Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pivotal Role of Heme Oxygenase Reaction—Salutary vs. Deleterious Effects
2.1. Beneficial Role of HO
2.2. Absense of Cytoprotection or Aggravation of Disease by Upregulated HO
2.3. Mechanisms Underlying Deleterious Effects of HO
3. Chemistry of Heme
4. Pro-Oxidative Properties of Heme
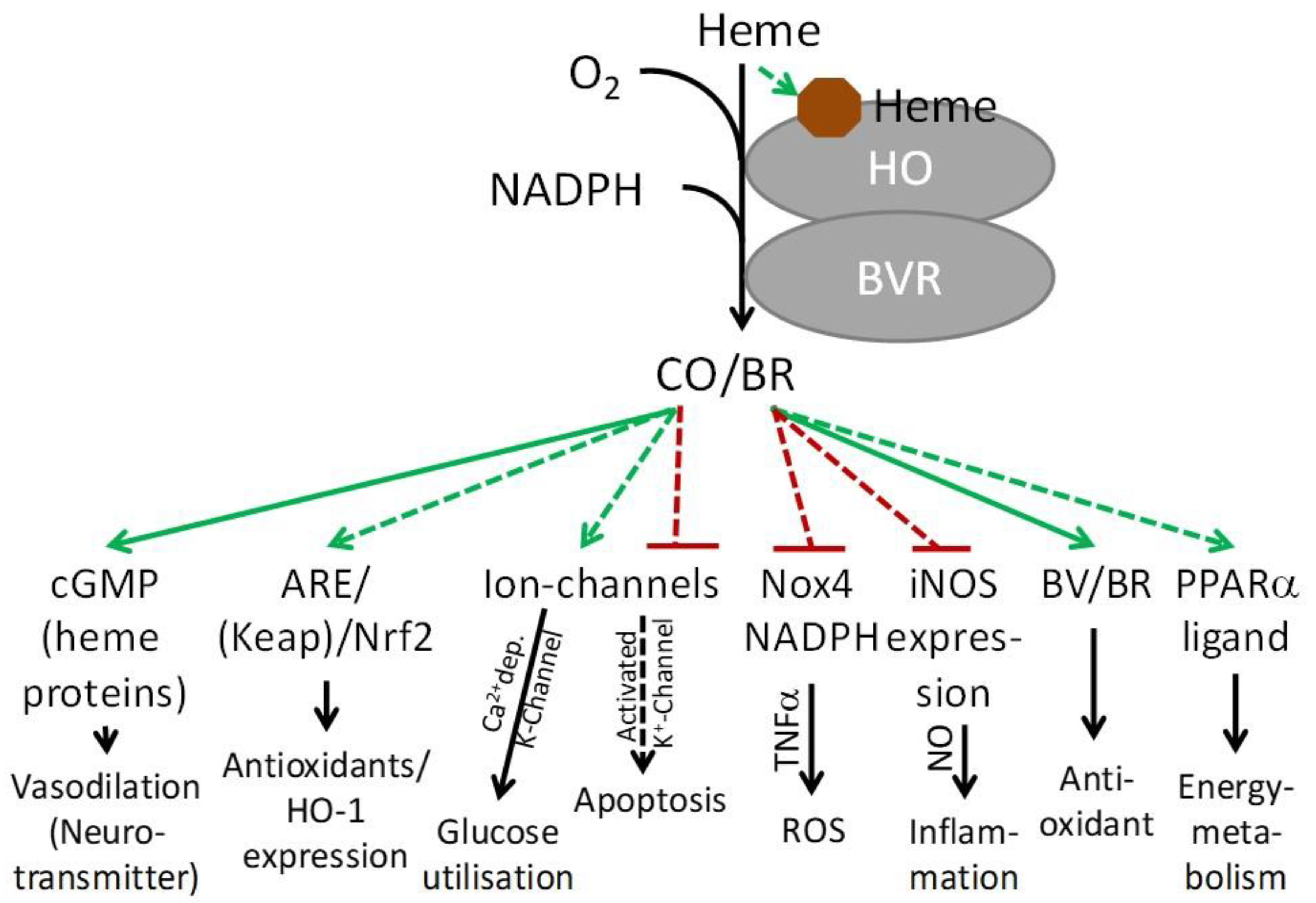
5. Heme and NO/CO and the Resulting Reactions
6. Heme Modulating Cellular Pathways—From Transcription to Differentiation
6.1. Heme Regulating Its Degradation and the Anti-oxidative Defense
6.2. Heme Regulating Erythrocyte Maturation and Differentiation
6.3. Heme Regulating Energy Metabolism and Glucose Utilisation
6.4. Heme Regulating Macrophage Function
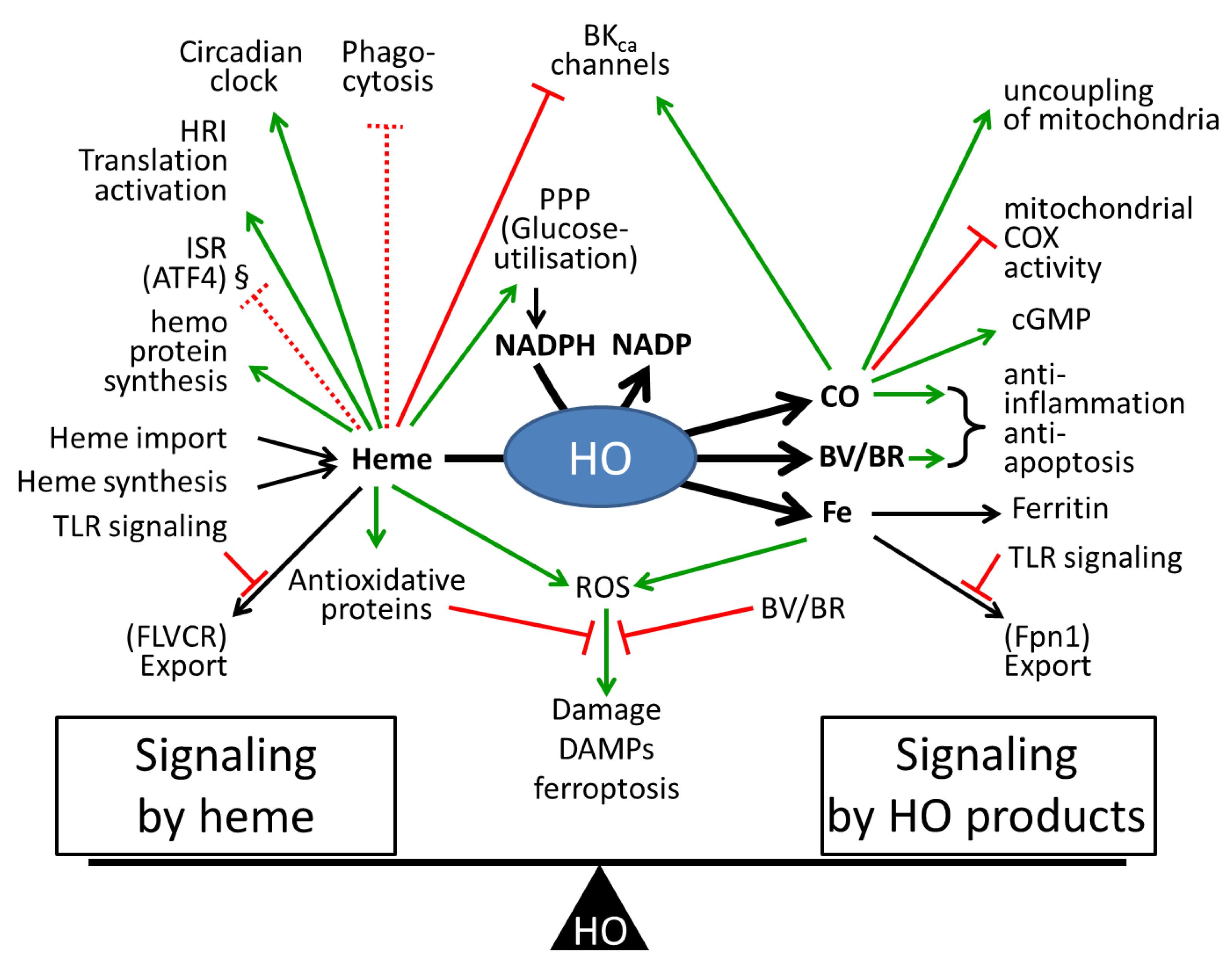
7. Modulation of Heme-Mediated Signaling Pathways by HO Activity
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef]
- McCoubrey, W.K.; Huang, T.J., Jr.; Maines, M.D. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur. J. Biochem. 1997, 247, 725–732. [Google Scholar] [CrossRef]
- Poss, K.D.; Thomas, M.J.; Ebralidze, A.K.; O’Dell, T.J.; Tonegawa, S. Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron 1995, 15, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [Green Version]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Wada, T.; Yachie, A. An interesting tetrad of asplenia, inflammation, hemolysis, and nephritis. Pediatr. Hematol. Oncol. 2011, 28, 723–726. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [Green Version]
- Ewing, J.F.; Maines, M.D. Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Brain Res. Protoc. 1997, 1, 165–174. [Google Scholar] [CrossRef]
- Boehning, D.; Snyder, S.H. Novel neural modulators. Annu. Rev. Neurosci. 2003, 26, 105–131. [Google Scholar] [CrossRef]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox. Signal. 2004, 6, 878–887. [Google Scholar] [CrossRef]
- Aztatzi-Santillan, E.; Nares-Lopez, F.E.; Marquez-Valadez, B.; Aguilera, P.; Chanez-Cardenas, M.E. The protective role of heme oxygenase-1 in cerebral ischemia. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 310–316. [Google Scholar] [CrossRef]
- Camara, N.O.; Soares, M.P. Heme oxygenase-1 (HO-1), a protective gene that prevents chronic graft dysfunction. Free Radic. Biol. Med. 2005, 38, 426–435. [Google Scholar] [CrossRef]
- Li, S.; Fujino, M.; Takahara, T.; Li, X.K. Protective role of heme oxygenase-1 in fatty liver ischemia-reperfusion injury. Med Mol. Morphol. 2019, 52, 61–72. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Soares, M.P.; Brouard, S.; Smith, R.N.; Bach, F.H. Heme oxygenase-1, a protective gene that prevents the rejection of transplanted organs. Immunol. Rev. 2001, 184, 275–285. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Bernardini, C.; Grilli, E.; Duvigneau, J.C.; Zannoni, A.; Tugnoli, B.; Gentilini, F.; Bertuzzi, T.; Spinozzi, S.; Camborata, C.; Bacci, M.L.; et al. Cellular stress marker alteration and inflammatory response in pigs fed with an ochratoxin contaminated diet. Res. Vet. Sci. 2014, 97, 244–250. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Piskernik, C.; Haindl, S.; Kloesch, B.; Hartl, R.T.; Huttemann, M.; Lee, I.; Ebel, T.; Moldzio, R.; Gemeiner, M.; et al. A novel endotoxin-induced pathway: Upregulation of heme oxygenase 1, accumulation of free iron, and free iron-mediated mitochondrial dysfunction. Lab. Invest. 2008, 88, 70–77. [Google Scholar] [CrossRef]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Postl, A.; Zifko, C.; Hartl, R.T.; Ebel, T.; Miller, I.; Moldzio, R.; Redl, H.; Kozlov, A.V.; Bahrami, S.; Duvigneau, J.C. Transient increase of free iron in rat livers following hemorrhagic-traumatic shock and reperfusion is independent of heme oxygenase 1 upregulation. Shock 2011, 36, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE 2018, 13, e0196455. [Google Scholar] [CrossRef] [PubMed]
- Sassa, S. Why heme needs to be degraded to iron, biliverdin IXalpha, and carbon monoxide? Antioxid. Redox. Signal. 2004, 6, 819–824. [Google Scholar] [CrossRef]
- Stocker, R. Antioxidant activities of bile pigments. Antioxid. Redox. Signal. 2004, 6, 841–849. [Google Scholar] [CrossRef]
- Maghzal, G.J.; Leck, M.C.; Collinson, E.; Li, C.; Stocker, R. Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 2009, 284, 29251–29259. [Google Scholar] [CrossRef]
- Basuroy, S.; Bhattacharya, S.; Leffler, C.W.; Parfenova, H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C422–C432. [Google Scholar] [CrossRef]
- Chen, J.; Tu, Y.; Connolly, E.C.; Ronnett, G.V. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2005, 2, 121–131. [Google Scholar] [CrossRef]
- Jangi, S.; Otterbein, L.; Robson, S. The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int. J Biochem. Cell Biol. 2013, 45, 2843–2851. [Google Scholar] [CrossRef]
- Longhi, M.S.; Vuerich, M.; Kalbasi, A.; Kenison, J.E.; Yeste, A.; Csizmadia, E.; Vaughn, B.; Feldbrugge, L.; Mitsuhashi, S.; Wegiel, B.; et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valaskova, P.; Dvorak, A.; Lenicek, M.; Zizalova, K.; Kutinova-Canova, N.; Zelenka, J.; Cahova, M.; Vitek, L.; Muchova, L. Hyperbilirubinemia in Gunn Rats is Associated with Decreased Inflammatory Response in LPS-Mediated Systemic Inflammation. Int. J. Mol. Sci. 2019, 20, 2306. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Smith, D.L.; Zucker, S.D. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 2004, 40, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D., Jr. RNA sequencing in human HepG2 hepatocytes reveals PPAR-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D.; Adeosun, S.O., Jr.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W. and Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- Hinds, T.D.; Burns, K.A., Jr.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; Alamodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Cimini, F.A.; Arena, A.; Barchetta, I.; Tramutola, A.; Ceccarelli, V.; Lanzillotta, C.; Fontana, M.; Bertoccini, L.; Leonetti, F.; Capoccia, D.; et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brann, D.W.; Bhat, G.K.; Lamar, C.A.; Mahesh, V.B. Gaseous transmitters and neuroendocrine regulation. Neuroendocrinology 1997, 65, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Telezhkin, V.; Brazier, S.P.; Mears, R.; Muller, C.T.; Riccardi, D.; Kemp, P.J. Cysteine residue 911 in C-terminal tail of human BK(Ca)alpha channel subunit is crucial for its activation by carbon monoxide. Pflug. Arch. 2011, 461, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, R.; Princ, F.; de Stein, M.L.; Fin, C.; Juknat, A.A.; Batile, A.; Izquierdo, I.; Medina, J.H. Evidence for the involvement of hippocampal CO production in the acquisition and consolidation of inhibitory avoidance learning. Neuroreport 1995, 6, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buechler, C.; Pohl, R.; Aslanidis, C. Pro-Resolving Molecules-New Approaches to Treat Sepsis? Int. J. Mol. Sci. 2017, 18, 476. [Google Scholar] [CrossRef]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox. Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef]
- Lee, H.; Choi, Y.K. Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. Int. J. Mol. Sci. 2018, 20, 78. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1029–L1037. [Google Scholar] [CrossRef] [PubMed]
- Li, V.G.; Tibullo, D.; Vanella, L.; Giallongo, C.; Di, R.F.; Forte, S.; Di, R.M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid. Redox. Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef]
- Podkalicka, P.; Mucha, O.; Jozkowicz, A.; Dulak, J.; Loboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. (Pozn. ) 2018, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism —Targeting HO-1 as an anti-tumor therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.S.; Weis, S.; Yang, G.; Zhuang, T.; Abate, A.; Dennery, P.A. Catalytic inactive heme oxygenase-1 protein regulates its own expression in oxidative stress. Free Radic. Biol. Med. 2008, 44, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Lornejad-Schafer, M.R.; Schoffl, H.; Flaccus, A.; Lambert, C.; Biesalski, H.K. Inhibition of heme oxygenase-1 increases responsiveness of melanoma cells to ALA-based photodynamic therapy. Int. J. Oncol. 2007, 31, 1539–1545. [Google Scholar] [CrossRef]
- Raffaele, M.; Pittala, V.; Zingales, V.; Barbagallo, I.; Salerno, L.; Li, V.G.; Romeo, G.; Carota, G.; Sorrenti, V.; Vanella, L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. Int. J. Mol. Sci. 2019, 20, 2593. [Google Scholar] [CrossRef]
- Salerno, L.; Romeo, G.; Modica, M.N.; Amata, E.; Sorrenti, V.; Barbagallo, I.; Pittala, V. Heme oxygenase-1: A new druggable target in the management of chronic and acute myeloid leukemia. Eur. J. Med. Chem. 2017, 142, 163–178. [Google Scholar] [CrossRef]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—the comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Takano, T.; Miyauchi, A.; Matsuzuka, F.; Yoshida, H.; Kuma, K.; Amino, N. Decreased expression of glutathione peroxidase mRNA in thyroid anaplastic carcinoma. Cancer Lett. 2002, 182, 69–74. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Takano, T.; Miyauchi, A.; Matsuzuka, F.; Yoshida, H.; Kuma, K.; Amino, N. Decreased expression of catalase mRNA in thyroid anaplastic carcinoma. Jpn. J. Clin. Oncol. 2003, 33, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ito, K.; Kohara, H.; Yamaguchi, Y.; Adachi, K.; Endo, H. Negative regulation of catalase gene expression in hepatoma cells. Mol. Cell Biol. 1992, 12, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Nakamura, S.; Kayasuga, A.; Isa, S.; Sato, K. Multiple elements for negative regulation of the rat catalase gene expression in dedifferentiated hepatoma cells. J. Biochem. 2000, 128, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Sawa, T.; Fang, J.; Tanaka, S.; Miyamoto, Y.; Akaike, T.; Maeda, H. Pegylated zinc protoporphyrin: A water-soluble heme oxygenase inhibitor with tumor-targeting capacity. Bioconjug. Chem. 2002, 13, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Song, D.M.; Niu, Y.H.; Wang, B.S. Inhibition of heme oxygenase-1 enhances the chemosensitivity of laryngeal squamous cell cancer Hep-2 cells to cisplatin. Apoptosis 2016, 21, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.S.; Li, H.S.; Qi, D.F.; Zhang, J.; Jiang, X.C.; Shi, K.; Zhang, X.J.; Zhang, X.H. Zinc protoporphyrin IX enhances chemotherapeutic response of hepatoma cells to cisplatin. World, J. Gastroenterol. 2014, 20, 8572–8582. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Sawa, T.; Akaike, T.; Akuta, T.; Sahoo, S.K.; Khaled, G.; Hamada, A.; Maeda, H. In vivo antitumor activity of pegylated zinc protoporphyrin: Targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003, 63, 3567–3574. [Google Scholar]
- Fang, J.; Tsukigawa, K.; Liao, L.; Yin, H.; Eguchi, K.; Maeda, H. Styrene-maleic acid-copolymer conjugated zinc protoporphyrin as a candidate drug for tumor-targeted therapy and imaging. J. Drug Target 2016, 24, 399–407. [Google Scholar] [CrossRef]
- Salerno, L.; Pittala, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di, G.C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.F.; Salerno, L.; Al, Z.R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [PubMed]
- Appleton, S.D.; Chretien, M.L.; McLaughlin, B.E.; Vreman, H.J.; Stevenson, D.K.; Brien, J.F.; Nakatsu, K.; Maurice, D.H.; Marks, G.S. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metab. Dispos. 1999, 27, 1214–1219. [Google Scholar] [PubMed]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronel, C.; Rochefort, G.Y.; Arlicot, N.; Bodard, S.; Chalon, S.; Antier, D. Oxidative stress is related to the deleterious effects of heme oxygenase-1 in an in vivo neuroinflammatory rat model. Oxid. Med. Cell Longev. 2013, 2013, 264935. [Google Scholar] [CrossRef]
- Hopper, C.P.; Meinel, L.; Steiger, C.; Otterbein, L.E. Where is the Clinical Breakthrough of Heme Oxygenase-1 / Carbon Monoxide Therapeutics? Curr. Pharm. Des. 2018, 24, 2264–2282. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Tsuchihashi, S.; Livhits, M.; Zhai, Y.; Busuttil, R.W.; Araujo, J.A.; Kupiec-Weglinski, J.W. Basal rather than induced heme oxygenase-1 levels are crucial in the antioxidant cytoprotection. J. Immunol. 2006, 177, 4749–4757. [Google Scholar] [CrossRef]
- Ohnishi, M.; Katsuki, H.; Unemura, K.; Izumi, Y.; Kume, T.; Takada-Takatori, Y.; Akaike, A. Heme oxygenase-1 contributes to pathology associated with thrombin-induced striatal and cortical injury in organotypic slice culture. Brain Res. 2010, 1347, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Song, W.; Schipper, H.M. Astroglia overexpressing heme oxygenase-1 predispose co-cultured PC12 cells to oxidative injury. J. Neurosci. Res. 2007, 85, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.; Leiva, E.; Arredondo-Olguin, M. Short repeats in the heme oxygenase 1 gene promoter is associated with increased levels of inflammation, ferritin and higher risk of type-2 diabetes mellitus. J. Trace Elem. Med. Biol. 2016, 37, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Ndisang, J.F. A critical and comprehensive insight on heme oxygenase and related products including carbon monoxide, bilirubin, biliverdin and ferritin in type-1 and type-2 diabetes. Curr. Pharm. Des. 2014, 20, 1370–1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Wang, G.G.; Li, W.; Jiang, Y.X.; Lu, X.H.; Zhou, P.P. Heme Oxygenase-1 Promotes Delayed Wound Healing in Diabetic Rats. J. Diabetes Res. 2016, 2016, 9726503. [Google Scholar] [CrossRef] [PubMed]
- Pagnin, E.; Maiolino, G.; Calo, L.A. Heme oxygenase-1 in type 2 diabetes: From cell first-line defense to early marker of diabetic nephropathy. Minerva Med 2016, 107, 123–124. [Google Scholar] [PubMed]
- Tsuji, M.H.; Yanagawa, T.; Iwasa, S.; Tabuchi, K.; Onizawa, K.; Bannai, S.; Toyooka, H.; Yoshida, H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999, 138, 53–59. [Google Scholar] [CrossRef]
- Tsai, J.R.; Wang, H.M.; Liu, P.L.; Chen, Y.H.; Yang, M.C.; Chou, S.H.; Cheng, Y.J.; Yin, W.H.; Hwang, J.J.; Chong, I.W. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol. 2012, 35, 461–471. [Google Scholar] [CrossRef]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef]
- Wang, T.Y.; Liu, C.L.; Chen, M.J.; Lee, J.J.; Pun, P.C.; Cheng, S.P. Expression of haem oxygenase-1 correlates with tumour aggressiveness and BRAF V600E expression in thyroid cancer. Histopathology 2015, 66, 447–456. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, Y.; Lu, J.; Xue, J.; Liu, P. High expression of HO-1 predicts poor prognosis of ovarian cancer patients and promotes proliferation and aggressiveness of ovarian cancer cells. Clin. Transl. Oncol. 2018, 20, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.; Dolle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides--small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L. Glutathione peroxidase: Fact and fiction. Ciba Found. Symp. 1978, 95–122. [Google Scholar]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro-Oncology 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergent, O.; Tomasi, A.; Ceccarelli, D.; Masini, A.; Nohl, H.; Cillard, P.; Cillard, J.; Vladimirov, Y.A.; Kozlov, A.V. Combination of iron overload plus ethanol and ischemia alone give rise to the same endogenous free iron pool. Biometals 2005, 18, 567–575. [Google Scholar] [CrossRef]
- Kagan, V.E.; Kozlov, A.V.; Tyurina, Y.Y.; Shvedova, A.A.; Yalowich, J.C. Antioxidant mechanisms of nitric oxide against iron-catalyzed oxidative stress in cells. Antioxid. Redox. Signal. 2001, 3, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lonn, M.E.; Xu, X.; Maghzal, G.J.; Frazer, D.M.; Thomas, S.R.; Halliwell, B.; Richardson, D.R.; Anderson, G.J.; Stocker, R. Sustained expression of heme oxygenase-1 alters iron homeostasis in nonerythroid cells. Free Radic. Biol. Med 2012, 53, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Lamb, N.J.; Quinlan, G.J.; Mumby, S.; Evans, T.W.; Gutteridge, J.M. Haem oxygenase shows pro-oxidant activity in microsomal and cellular systems: Implications for the release of low-molecular-mass iron. Biochem. J. 1999, 344 Pt 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Rachmilewitz, E.A. Iron overload in hematological disorders. Presse Med. 2017, 46, e296–e305. [Google Scholar] [CrossRef] [PubMed]
- Puntarulo, S. Iron, oxidative stress and human health. Mol. Asp. Med. 2005, 26, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.Z. Hepcidin-ferroportin axis in health and disease. Vitam. Horm. 2019, 110, 17–45. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Perry, G. Nucleic acid oxidative damage in Alzheimer’s disease-explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J. Trace Elem. Med. Biol. 2016, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatic iron overload and hepatocellular carcinoma. Cancer Lett. 2009, 286, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidinger, A.; Dungel, P.; Perlinger, M.; Singer, K.; Ghebes, C.; Duvigneau, J.C.; Mullebner, A.; Schafer, U.; Redl, H.; Kozlov, A.V. Experimental data suggesting that inflammation mediated rat liver mitochondrial dysfunction results from secondary hypoxia rather than from direct effects of inflammatory mediators. Front. Physiol. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, B.C.; Krause, G.S.; Aust, S.D.; Eyster, G.E. Postischemic tissue injury by iron-mediated free radical lipid peroxidation. Ann. Emerg. Med. 1985, 14, 804–809. [Google Scholar] [CrossRef]
- Badylak, S.F.; Simmons, A.; Turek, J.; Babbs, C.F. Protection from reperfusion injury in the isolated rat heart by postischaemic deferoxamine and oxypurinol administration. Cardiovasc. Res. 1987, 21, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, M.S.; Hedlund, B.E. Role of iron in postischemic renal injury in the rat. Kidney Int. 1988, 34, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.A.; Grisham, M.B.; Granger, D.N. A role for iron in oxidant-mediated ischemic injury to intestinal microvasculature. Am. J. Physiol. 1987, 253, G49–G53. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; Paliakkara, J.J.; Melman, G.; Melman, A. Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication. Pharmaceuticals 2018, 11, 120. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr. Pharm. Des. 2011, 17, 3460–3473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Du, J.L.; Duan, Z.J.; Guo, S.B.; Sun, X.Y.; Liu, Z. Inhibiting heme oxygenase-1 attenuates rat liver fibrosis by removing iron accumulation. World J. Gastroenterol. 2013, 19, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Reichen, J.; Hoilien, C.; Sheldon, G.F.; Kirshenbaum, G. A novel method for continuous monitoring of bilirubin production in unstressed rats. Am. J. Physiol. 1983, 244, G336–G340. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Quan, S.; Mieyal, P.A.; Yang, L.; Burke-Wolin, T.; Mingone, C.J.; Goodman, A.I.; Nasjletti, A.; Wolin, M.S. Modulation of cGMP by human HO-1 retrovirus gene transfer in pulmonary microvessel endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L1117–L1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Zhang, Y.Z.; Furuyama, K.; Ogawa, K.; Igarashi, K.; Shibahara, S. Down-regulation of heme oxygenase-2 is associated with the increased expression of heme oxygenase-1 in human cell lines. FEBS J. 2006, 273, 5333–5346. [Google Scholar] [CrossRef] [PubMed]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Kaneko, K.; Vargas, P.D. Heme as a magnificent molecule with multiple missions: Heme determines its own fate and governs cellular homeostasis. Tohoku J. Exp. Med. 2007, 213, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Tissue-specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [PubMed]
- Reeder, B.J. The redox activity of hemoglobins: From physiologic functions to pathologic mechanisms. Antioxid. Redox. Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, T.; Sassa, S.; Kappas, A. The oxidative degradation of heme c by the microsomal heme oxygenase system. J. Biol. Chem. 1982, 257, 7803–7807. [Google Scholar] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.H.; Muller-Eberhard, U. A protein of the Z class of liver cytosolic proteins in the rat that preferentially binds heme. J. Biol. Chem. 1985, 260, 14521–14528. [Google Scholar] [PubMed]
- Taketani, S.; Adachi, Y.; Kohno, H.; Ikehara, S.; Tokunaga, R.; Ishii, T. Molecular characterization of a newly identified heme-binding protein induced during differentiation of urine erythroleukemia cells. J. Biol. Chem. 1998, 273, 31388–31394. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.; Smith, A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem. J. 1988, 256, 861–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Simoes, R.L.; Arruda, M.A.; Canetti, C.; Serezani, C.H.; Fierro, I.M.; Barja-Fidalgo, C. Proinflammatory responses of heme in alveolar macrophages: Repercussion in lung hemorrhagic episodes. Mediators. Inflamm. 2013, 2013, 946878. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.L.; Dutra, F.F.; Alves, L.; Figueiredo, R.T.; Mourao-Sa, D.; Fortes, G.B.; Bergstrand, S.; Lonn, D.; Cevallos, R.R.; Pereira, R.M.; et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J. Biol. Chem. 2010, 285, 32844–32851. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [PubMed]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinchi, F.; Costa da, S.M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Hauser, C.J.; Otterbein, L.E. Heme as a danger molecule in pathogen recognition. Free Radic. Biol. Med. 2015, 89, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Maier, J.; Gorki, A.D.; Huber, K.V.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.T.; Phelps, S.R.; Shadle, C.; Sanchez-Bonilla, M.; Keel, S.B.; Abkowitz, J.L. Coordinate expression of heme and globin is essential for effective erythropoiesis. J. Clin. Investig. 2015, 125, 4681–4691. [Google Scholar] [CrossRef] [Green Version]
- Klouche, K.; Morena, M.; Canaud, B.; Descomps, B.; Beraud, J.J.; Cristol, J.P. Mechanism of in vitro heme-induced LDL oxidation: Effects of antioxidants. Eur. J. Clin. Investig. 2004, 34, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Toppo, S.; Flohe, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungel, P.; Perlinger, M.; Weidinger, A.; Redl, H.; Kozlov, A.V. The cytoprotective effect of nitrite is based on the formation of dinitrosyl iron complexes. Free Radic. Biol. Med. 2015, 89, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Fotiou, S.; Fotiou, D.; Deliconstantinos, G. Formation of heme-iron complexes with nitric oxide (NO) and peroxynitrite (ONOO-) after ultraviolet radiation as a protective mechanism in rat skin. In Vivo 2009, 23, 281–286. [Google Scholar]
- Sahni, S.; Hickok, J.R.; Thomas, D.D. Nitric oxide reduces oxidative stress in cancer cells by forming dinitrosyliron complexes. Nitric Oxide 2018, 76, 37–44. [Google Scholar] [CrossRef]
- Deliconstantinos, G.; Villiotou, V.; Stavrides, J.C. Scavenging effects of hemoglobin and related heme containing compounds on nitric oxide, reactive oxidants and carcinogenic volatile nitrosocompounds of cigarette smoke. A new method for protection against the dangerous cigarette constituents. Anticancer Res. 1994, 14, 2717–2726. [Google Scholar]
- Willis, D.; Tomlinson, A.; Frederick, R.; Paul-Clark, M.J.; Willoughby, D.A. Modulation of heme oxygenase activity in rat brain and spleen by inhibitors and donors of nitric oxide. Biochem. Biophys. Res. Commun. 1995, 214, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta 1999, 1411, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Rus, A.; Molina, F.; Peinado, M.A.; Del Moral, M.L. Endogenous nitric oxide can act as beneficial or deleterious in the hypoxic lung depending on the reoxygenation time. Anat. Rec. 2010, 293, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- van Faassen, E.E.; Bahrami, S.; Feelisch, M.; Hogg, N.; Kelm, M.; Kim-Shapiro, D.B.; Kozlov, A.V.; Li, H.; Lundberg, J.O.; Mason, R.; et al. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med. Res. Rev. 2009, 29, 683–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and nitrite in biology, nutrition and therapeutics1. Nat. Chem. Biol. 2009, 5, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Numazawa, S.; Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic. Biol. Med. 2005, 38, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Conditional expression of the ubiquitous transcription factor MafK induces erythroleukemia cell differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 7445–7449. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Tahara, T.; Sun, J.; Nakanishi, K.; Yamamoto, M.; Mori, H.; Saito, T.; Fujita, H.; Igarashi, K.; Taketani, S. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J. Biol. Chem. 2004, 279, 5480–5487. [Google Scholar] [CrossRef]
- Warnatz, H.J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef]
- Hira, S.; Tomita, T.; Matsui, T.; Igarashi, K.; Ikeda-Saito, M. Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB Life 2007, 59, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. Embo J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T. Binding of cysteine thiolate to the Fe(III) heme complex is critical for the function of heme sensor proteins. J. Inorg. Biochem. 2012, 108, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Wissbrock, A.; Paul George, A.A.; Brewitz, H.H.; Kuhl, T.; Imhof, D. The molecular basis of transient heme-protein interactions: Analysis, concept and implementation. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, C.; Tashiro, S.; Nishito, Y.; Sueda, T.; Igarashi, K. Dynamic cytoplasmic anchoring of the transcription factor Bach1 by intracellular hyaluronic acid binding protein IHABP. J. Biochem. 2005, 137, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Busche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Fureder, W.; Bettelheim, P.; Pfeilstocker, M.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Koury, M.J.; Bondurant, M.C. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science 1990, 248, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Felsenfeld, G. The erythroid-specific transcription factor Eryf1: A new finger protein. Cell 1989, 58, 877–885. [Google Scholar] [CrossRef]
- Sadlon, T.J.; Dell’Oso, T.; Surinya, K.H.; May, B.K. Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int. J. Biochem. Cell Biol. 1999, 31, 1153–1167. [Google Scholar] [CrossRef]
- Tanimura, N.; Miller, E.; Igarashi, K.; Yang, D.; Burstyn, J.N.; Dewey, C.N.; Bresnick, E.H. Mechanism governing heme synthesis reveals a GATA factor/heme circuit that controls differentiation. Embo Rep. 2016, 17, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De, D.I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef]
- Mercurio, S.; Petrillo, S.; Chiabrando, D.; Bassi, Z.I.; Gays, D.; Camporeale, A.; Vacaru, A.; Miniscalco, B.; Valperga, G.; Silengo, L.; et al. The heme exporter Flvcr1 regulates expansion and differentiation of committed erythroid progenitors by controlling intracellular heme accumulation. Haematologica 2015, 100, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Quigley, J.G. Heme and FLVCR-related transporter families SLC48 and SLC49. Mol. Asp. Med. 2013, 34, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Kato, H.; Hada, H.; Itoh-Nakadai, A.; Fujiwara, T.; Muto, A.; Inoguchi, Y.; Ichiyanagi, K.; Hojo, W.; Tomosugi, N.; et al. Iron-heme-Bach1 axis is involved in erythroblast adaptation to iron deficiency. Haematologica 2017, 102, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. Embo J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.; Zachariah, R.S.; Velazquez, J.G.; Liu, S.; Sun, C.W.; Townes, T.M.; Chen, J.J. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 2012, 119, 5276–5284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Macias-Garcia, A.; Ulirsch, J.C.; Velazquez, J.; Butty, V.L.; Levine, S.S.; Sankaran, V.G.; Chen, J.J. HRI coordinates translation necessary for protein homeostasis and mitochondrial function in erythropoiesis. Elife 2019, 8, e46976. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wu, N.; Curtin, J.C.; Qatanani, M.; Szwergold, N.R.; Reid, R.A.; Waitt, G.M.; Parks, D.J.; Pearce, K.H.; Wisely, G.B.; et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science 2007, 318, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Takeda, K.; Ishikawa, K.; Yoshizawa, M.; Sato, M.; Shibahara, S.; Furuyama, K. Coordinated expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 and heme oxygenase 2: Evidence for a regulatory link between glycolysis and heme catabolism. Tohoku J. Exp. Med. 2012, 228, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Clark, J.; Klarer, A.C.; Imbert-Fernandez, Y.; Lane, A.N.; Telang, S. Fructose-2,6-bisphosphate synthesis by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) is required for the glycolytic response to hypoxia and tumor growth 1. Oncotarget 2014, 5, 6670–6686. [Google Scholar] [CrossRef]
- Padmanaban, G.; Venkateswar, V.; Rangarajan, P.N. Haem as a multifunctional regulator. Trends Biochem. Sci. 1989, 14, 492–496. [Google Scholar] [CrossRef]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef]
- Kimpara, T.; Takeda, A.; Yamaguchi, T.; Arai, H.; Okita, N.; Takase, S.; Sasaki, H.; Itoyama, Y. Increased bilirubins and their derivatives in cerebrospinal fluid in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 551–554. [Google Scholar] [CrossRef]
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.M.; Ying, L.; Burris, T.P. Relationship between circadian oscillations of Rev-erbalpha expression and intracellular levels of its ligand, heme. Biochem. Biophys. Res. Commun. 2008, 368, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Udoh, U.S.; Young, M.E. Circadian regulation of metabolism. J. Endocrinol. 2014, 222, R75–R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, J.S. Molecular components of the circadian clock in mammals. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 6–11. [Google Scholar] [CrossRef]
- Raghuram, S.; Stayrook, K.R.; Huang, P.; Rogers, P.M.; Nosie, A.K.; McClure, D.B.; Burris, L.L.; Khorasanizadeh, S.; Burris, T.P.; Rastinejad, F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat. Struct. Mol. Biol. 2007, 14, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yin, L.; Hanniman, E.A.; Joshi, S.; Lazar, M.A. Negative feedback maintenance of heme homeostasis by its receptor, Rev-erbalpha. Genes Dev. 2009, 23, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Scrima, R.; Cela, O.; Merla, G.; Augello, B.; Rubino, R.; Quarato, G.; Fugetto, S.; Menga, M.; Fuhr, L.; Relogio, A.; et al. Clock-genes and mitochondrial respiratory activity: Evidence of a reciprocal interplay. Biochim. Biophys. Acta 2016, 1857, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Mullebner, A.; Dorighello, G.G.; Kozlov, A.V.; Duvigneau, J.C. Interaction between Mitochondrial Reactive Oxygen Species, Heme Oxygenase, and Nitric Oxide Synthase Stimulates Phagocytosis in Macrophages. Front. Med. (Lausanne) 2017, 4, 252. [Google Scholar] [CrossRef]
- Zhang, T.; Bu, P.; Zeng, J.; Vancura, A. Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 2017, 292, 16942–16954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.H.; Chiang, M.T.; Chau, L.Y. Ubiquitin-proteasome system mediates heme oxygenase-1 degradation through endoplasmic reticulum-associated degradation pathway. Biochim. Biophys. Acta 2008, 1783, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Broca, C.; Varin, E.; Armanet, M.; Tourrel-Cuzin, C.; Bosco, D.; Dalle, S.; Wojtusciszyn, A. Proteasome dysfunction mediates high glucose-induced apoptosis in rodent beta cells and human islets. PLoS ONE 2014, 9, e92066. [Google Scholar] [CrossRef]
- Dong, C.; Zheng, H.; Huang, S.; You, N.; Xu, J.; Ye, X.; Zhu, Q.; Feng, Y.; You, Q.; Miao, H.; et al. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp. Cell Res. 2015, 337, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.S.; Figueiredo-Pereira, C.; Vieira, H.L. Carbon monoxide and mitochondria-modulation of cell metabolism, redox response and cell death. Front. Physiol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braud, L.; Pini, M.; Muchova, L.; Manin, S.; Kitagishi, H.; Sawaki, D.; Czibik, G.; Ternacle, J.; Derumeaux, G.; Foresti, R.; et al. Carbon monoxide-induced metabolic switch in adipocytes improves insulin resistance in obese mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Yoshida, T.; Kikuchi, G. Purification and properties of heme oxygenase from pig spleen microsomes. J. Biol. Chem. 1978, 253, 4224–4229. [Google Scholar]
- Wagner, G.; Lindroos-Christensen, J.; Einwallner, E.; Husa, J.; Zapf, T.C.; Lipp, K.; Rauscher, S.; Groger, M.; Spittler, A.; Loewe, R.; et al. HO-1 inhibits preadipocyte proliferation and differentiation at the onset of obesity via ROS dependent activation of Akt2. Sci. Rep. 2017, 7, 40881. [Google Scholar] [CrossRef]
- Gordon, S.; Pluddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a Target for Therapeutic Interventions. Front. Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, T.D.; Agarwal, A.; George, J.F. The mononuclear phagocyte system in homeostasis and disease: A role for heme oxygenase-1. Antioxid. Redox. Signal. 2014, 20, 1770–1788. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Chau, L.Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Wagener, F.A.; Yachie, A.; Wiegerinck, E.T.; Kemna, E.H.; Swinkels, D.W. Hepcidin suppression and defective iron recycling account for dysregulation of iron homeostasis in heme oxygenase-1 deficiency. J. Cell Mol. Med. 2009, 13, 3091–3102. [Google Scholar] [CrossRef]
- Puri, N.; Arefiev, Y.; Chao, R.; Sacerdoti, D.; Chaudry, H.; Nichols, A.; Srikanthan, K.; Nawab, A.; Sharma, D.; Lakhani, V.H.; et al. Heme Oxygenase Induction Suppresses Hepatic Hepcidin and Rescues Ferroportin and Ferritin Expression in Obese Mice. J. Nutr. Metab. 2017, 2017, 4964571. [Google Scholar] [CrossRef]
- Philip, M.; Chiu, E.Y.; Hajjar, A.M.; Abkowitz, J.L. TLR Stimulation Dynamically Regulates Heme and Iron Export Gene Expression in Macrophages. J. Immunol. Res. 2016, 2016, 4039038. [Google Scholar] [CrossRef]
- Khan, A.A.; Quigley, J.G. Control of intracellular heme levels: Heme transporters and heme oxygenases. Biochim. Biophys. Acta 2011, 1813, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Kortman, G.A.; Mulder, M.L.; Richters, T.J.; Shanmugam, N.K.; Trebicka, E.; Boekhorst, J.; Timmerman, H.M.; Roelofs, R.; Wiegerinck, E.T.; Laarakkers, C.M.; et al. Low dietary iron intake restrains the intestinal inflammatory response and pathology of enteric infection by food-borne bacterial pathogens. Eur. J. Immunol. 2015, 45, 2553–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Mosser, D.M. Macrophage activation by endogenous danger signals. J. Pathol. 2008, 214, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed]
- McCoubrey, W.K.; Eke, B., Jr.; Maines, M.D. Multiple transcripts encoding heme oxygenase-2 in rat testis: Developmental and cell-specific regulation of transcripts and protein. Biol. Reprod. 1995, 53, 1330–1338. [Google Scholar] [CrossRef]
- McCoubrey, W.K.; Huang, T.J., Jr.; Maines, M.D. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J. Biol. Chem. 1997, 272, 12568–12574. [Google Scholar] [CrossRef]
- Ragsdale, S.W.; Yi, L. Thiol/Disulfide redox switches in the regulation of heme binding to proteins. Antioxid. Redox. Signal. 2011, 14, 1039–1047. [Google Scholar] [CrossRef]
- Yi, L.; Jenkins, P.M.; Leichert, L.I.; Jakob, U.; Martens, J.R.; Ragsdale, S.W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 2009, 284, 20556–20561. [Google Scholar] [CrossRef]
- Boehning, D.; Moon, C.; Sharma, S.; Hurt, K.J.; Hester, L.D.; Ronnett, G.V.; Shugar, D.; Snyder, S.H. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron 2003, 40, 129–137. [Google Scholar] [CrossRef]
- Boehning, D.; Sedaghat, L.; Sedlak, T.W.; Snyder, S.H. Heme oxygenase-2 is activated by calcium-calmodulin. J. Biol. Chem. 2004, 279, 30927–30930. [Google Scholar] [CrossRef]
- Weng, Y.H.; Yang, G.; Weiss, S.; Dennery, P.A. Interaction between heme oxygenase-1 and -2 proteins. J. Biol. Chem. 2003, 278, 50999–51005. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di, D.F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef] [Green Version]
- Kinobe, R.; Ji, Y.; Nakatsu, K. Peroxynitrite-mediated inactivation of heme oxygenases. BMC Pharmacol. 2004, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: Tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA 1987, 84, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef]
- Chang, E.F.; Wong, R.J.; Vreman, H.J.; Igarashi, T.; Galo, E.; Sharp, F.R.; Stevenson, D.K.; Noble-Haeusslein, L.J. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J. Neurosci. 2003, 23, 3689–3696. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways. Antioxidants 2019, 8, 475. https://doi.org/10.3390/antiox8100475
Duvigneau JC, Esterbauer H, Kozlov AV. Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways. Antioxidants. 2019; 8(10):475. https://doi.org/10.3390/antiox8100475
Chicago/Turabian StyleDuvigneau, J. Catharina, Harald Esterbauer, and Andrey V. Kozlov. 2019. "Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways" Antioxidants 8, no. 10: 475. https://doi.org/10.3390/antiox8100475