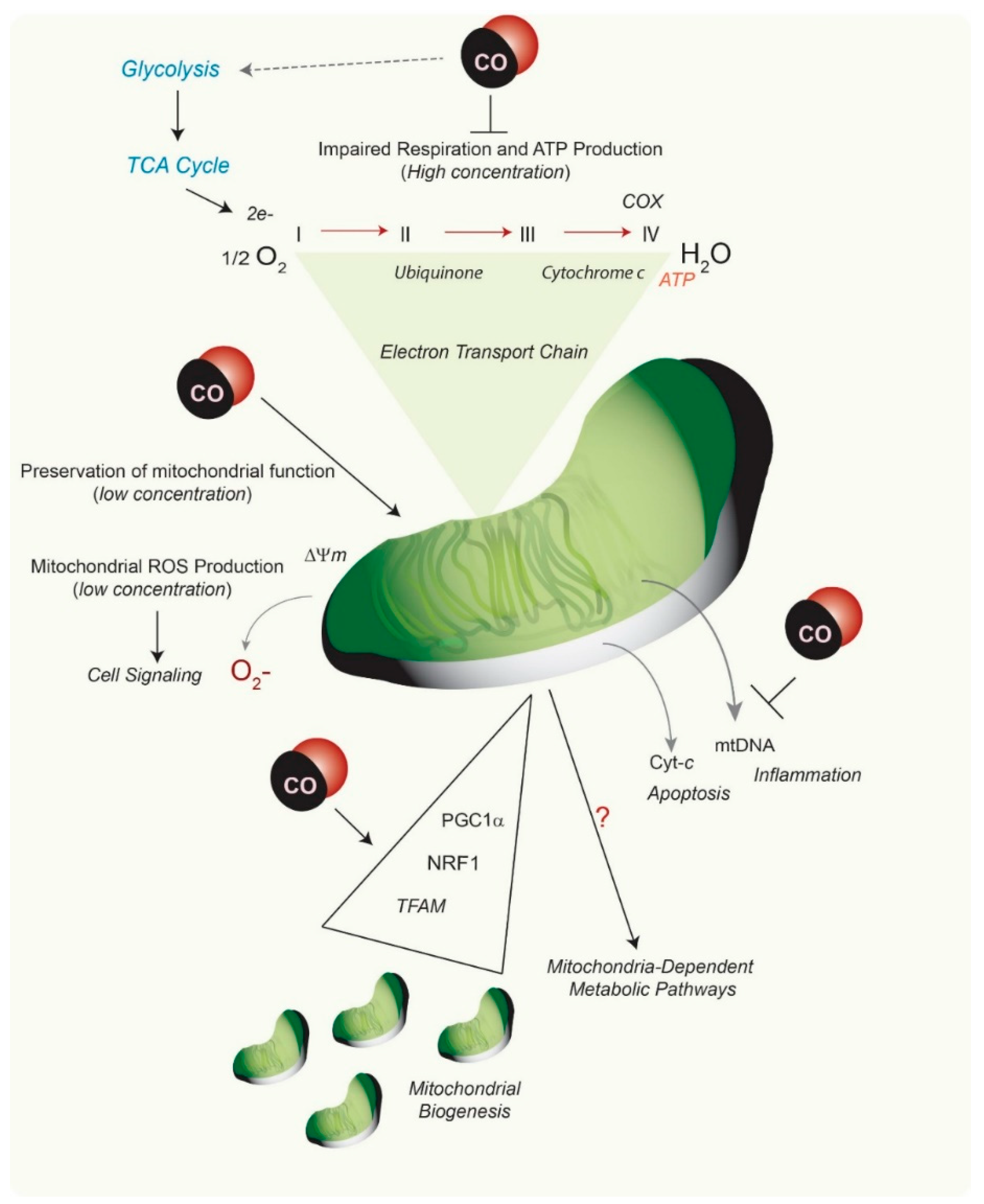{kind=link}
| CORM | Chemical Formula | Properties | Structure | Refs. |
|---|---|---|---|---|
| CORM2 | Tricarbonyl dichloro Ruthenium (II) dimer. [Ru(CO3)Cl2]2 | Fast CO release kinetics. Activatable by ligand substitution. Lipid/ DMSO soluble. |  | [89] |
| CORM3 | Tricarbonylchloro (glycinato) ruthenium (II). C5H4ClNO5Ru | Fast CO release kinetics. Activatable by ligand substitution. Water soluble. |  | [78,89] |
| CORM-A1 | Sodium boranocarbonate. CH3BNa2O2 | Slow CO release kinetics. Activatable by pH changes. Water soluble. |  | [90] |
| CORM-401 | Tetracarbonyl[N-(dithiocarboxy-?S,?S′)-N-methylglycine] manganate. (Mn(CO)4(S2CNMe(CH2CO2H))) | Releases 3 mol CO per molecule. Activatable by oxidants, Soluble in DMSO. |  | [81,91] |
| CORM-S1 | Dicarbonyl-bis(cysteamine) iron(II). Fe(SCH2CH2NH2)2 (CO)2 | Photoactivatableby visible light (>400 nm). Prototype “PhotoCORM” |  | [92,93] |
2. Protective Roles of HO-1/CO in Inflammatory Conditions
| Injury/Disease Model | Species | Dose | Observation | Ref. |
|---|---|---|---|---|
| LPS | Mice | 250 ppm [100–500 ppm] CO | Anti-inflammatory effect, reduction of pro-inflammatory cytokines (TNFα, IL-6), p38 MAPK, JNK, IL-6 dependent. Upregulation of serum IL-10. | [58,114,115,116] |
| “ | Pig | 250 ppm | Reduced disseminated intravascular coagulation; Reduction of pro-inflammatory cytokines (IL-1β) and endothelial ICAM expression. Upregulation of IL-10 | [94] |
| “ | Macaque | 500 ppm CO | Reduction of pro-inflammatory cytokine (TNFα), >30% CO-Hb | [95] |
| Pneumonia-ALI | Baboon | 100–300 ppm CO | Reduction of lung inflammation, improved histology | [96] |
| Hyperoxia | Rat | 250 ppm CO | Reduction of lung edema, inflammation, fibrin deposition, and apoptosis; improved histology. | [59] |
| “ | Mice | 250 ppm CO | Reduction of lung inflammation; p38 MAPK-dependent | [60] |
| VILI | Rat | 250 ppm CO | Reduction of lung inflammation, Reduced neutrophil influx | [103] |
| “ | Mice | 250 ppm CO | Reduction of lung inflammation, Improved lung histology | [101,102] |
| MA-ALI/ECM | Mice | CORM (ALF-492 36.7 mg/kg) 50 ppm CO | Improved lung histology and survival/Protection in CM, reduced neuroinflammation. | [117,118,119,120,121] |
| Hemorrhagic shock | Mice | 250 ppm CO CORM3 (4 mg/kg) | Reduced inflammation, apoptosis. Upregulation of IL-10. | [97,98,99] |
| Sepsis/CLP | Mice | 250 ppm CO | Increased autophagy, improved bacterial clearance | [67,100] |
| “ | CORM2 (30 mg/kg) | Reduced HMGB1 expression, Improved survival in sepsis | [79] | |
| Lung I/R | Mice | 250 ppm CO | p38 MAPK-dependent anti-inflammatory effects | [66] |
| Lung transplant | Rat | 250–500 ppm | Improved histology, reduction of IRI and pro-inflammatory cytokines (i.e., IL-6); p38 MAPK dependent | [104,105,106] |
| Kidney transplant | Rat | 20 ppm; 250 ppm CO | Improved graft function, reduction of IRI and pro-inflammatory cytokines, improved blood flow. Reduced chronic allograft nephropathy | [108,109,110] |
| Acute compartment syndrome | Pig | CORM3 | Reduced tissue injury, apoptosis, leukocyte activation. | [122] |
3. Therapeutic Modulation of HO-1 in Human Disease
4. Inhaled Carbon Monoxide (iCO): Summary of Current Clinical Progress
5. Conclusions
Funding
Conflicts of Interest
References
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [PubMed]
- Tenhunen, R.; Ross, M.E.; Marver, H.S.; Schmid, R. Reduced nicotinamide-adenine dinucleotide phosphate dependent biliverdin reductase: Partial purification and characterization. Biochemistry 1970, 9, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar]
- Cruse, I.; Maines, M.D. Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 1988, 263, 3348–3353. [Google Scholar]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a target for therapeutic interventions. Front. Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2019, 133, 88–100. [Google Scholar] [CrossRef]
- Kumar, A.; Ganini, D.; Deterding, L.J.; Ehrenshaft, M.; Chatterjee, S.; Mason, R.P. Immuno-spin trapping of heme-induced protein radicals: Implications for heme oxygenase-1 induction and heme degradation. Free Radic. Biol. Med. 2013, 61, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [Green Version]
- James, J.; Srivastava, A.; Varghese, M.V.; Eccles, C.A.; Zemskova, M.; Rafikova, O.; Rafikov, R. Heme induces rapid endothelial barrier dysfunction via the MKK3/p38MAPK axis. Blood J. Am. Soc. Hematol. 2020, 136, 749–754. [Google Scholar]
- Englert, F.A.; Seidel, R.A.; Galler, K.; Gouveia, Z.; Soares, M.P.; Neugebauer, U.; Clemens, M.G.; Sponholz, C.; Heinemann, S.H.; Pohnert, G.; et al. Labile heme impairs hepatic microcirculation and promotes hepatic injury. Arch. Biochem. Biophys. 2019, 672, 108075. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.L.M.; Marinho, C.R.F.; Epiphanio, S. Could heme oxygenase-1 be a new target for therapeutic intervention in malaria-associated acute lung injury/acute respiratory distress syndrome? Front. Cell Infect. Microbiol. 2018, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, E.; Belcher, J.D.; Otterbein, L.E.; Coates, T.D.; Wood, J.; Skolnick, B.E.; Levy, H.; Vercellotti, G.M. The role of carbon monoxide and heme oxygenase in the prevention of sickle cell disease vaso-occlusive crises. Am. J. Hematol. 2017, 92, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Morita, T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1786–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar]
- Keyse, S.M.; Applegate, L.A.; Tromvoukis, Y.; Tyrrell, R.M. Oxidant stress leads to transcriptional activation of the human heme oxygenase gene in cultured skin fibroblasts. Mol. Cell. Biol. 1990, 10, 4967–4969. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Vile, G.F.; Tyrrell, R.M. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J. Biol. Chem. 1993, 268, 14678–14681. [Google Scholar] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Ames, B.N. Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc. Natl. Acad. Sci. USA 1987, 84, 8130–8134. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease. Am. J. Physiol. Cell Physiol. 2018, 314, C211–C227. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933. [Google Scholar]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Li Volti, G.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme oxygenase-1 in central nervous system malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef]
- Morse, D.; Lin, L.; Choi, A.M.; Ryter, S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic. Biol. Med. 2009, 47, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Drummond, H.A.; Mitchell, Z.L.; Abraham, N.G.; Stec, D.E. Targeting heme oxygenase-1 in cardiovascular and kidney disease. Antioxidants 2019, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme Oxygenase 1 as a therapeutic target in acute kidney injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Rong, J. Therapeutic potential of heme oxygenase-1/carbon monoxide system against ischemia-reperfusion injury. Curr. Pharm. Des. 2017, 23, 3884–3898. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, S.; Fondevila, C.; Kupiec-Weglinski, J.W. Heme oxygenase system in ischemia and reperfusion injury. Ann. Transplant. 2004, 9, 84–87. [Google Scholar] [PubMed]
- Hirao, H.; Dery, K.J.; Kageyama, S.; Nakamura, K.; Kupiec-Weglinski, J.W. Heme Oxygenase-1 in liver transplant ischemia-reperfusion injury: From bench-to-bedside. Free Radic. Biol. Med. 2020, 157, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The protective role of heme oxygenase-1 in atherosclerotic diseases. Int. J. Mol. Sci. 2019, 20, 3628. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Choi, S. Therapeutic aspects of carbon monoxide in cardiovascular disease. Int. J. Mol. Sci. 2018, 19, 2381. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Joe, Y.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Similarities and distinctions in the effects of metformin and carbon monoxide in immunometabolism. Mol. Cells. 2019, 42, 292–300. [Google Scholar]
- Waldman, M.; Arad, M.; Abraham, N.G.; Hochhauser, E. The peroxisome proliferator-activated receptor-gamma coactivator-1α-heme oxygenase 1 axis, a powerful antioxidative pathway with potential to attenuate diabetic cardiomyopathy. Antioxid. Redox Signal. 2020, 32, 1273–1290. [Google Scholar] [CrossRef]
- Peterson, S.J.; Dave, N.; Kothari, J. The effects of heme oxygenase upregulation on obesity and the metabolic syndrome. Antioxid. Redox Signal. 2020, 32, 1061–1070. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podkalicka, P.; Mucha, O.; Józkowicz, A.; Dulak, J.; Łoboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. 2018, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Coburn, R.F.; Blakemore, W.S.; Forster, R.E. Endogenous carbon monoxide production in man. J. Clin. Investig. 1963, 42, 1172–1178. [Google Scholar] [CrossRef] [Green Version]
- Sjostrand, T. Endogenous production of carbon monoxide in man under normal and pathophysiological conditions. Scand. J. Clin. Lab. Investig. 1949, 1, 201–214. [Google Scholar] [CrossRef]
- Sjostrand, T. The formation of carbon monoxide by the decomposition of hemoglobin in vivo. Acta Physiol. Scand. 1952, 26, 338–344. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Mantell, L.L.; Choi, A.M. Carbon monoxide provides protection against hyperoxic lung injury. Am. J. Physiol. 1999, 276, L688–L694. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Otterbein, S.L.; Ifedigbo, E.; Liu, F.; Morse, D.E.; Fearns, C.; Ulevitch, R.J.; Knickelbein, R.; Flavell, R.A.; Choi, A.M. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am. J. Pathol. 2003, 163, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J. Immunol. 2009, 182, 3809–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Otterbein, L.E.; Alam, J.; Flavell, R.A.; Davis, R.J.; Choi, A.M.; Lee, P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003, 278, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442. [Google Scholar] [CrossRef]
- Willis, D.; Moore, A.R.; Frederick, R.; Willoughby, D.A. Heme oxygenase: A novel target for the modulation of the inflammatory response. Nat. Med. 1996, 2, 87–90. [Google Scholar] [CrossRef]
- Chiang, N.; Shinohara, M.; Dalli, J.; Mirakaj, V.; Kibi, M.; Choi, A.M.; Serhan, C.N. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J. Immunol. 2013, 190, 6378–6388. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Kraft, B.D.; Colas, R.A.; Shinohara, M.; Fredenburgh, L.E.; Hess, D.R.; Chiang, N.; Welty-Wolf, K.E.; Choi, A.M.; Piantadosi, C.A.; et al. The regulation of pro-resolving lipid mediator profiles in baboon pneumonia by inhaled carbon monoxide. Am. J. Respir. Cell Mol. Biol. 2015, 53, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.L.; Berka, V.; Martin, E.; Olson, J.S. A “sliding scale rule” for selectivity among NO, CO, and O₂ by heme protein sensors. Biochemistry 2012, 51, 172–186. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Erecinska, M.; Wagner, M. Mitochondrial responses to carbon monoxide toxicity. Ann. N. Y. Acad. Sci. 1970, 174, 1970. [Google Scholar] [CrossRef] [PubMed]
- Bilban, M.; Bach, F.H.; Otterbein, S.L.; Ifedigbo, E.; d’Avila, J.C.; Esterbauer, H.; Chin, B.Y.; Usheva, A.; Robson, S.C.; Wagner, O.; et al. Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 2006, 24, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 867–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo-Pereira, C.; Dias-Pedroso, D.; Soares, N.L.; Vieira, H.L.A. CO-mediated cytoprotection is dependent on cell metabolism modulation. Redox Biol. 2020, 32, 101470. [Google Scholar] [CrossRef]
- Chin, B.Y.; Jiang, G.; Wegiel, B.; Wang, H.J.; Macdonald, T.; Zhang, X.C.; Gallo, D.; Cszimadia, E.; Bach, F.H.; Lee, P.J.; et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl. Acad. Sci. USA 2007, 104, 5109–5114. [Google Scholar] [CrossRef] [Green Version]
- Lo Iacono, L.; Boczkowski, J.; Zini, R.; Salouage, I.; Berdeaux, A.; Motterlini, R.; Morin, D. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Tsoyi, K.; Lee, T.Y.; Lee, Y.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. Heme-oxygenase-1 induction and carbon monoxide-releasing molecule inhibit lipopolysaccharide (LPS)-induced high-mobility group box 1 release in vitro and improve survival of mice in LPS- and cecal ligation and puncture-induced sepsis model in vivo. Mol. Pharmacol. 2009, 76, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Stucki, D.; Steinhausen, J.; Westhoff, P.; Krahl, H.; Brilhaus, D.; Massenberg, A.; Weber, A.P.M.; Reichert, A.S.; Brenneisen, P.; Stahl, W. Endogenous carbon monoxide signaling modulates mitochondrial function and intracellular glucose utilization: Impact of the heme oxygenase substrate hemin. Antioxidants 2020, 9, 652. [Google Scholar] [CrossRef]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Yu, J.; Zhang, Y.; Wu, L.; Dong, S.; Wu, L.; Wu, L.; Du, S.; Zhang, Y.; Ma, D. PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab. Investig. 2019, 99, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, Y.; Li, Z.; Dong, S.; Wang, D.; Gong, L.; Shi, J.; Zhang, Y.; Liu, D.; Mu, R. Effect of heme oxygenase-1 on mitofusin-1 protein in LPS-induced ALI/ARDS in rats. Sci. Rep. 2016, 6, 36530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Shi, J.; Wang, D.; Dong, S.; Zhang, Y.; Wang, M.; Gong, L.; Fu, Q.; Liu, D. Heme oxygenase-1/carbon monoxide-regulated mitochondrial dynamic equilibrium contributes to the attenuation of endotoxin-induced acute lung injury in rats and in lipopolysaccharide-activated macrophages. Anesthesiology 2016, 125, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Carraway, M.S.; Ali, A.S.; Reynolds, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Investig. 2007, 117, 3730–3741. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Carraway, M.S.; Tatro, L.G.; Piantadosi, C.A. A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 2007, 120, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Joe, Y.; Rah, S.Y.; Kim, S.K.; Park, S.U.; Park, J.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; et al. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018, 9, 1060. [Google Scholar] [CrossRef]
- Motterlini, R. Carbon monoxide-releasing molecules (CO-RMs): Vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem. Soc. Trans. 2007, 35 Pt 5, 1142–1146. [Google Scholar] [CrossRef]
- Motterlini, R.; Sawle, P.; Hammad, J.; Bains, S.; Alberto, R.; Foresti, R.; Green, C.J. CORM-A1: A new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005, 19, 284–286. [Google Scholar] [CrossRef] [Green Version]
- Fayad-Kobeissi, S.; Ratovonantenaina, J.; Dabiré, H.; Wilson, J.L.; Rodriguez, A.M.; Berdeaux, A.; Dubois-Randé, J.L.; Mann, B.E.; Motterlini, R.; Foresti, R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem. Pharmacol. 2016, 102, 64–77. [Google Scholar] [CrossRef]
- Kretschmer, R.; Gessner, G.; Görls, H.; Heinemann, S.H.; Westerhausen, M. Dicarbonyl-bis(cysteamine)iron(II): A light induced carbon monoxide releasing molecule based on iron (CORM-S1). J. Inorg. Biochem. 2011, 105, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.A.; Wright, J.A. PhotoCORMs: CO release moves into the visible. Dalton Trans. 2016, 45, 6801–6811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzola, S.; Forni, M.; Albertini, M.; Bacci, M.L.; Zannoni, A.; Gentilini, F.; Lavitrano, M.; Bach, F.H.; Otterbein, L.E.; Clement, M.G. Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J. 2005, 19, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.A.; Channell, M.M.; Royer, C.M.; Ryter, S.W.; Choi, A.M.; McDonald, J.D. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L891–L897. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Kraft, B.D.; Hess, D.R.; Harris, R.S.; Wolf, M.A.; Suliman, H.B.; Roggli, V.L.; Davies, J.D.; Winkler, T.; Stenzler, A.; et al. Effects of inhaled CO administration on acute lung injury in baboons with pneumococcal pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L834–L846. [Google Scholar] [CrossRef]
- Kawanishi, S.; Takahashi, T.; Morimatsu, H.; Shimizu, H.; Omori, E.; Sato, K.; Matsumi, M.; Maeda, S.; Nakao, A.; Morita, K. Inhalation of carbon monoxide following resuscitation ameliorates hemorrhagic shock-induced lung injury. Mol. Med. Rep. 2013, 7, 3–10. [Google Scholar] [CrossRef]
- Kanagawa, F.; Takahashi, T.; Inoue, K.; Shimizu, H.; Omori, E.; Morimatsu, H.; Maeda, S.; Katayama, H.; Nakao, A.; Morita, K. Protective effect of carbon monoxide inhalation on lung injury after hemorrhagic shock/resuscitation in rats. J. Trauma 2010, 69, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Kumada, Y.; Takahashi, T.; Shimizu, H.; Nakamura, R.; Omori, E.; Inoue, K.; Morimatsu, H. Therapeutic effect of carbon monoxide-releasing molecule-3 on acute lung injury after hemorrhagic shock and resuscitation. Exp. Ther. Med. 2019, 17, 3429–3440. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Investig. 2008, 118, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Hoetzel, A.; Schmidt, R.; Vallbracht, S.; Goebel, U.; Dolinay, T.; Kim, H.P.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med. 2009, 37, 1708–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoetzel, A.; Dolinay, T.; Vallbracht, S.; Zhang, Y.; Kim, H.P.; Ifedigbo, E.; Alber, S.; Kaynar, A.M.; Schmidt, R.; Ryter, S.W.; et al. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am. J. Respir. Crit. Care Med. 2008, 177, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinay, T.; Szilasi, M.; Liu, M.; Choi, A.M. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Kubo, M.; Morse, D.; Zhou, Z.; Zhang, X.; Dauber, J.H.; Fabisiak, J.; Alber, S.M.; Watkins, S.C.; Zuckerbraun, B.S.; et al. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am. J. Pathol. 2003, 163, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Kohmoto, J.; Nakao, A.; Kaizu, T.; Tsung, A.; Ikeda, A.; Tomiyama, K.; Billiar, T.R.; Choi, A.M.; Murase, N.; McCurry, K.R. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 2006, 140, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kohmoto, J.; Nakao, A.; Stolz, D.B.; Kaizu, T.; Tsung, A.; Ikeda, A.; Shimizu, H.; Takahashi, T.; Tomiyama, K.; Sugimoto, R.; et al. Carbon monoxide protects rat lung transplants from ischemia-reperfusion injury via a mechanism involving p38 MAPK pathway. Am. J. Transplant. 2007, 7, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Kohmoto, J.; Nakao, A.; Sugimoto, R.; Wang, Y.; Zhan, J.; Ueda, H.; McCurry, K.R. Carbon monoxide saturated preservation solution protects lung grafts from ischemia reperfusion injury. J. Thorac. Cardiovasc. Surg. 2008, 136, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Neto, J.S.; Nakao, A.; Toyokawa, H.; Nalesnik, M.A.; Romanosky, A.J.; Kimizuka, K.; Kaizu, T.; Hashimoto, N.; Azhipa, O.; Stolz, D.B.; et al. Low dose carbon monoxide inhalation prevents development of chronic allograft nephropathy. Am. J. Physiol. Renal Physiol. 2006, 290, F324–F334. [Google Scholar] [CrossRef] [Green Version]
- Neto, J.S.; Nakao, A.; Kimizuka, K.; Romanosky, A.J.; Stolz, D.B.; Uchiyama, T.; Nalesnik, M.A.; Otterbein, L.E.; Murase, N. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am. J. Physiol. Renal Physiol. 2004, 287, F979–F989. [Google Scholar] [CrossRef]
- Nakao, A.; Faleo, G.; Nalesnik, M.A.; Seda-Neto, J.; Kohmoto, J.; Murase, N. Low-dose carbon monoxide inhibits progressive chronic allograft nephropathy and restores renal allograft function. Am. J. Physiol. Renal Physiol. 2009, 297, F19–F26. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Toyoda, Y. Application of carbon monoxide for transplantation. Curr. Pharm. Biotechnol. 2012, 13, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Hashiba, T.; Tsuburai, T.; Ikehara, K.; Matsuse, T.; Ishigatsubo, Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum. Gene Ther. 2001, 12, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, T.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Inoue, S.; Tsuburai, T.; Matsuse, T.; Ishigatubo, Y. Adenovirus-mediated transfer of heme oxygenase-1 cDNA attenuates severe lung injury induced by the influenza virus in mice. Gene Ther. 2001, 8, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, D.; Pischke, S.E.; Zhou, Z.; Davis, R.J.; Flavell, R.A.; Loop, T.; Otterbein, S.L.; Otterbein, L.E.; Choi, A.M. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 2003, 278, 36993–36998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.P.; Wang, X.; Zhang, J.; Suh, G.Y.; Benjamin, I.; Ryter, S.W.; Choi, A.M. Heat shock factor-1 mediates the cytoprotective effect of carbon monoxide. J. Immunol. 2005, 175, 2622–2629. [Google Scholar] [CrossRef] [Green Version]
- Sarady, J.K.; Zuckerbraun, B.S.; Bilban, M.; Wagner, O.; Usheva, A.; Liu, F.; Ifedigbo, E.; Zamora, R.; Choi, A.M.; Otterbein, L.E. Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J. 2004, 18, 854–856. [Google Scholar] [CrossRef]
- Seixas, E.; Gozzelino, R.; Chora, A.; Ferreira, A.; Silva, G.; Larsen, R.; Rebelo, S.; Penido, C.; Smith, N.R.; Coutinho, A.; et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. USA 2009, 106, 15837–15842. [Google Scholar] [CrossRef] [Green Version]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Pereira, M.L.; Ortolan, L.S.; Sercundes, M.K.; Debone, D.; Murillo, O.; Lima, F.A.; Marinho, C.R.; Epiphanio, S. Association of heme oxygenase 1 with lung protection in malaria-associated ALI/ARDS. Mediators Inflamm. 2016, 2016, 4158698. [Google Scholar] [CrossRef] [Green Version]
- Epiphanio, S.; Campos, M.G.; Pamplona, A.; Carapau, D.; Pena, A.C.; Ataíde, R.; Monteiro, C.A.; Félix, N.; Costa-Silva, A.; Marinho, C.R.; et al. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 2010, 6, e1000916. [Google Scholar] [CrossRef]
- Pena, A.C.; Penacho, N.; Mancio-Silva, L.; Neres, R.; Seixas, J.D.; Fernandes, A.C.; Romão, C.C.; Mota, M.M.; Bernardes, G.J.; Pamplona, A. A novel carbon monoxide-releasing molecule fully protects mice from severe malaria. Antimicrob. Agents Chemother. 2012, 56, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bihari, A.; Cepinskas, G.; Sanders, D.; Lawendy, A.R. Systemic administration of carbon monoxide-releasing molecule-3 protects the skeletal muscle in porcine model of compartment syndrome. Crit. Care Med. 2018, 46, e469–e472. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Perrella, M.A.; Barragan-Bradford, D.; Hess, D.R.; Peters, E.; Welty-Wolf, K.E.; Kraft, B.D.; Harris, R.S.; Maurer, R.; Nakahira, K.; et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight. 2018, 3, e124039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. Medical progress: The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Mumby, S.; Upton, R.L.; Chen, Y.; Stanford, S.J.; Quinlan, G.J.; Nicholson, A.G.; Gutteridge, J.M.; Lamb, N.J.; Evans, T.W. Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit. Care Med. 2004, 32, 1130–1135. [Google Scholar] [CrossRef]
- Lagan, A.L.; Melley, D.D.; Evans, T.W.; Quinlan, G.J. Pathogenesis of the systemic inflammatory syndrome and acute lung injury: Role of iron mobilization and decompartmentalization. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L161–L174. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Furguson, N.; Laydon, D.; Nedjati-Gilani, G.; Imai, N.; Ainslie, K.; Baguelin, M.; Bhatia, S.; Boonyasiri, A.; Cucunubá, Z.; Cuomo-Dannenburg, G.; et al. Imperial College COVID-19 Response Team. Report 9: Impact of Non-Pharmaceutical Interventions (NPIs) to Reduce COVID19 Mortality and Healthcare Demand. Available online: https://www.imperial.ac.uk/media/imperial-college/medicine/sph/ide/gida-fellowships/Imperial-College-COVID19-NPI-modelling-16-03-2020.pdf (accessed on 19 September 2020).
- Wagener, F.A.D.T.G.; Pickkers, P.; Peterson, S.J.; Immenschuh, S.; Abraham, N.G. Targeting the heme-heme oxygenase system to prevent severe complications following COVID-19 infections. Antioxidants 2020, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020, 25, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, R.; Hung, C.C.; Hall, S.R.; Fukunaga, K.; Nagaishi, T.; Maeno, T.; Owen, C.; Macias, A.A.; Fredenburgh, L.E.; Ishizaka, A.; et al. High-mobility group box 1 contributes to lethality of endotoxemia in heme oxygenase-1-deficient mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, R.; Maier, J.; Gorki, A.D.; Huber, K.V.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Choi, H.S.; Yim, S.Y.; Lee, S.M. Heme oxygenase-1 protects the liver from septic injury by modulating TLR4-mediated mitochondrial quality control in mice. Shock 2018, 50, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Onyiah, J.C.; Sheikh, S.Z.; Maharshak, N.; Steinbach, E.C.; Russo, S.M.; Kobayashi, T.; Mackey, L.C.; Hansen, J.J.; Moeser, A.J.; Rawls, J.F.; et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 2013, 144, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Nakahira, K.; Dalli, J.; Siempos, I.I.; Norris, P.C.; Colas, R.A.; Moon, J.S.; Shinohara, M.; Hisata, S.; Howrylak, J.A.; et al. NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am. J. Respir. Crit. Care Med. 2017, 196, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tao, A.; Lan, T.; Cepinskas, G.; Kao, R.; Martin, C.M.; Rui, T. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res. Cardiol. 2017, 112, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, J.; Li, Y.; Chang, R.; Wu, H.; Lin, J.; Huang, Z. Exogenous carbon monoxide decreases sepsis-induced acute kidney injury and inhibits NLRP3 inflammasome activation in rats. Int. J. Mol. Sci. 2015, 16, 20595–20608. [Google Scholar] [CrossRef] [Green Version]
- Takaki, S.; Takeyama, N.; Kajita, Y.; Yabuki, T.; Noguchi, H.; Miki, Y.; Inoue, Y.; Nakagawa, T.; Noguchi, H. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010, 36, 42–48. [Google Scholar] [CrossRef]
- Lee, J.C.; Christie, J.D. Primary graft dysfunction. Proc. Am. Thorac. Soc. 2009, 6, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharat, A.; Kreisel, D. Immunopathogenesis of primary graft dysfunction after lung transplantation. Ann. Thorac. Surg. 2018, 105, 671–674. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; de Caestecker, M.; Otterbein, L.E.; Wang, B. Carbon monoxide: An emerging therapy for acute kidney injury. Med. Res. Rev. 2020, 40, 1147–1177. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Vreman, H.J.; Schulz, S.; Kalish, F.S.; Pierce, N.W.; Stevenson, D.K. In vitro inhibition of heme oxygenase isoenzymes by metalloporphyrins. J. Perinatol. 2011, 31 (Suppl. 1), S35–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinobe, R.T.; Dercho, R.A.; Nakatsu, K. Inhibitors of the heme oxygenase—Carbon monoxide system: On the doorstep of the clinic? Can. J. Physiol. Pharmacol. 2008, 86, 577–599. [Google Scholar] [CrossRef] [PubMed]
- Kinobe, R.T.; Vlahakis, J.Z.; Vreman, H.J.; Stevenson, D.K.; Brien, J.F.; Szarek, W.A.; Nakatsu, K. Selectivity of imidazole-dioxolane compounds for in vitro inhibition of microsomal haem oxygenase isoforms. Br. J. Pharmacol. 2006, 147, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—the comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Greenberg, M.; Glick, Y.; Singh, S.P.; Bellner, L.; Favero, G.; Rezzani, R.; Rodella, L.F.; Agostinucci, K.; Shapiro, J.I.; et al. Adipocyte specific HO-1 gene therapy is effective in antioxidant treatment of insulin resistance and vascular function in an obese mice model. Antioxidants 2020, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Lee, J.H.; Chung, H.T.; Pae, H.O. Therapeutic roles of heme oxygenase-1 in metabolic diseases: Curcumin and resveratrol analogues as possible inducers of heme oxygenase-1. Oxid. Med. Cell. Longev. 2013, 2013, 639541. [Google Scholar] [CrossRef] [Green Version]
- Hammad, A.S.A.; Ahmed, A.F.; Heeba, G.H.; Taye, A. Heme oxygenase-1 contributes to the protective effect of resveratrol against endothelial dysfunction in STZ-induced diabetes in rats. Life Sci. 2019, 239, 117065. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Rios, M.; Fernández-Fierro, A.; Covián, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally derived heme-oxygenase 1 inducers and their therapeutic application to immune-mediated diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Goebel, U.; Wollborn, J. Carbon monoxide in intensive care medicine-time to start the therapeutic application?! Intensive Care Med. Exp. 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Hopper, C.P.; Meinel, L.; Steiger, C.; Otterbein, L.E. Where is the clinical breakthrough of heme oxygenase-1 / carbon monoxide therapeutics? Curr. Pharm. Des. 2018, 24, 2264–2282. [Google Scholar] [CrossRef]
- Mayr, F.B.; Spiel, A.; Leitner, J.; Marsik, C.; Germann, P.; Ullrich, R.; Wagner, O.; Jilma, B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am. J. Respir. Crit. Care Med. 2006, 171, 354–360. [Google Scholar] [CrossRef]
- Bathoorn, E.; Slebos, D.J.; Postma, D.S.; Koeter, G.H.; van Oosterhout, A.J.; van der Toorn, M.; Boezen, H.M.; Kerstjens, H.A. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: A pilot study. Eur. Respir. J. 2007, 30, 1131–1137. [Google Scholar] [CrossRef]
- Rosas, I.O.; Rosas, I.O.; Goldberg, H.J.; Collard, H.R.; El-Chemaly, S.; Flaherty, K.; Hunninghake, G.M.; Lasky, J.A.; Lederer, D.J.; Machado, R.; et al. A Phase II clinical trial of low-dose inhaled carbon monoxide in idiopathic pulmonary fibrosis. Chest 2018, 153, 94–104. [Google Scholar] [CrossRef]
- Romão, C.C.; Blättler, W.A.; Seixas, J.D.; Bernardes, G.J. Developing drug molecules for therapy with carbon monoxide. Chem. Soc. Rev. 2012, 41, 3571–3583. [Google Scholar] [CrossRef]
- Ling, K.; Men, F.; Wang, W.C.; Zhou, Y.Q.; Zhang, H.W.; Ye, D.W. Carbon monoxide and its controlled release: Therapeutic application, detection, and development of carbon monoxide releasing molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef]
- Steiger, C.; Hermann, C.; Meinel, L. Localized delivery of carbon monoxide. Eur. J. Pharm. Biopharm. 2017, 118, 3–12. [Google Scholar] [CrossRef]
- Lazarus, L.S.; Benninghoff, A.D.; Berreau, L.M. Development of triggerable, trackable, and targetable carbon monoxide releasing molecules. Acc. Chem Res. 2020, 53, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryter, S.W. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants 2020, 9, 1153. https://doi.org/10.3390/antiox9111153
Ryter SW. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants. 2020; 9(11):1153. https://doi.org/10.3390/antiox9111153
Chicago/Turabian StyleRyter, Stefan W. 2020. "Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders" Antioxidants 9, no. 11: 1153. https://doi.org/10.3390/antiox9111153